
The Therapeutic Strategies Targeting Mitochondrial Metabolism in Cardiovascular Disease


Abstract
:1. Introduction
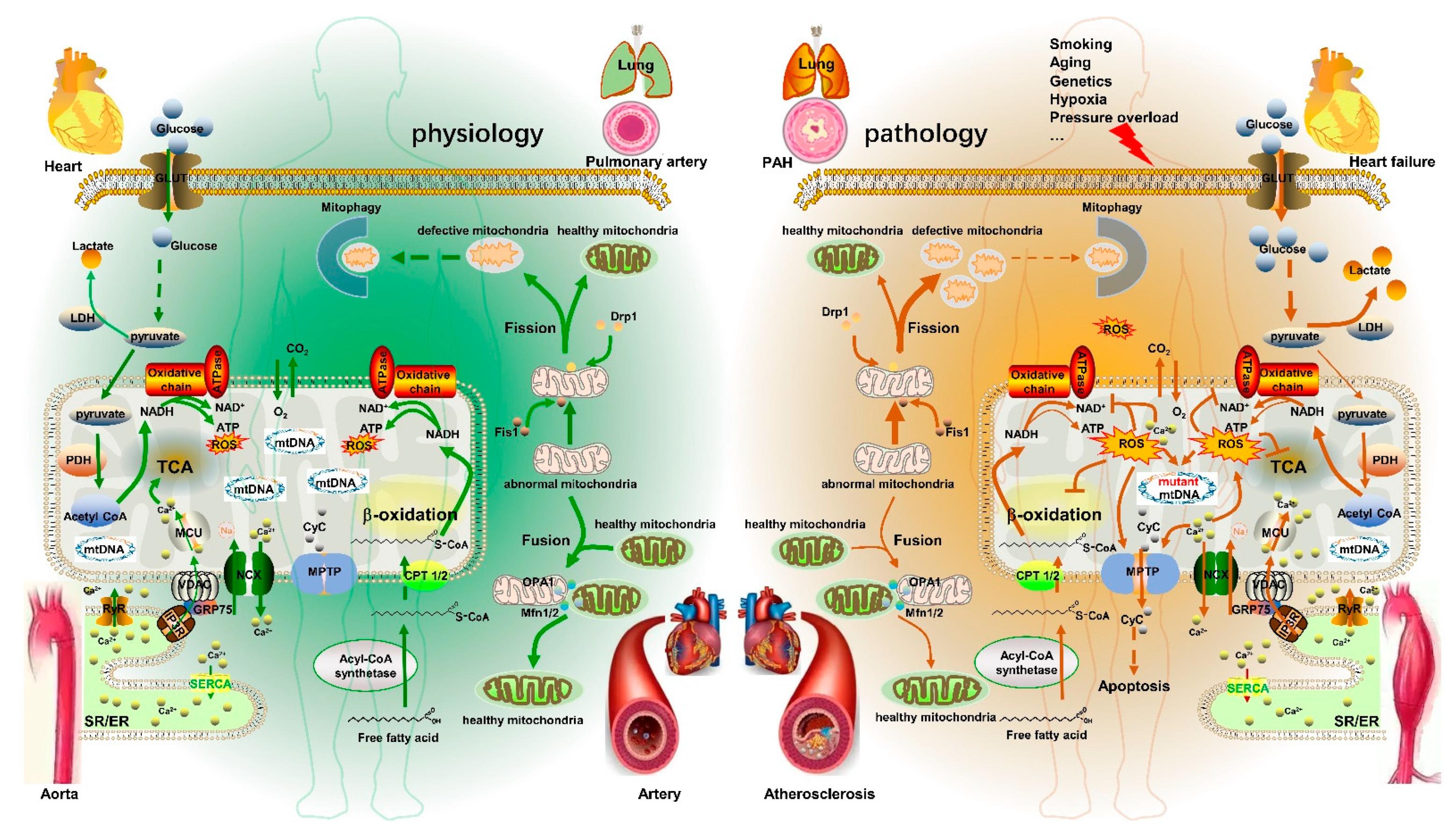
2. Mitochondrial Metabolism Dysfunction in CVD
3. Major Factors in Mitochondrial Metabolism
3.1. Mitochondrial DNA
3.2. Mitochondrial Dynamics
3.2.1. Mitochondrial Fusion
3.2.2. Mitochondrial Fission
3.3. Mitochondrial Calcium
3.3.1. Mitochondrial Calcium Transport
3.3.2. [Ca2+]m and Mitochondrial Metabolism
3.4. ROS
4. Mitochondrial Metabolism Disorder and CVD
4.1. PAH
4.2. Aortic Aneurysm
4.3. Atherosclerosis
4.4. Heart Failure
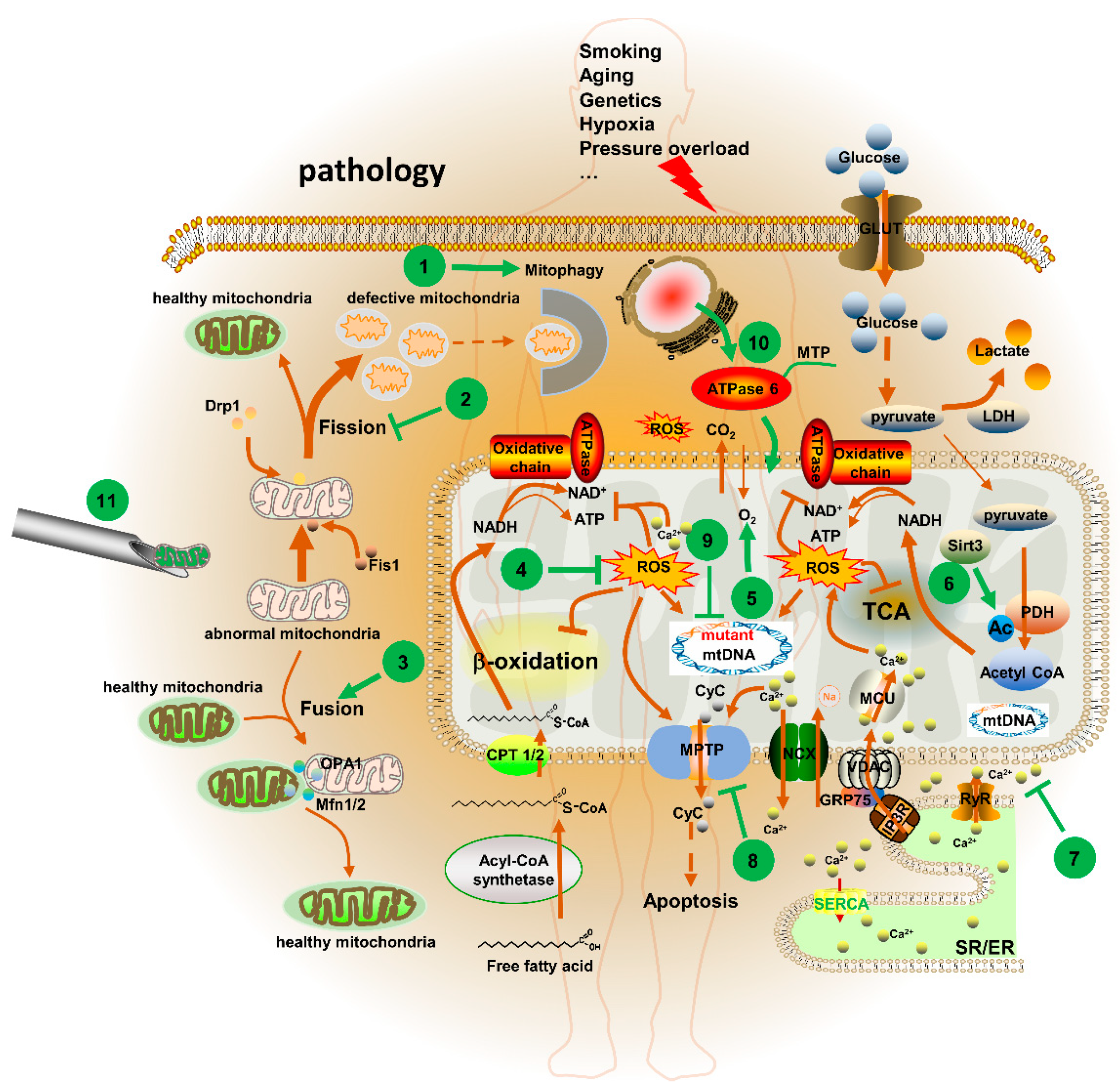
5. Mitochondria-Targeted Therapeutic Agents and Strategies in CVD
5.1. Potential Mitochondrial Targets for CVD
5.1.1. MPTP
5.1.2. Sirtuin 3 (Sirt3)
5.1.3. [Ca2+]m
5.1.4. Mitochondrial Dynamics
5.2. Mitochondria-Targeted Agents for CVD
5.2.1. CoQ10 and MitoQ
5.2.2. Melatonin
5.2.3. SS-31
5.2.4. MitoTEMPOL
5.2.5. MitoSNO
5.3. Mitochondria-Targeted Gene Therapy Strategies
5.3.1. Mitochondria-Targeted Gene Editing Technologies
5.3.2. Ectopic Expression of Mitochondrial Proteins
5.3.3. Mitochondrial Replacement Therapy (MRT) Technologies
6. Prospection
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: Update from the GBD 2019 study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Javadov, S. Association between ROS production, swelling and the respirasome integrity in cardiac mitochondria. Arch. Biochem. Biophys. 2017, 630, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, P.; Luo, J.; Huang, Y.; He, L.; Yang, H.; Li, Q.; Wu, S.; Zhelyabovska, O.; Yang, Q. Peroxisome proliferator-activated receptor β/δ activation in adult hearts facilitates mitochondrial function and cardiac performance under pressure-overload condition. Hypertension 2011, 57, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, P.; Dunmore, B.J.; Schlosser, K.; Schoors, S.; Dos Santos, C.; Perez-Iratxeta, C.; Lavoie, J.R.; Zhang, H.; Long, L.; Flockton, A.R.; et al. Identification of MicroRNA-124 as a major regulator of enhanced endothelial cell glycolysis in pulmonary arterial hypertension via PTBP1 (polypyrimidine tract binding protein) and pyruvate kinase M2. Circulation 2017, 136, 2451–2467. [Google Scholar] [CrossRef]
- Oller, J.; Gabandé-Rodríguez, E.; Roldan-Montero, R.; Ruiz-Rodríguez, M.J.; Redondo, J.M.; Martín-Ventura, J.L.; Mittelbrunn, M. Rewiring vascular metabolism prevents sudden death due to aortic ruptures-brief report. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 462–469. [Google Scholar] [CrossRef]
- Matsuura, Y.; Shimizu-Albergine, M.; Barnhart, S.; Kramer, F.; Hsu, C.-C.; Kothari, V.; Tang, J.; Gharib, S.A.; Kanter, J.E.; Abel, E.D.; et al. Diabetes suppresses glucose uptake and glycolysis in macrophages. Circ. Res. 2022, 130, 779–781. [Google Scholar] [CrossRef]
- Wei, Y.; Corbalán-Campos, J.; Gurung, R.; Natarelli, L.; Zhu, M.; Exner, N.; Erhard, F.; Greulich, F.; Geißler, C.; Uhlenhaut, N.H.; et al. Dicer in macrophages prevents atherosclerosis by promoting mitochondrial oxidative metabolism. Circulation 2018, 138, 2007–2020. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Energy conversion: Mitochondria and chloroplasts. In The Molecular Biology of the Cell, 4th ed.; Gibbs, S., Ed.; Garland Science: New York, NY, USA, 2002; p. 769. [Google Scholar]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing mitochondrial dysfunction in disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef]
- Fijalkowska, I.; Xu, W.; Comhair, S.A.A.; Janocha, A.J.; Mavrakis, L.A.; Krishnamachary, B.; Zhen, L.; Mao, T.; Richter, A.; Erzurum, S.C.; et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am. J. Pathol. 2010, 176, 1130–1138. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, W.; Lian, G.; Huang, B.; Du, A.; Gong, J.; Xiao, G.; Xu, C.; Wang, H.; Xie, L. CPT1 regulates the proliferation of pulmonary artery smooth muscle cells through the AMPK-p53-p21 pathway in pulmonary arterial hypertension. Mol. Cell. Biochem. 2019, 455, 169–183. [Google Scholar] [CrossRef]
- Wall, V.Z.; Barnhart, S.; Kanter, J.E.; Kramer, F.; Shimizu-Albergine, M.; Adhikari, N.; Wight, T.N.; Hall, J.L.; Bornfeldt, K.E. Smooth muscle glucose metabolism promotes monocyte recruitment and atherosclerosis in a mouse model of metabolic syndrome. JCI Insight 2018, 3, e96544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Houston, B. A comprehensive review of the bioenergetics of fatty acid and glucose metabolism in the healthy and failing heart in nondiabetic condition. Heart Fail. Rev. 2017, 22, 825–842. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef]
- Kauppila, J.H.K.; Stewart, J.B. Mitochondrial DNA: Radically free of free-radical driven mutations. Biochim. Biophys. Acta 2015, 1847, 1354–1361. [Google Scholar] [CrossRef] [Green Version]
- Kauppila, J.H.K.; Bonekamp, N.A.; Mourier, A.; Isokallio, M.A.; Just, A.; Kauppila, T.E.S.; Stewart, J.B.; Larsson, N.-G. Base-excision repair deficiency alone or combined with increased oxidative stress does not increase mtDNA point mutations in mice. Nucleic Acids Res. 2018, 46, 6642–6669. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Sainz, A.G.; Shadel, G.S. Mitochondrial DNA: Cellular genotoxic stress sentinel. Trends Biochem. Sci. 2021, 46, 812–821. [Google Scholar] [CrossRef]
- Kiyooka, T.; Ohanyan, V.; Yin, L.; Pung, Y.F.; Chen, Y.-R.; Chen, C.-L.; Kang, P.T.; Hardwick, J.P.; Yun, J.; Janota, D.; et al. Mitochondrial DNA integrity and function are critical for endothelium-dependent vasodilation in rats with metabolic syndrome. Basic Res. Cardiol. 2022, 117, 3. [Google Scholar] [CrossRef]
- Lechuga-Vieco, A.V.; Latorre-Pellicer, A.; Calvo, E.; Torroja, C.; Pellico, J.; Acín-Pérez, R.; García-Gil, M.L.; Santos, A.; Bagwan, N.; Bonzon-Kulichenko, E.; et al. Heteroplasmy of wild-type mitochondrial DNA variants in mice causes metabolic heart disease with pulmonary hypertension and frailty. Circulation 2022, 145, 1084–1101. [Google Scholar] [CrossRef]
- Fazzini, F.; Lamina, C.; Raftopoulou, A.; Koller, A.; Fuchsberger, C.; Pattaro, C.; Del Greco, F.M.; Döttelmayer, P.; Fendt, L.; Fritz, J.; et al. Association of mitochondrial DNA copy number with metabolic syndrome and type 2 diabetes in 14,176 individuals. J. Intern. Med. 2021, 290, 190–202. [Google Scholar] [CrossRef]
- Hong, Y.S.; Longchamps, R.J.; Zhao, D.; Castellani, C.A.; Loehr, L.R.; Chang, P.P.; Matsushita, K.; Grove, M.L.; Boerwinkle, E.; Arking, D.E.; et al. Mitochondrial DNA copy number and incident heart failure: The atherosclerosis risk in communities (ARIC) study. Circulation 2020, 141, 1823–1825. [Google Scholar] [CrossRef]
- Koller, A.; Fazzini, F.; Lamina, C.; Rantner, B.; Kollerits, B.; Stadler, M.; Klein-Weigel, P.; Fraedrich, G.; Kronenberg, F. Mitochondrial DNA copy number is associated with all-cause mortality and cardiovascular events in patients with peripheral arterial disease. J. Intern. Med. 2020, 287, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashar, F.N.; Zhang, Y.; Longchamps, R.J.; Lane, J.; Moes, A.; Grove, M.L.; Mychaleckyj, J.C.; Taylor, K.D.; Coresh, J.; Rotter, J.I.; et al. Association of mitochondrial DNA copy number with cardiovascular disease. JAMA Cardiol. 2017, 2, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021, 178, 2060–2076. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, F.; Yan, P.; Zhang, S.; Lou, Y.; Geng, Z.; Li, Z.; Zhang, Y.; Xu, Y.; Lu, Y.; et al. LARP7 protects against heart failure by enhancing mitochondrial biogenesis. Circulation 2021, 143, 2007–2022. [Google Scholar] [CrossRef]
- Zhuang, L.; Jia, K.; Chen, C.; Li, Z.; Zhao, J.; Hu, J.; Zhang, H.; Fan, Q.; Huang, C.; Xie, H.; et al. DYRK1B-STAT3 drives cardiac hypertrophy and heart failure by impairing mitochondrial bioenergetics. Circulation 2022, 145, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Kalkhoran, S.B.; Kriston-Vizi, J.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Rosdah, A.A.; Lees, J.G.; Da Costa, J.R.S.; Ling, N.X.Y.; Holien, J.K.; Samangouei, P.; et al. Hydralazine protects the heart against acute ischaemia/reperfusion injury by inhibiting Drp1-mediated mitochondrial fission. Cardiovasc. Res. 2022, 118, 282–294. [Google Scholar] [CrossRef]
- Cooper, H.A.; Cicalese, S.; Preston, K.J.; Kawai, T.; Okuno, K.; Choi, E.T.; Kasahara, S.; Uchida, H.A.; Otaka, N.; Scalia, R.; et al. Targeting mitochondrial fission as a potential therapeutic for abdominal aortic aneurysm. Cardiovasc. Res. 2021, 117, 971–982. [Google Scholar] [CrossRef]
- Chen, K.-H.; Dasgupta, A.; Lin, J.; Potus, F.; Bonnet, S.; Iremonger, J.; Fu, J.; Mewburn, J.; Wu, D.; Dunham-Snary, K.; et al. Epigenetic dysregulation of the dynamin-related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and promotes pulmonary arterial hypertension: Mechanistic and therapeutic implications. Circulation 2018, 138, 287–304. [Google Scholar] [CrossRef]
- Xiong, P.Y.; Tian, L.; Dunham-Snary, K.J.; Chen, K.-H.; Mewburn, J.D.; Neuber-Hess, M.; Martin, A.; Dasgupta, A.; Potus, F.; Archer, S.L. Biventricular increases in mitochondrial fission mediator (MiD51) and proglycolytic pyruvate kinase (PKM2) isoform in experimental group 2 pulmonary hypertension-novel mitochondrial abnormalities. Front. Cardiovasc. Med. 2018, 5, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendin, D.; Filadi, R.; Pizzo, P. The concerted action of mitochondrial dynamics and positioning: New characters in cancer onset and progression. Front. Oncol. 2017, 7, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wai, T.; Langer, T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Li, T.; Han, J.; Jia, L.; Hu, X.; Chen, L.; Wang, Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell 2019, 10, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem. Sci. 2001, 26, 23–29. [Google Scholar] [CrossRef]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [Green Version]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.-C. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 2008, 3, e3257. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Long, Q.; Liu, J.; Tang, H.; Li, Y.; Bao, F.; Qin, D.; Pei, D.; Liu, X. Mitochondrial fusion provides an ‘initial metabolic complementation’ controlled by mtDNA. Cell. Mol. Life Sci. 2015, 72, 2585–2598. [Google Scholar] [CrossRef]
- Delerue, T.; Tribouillard-Tanvier, D.; Daloyau, M.; Khosrobakhsh, F.; Emorine, L.J.; Friocourt, G.; Belenguer, P.; Blondel, M.; Arnauné-Pelloquin, L. A yeast-based screening assay identifies repurposed drugs that suppress mitochondrial fusion and mtDNA maintenance defects. Dis. Model. Mech. 2019, 12, dmm036558. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhao, X.; Li, X.; Lv, S.; Ma, R.; Qi, Y.; Abulikemu, A.; Duan, H.; Guo, C.; Li, Y.; et al. PM2.5 triggered apoptosis in lung epithelial cells through the mitochondrial apoptotic way mediated by a ROS-DRP1-mitochondrial fission axis. J. Hazard. Mater. 2020, 397, 122608. [Google Scholar] [CrossRef]
- Chan, C.-M.; Sekar, P.; Huang, D.-Y.; Hsu, S.-H.; Lin, W.-W. Different effects of metformin and A769662 on sodium iodate-induced cytotoxicity in retinal pigment epithelial cells: Distinct actions on mitochondrial fission and respiration. Antioxidants 2020, 9, 1057. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.; Grimm, A.; Dallmann, R.; Oettinghaus, B.; Restelli, L.M.; Witzig, M.; Ishihara, N.; Mihara, K.; Ripperger, J.A.; Albrecht, U.; et al. Circadian control of DRP1 activity regulates mitochondrial dynamics and bioenergetics. Cell Metab. 2018, 27, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Han, Y.; Gu, X.; Li, M.; Du, Y.; Feng, N.; Li, J.; Zhang, S.; Maslov, L.N.; Wang, G.; et al. Paeonol promotes Opa1-mediated mitochondrial fusion via activating the CK2α-Stat3 pathway in diabetic cardiomyopathy. Redox Biol. 2021, 46, 102098. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Rajapurohitam, V.; Kilić, A.; Hunter, J.C.; Zeidan, A.; Said Faruq, N.; Escobales, N.; Karmazyn, M. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: The effect of NHE-1 inhibition. Basic Res. Cardiol. 2011, 106, 99–109. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbah, H.N.; Gupta, R.C.; Singh-Gupta, V.; Zhang, K.; Lanfear, D.E. Abnormalities of mitochondrial dynamics in the failing heart: Normalization following long-term therapy with elamipretide. Cardiovasc. Drugs Ther. 2018, 32, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Simpson, A.W.; Brini, M.; Pozzan, T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 1992, 358, 325–327. [Google Scholar] [CrossRef]
- Young, M.J.; Bay, D.C.; Hausner, G.; Court, D.A. The evolutionary history of mitochondrial porins. BMC Evol. Biol. 2007, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; de Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca2+ fluxes and cancer. Mol. Cell 2020, 78, 1055–1069. [Google Scholar] [CrossRef]
- Vais, H.; Payne, R.; Paudel, U.; Li, C.; Foskett, J.K. Coupled transmembrane mechanisms control MCU-mediated mitochondrial Ca2+ uptake. Proc. Natl. Acad. Sci. USA 2020, 117, 21731–21739. [Google Scholar] [CrossRef] [PubMed]
- Magi, S.; Piccirillo, S.; Preziuso, A.; Amoroso, S.; Lariccia, V. Mitochondrial localization of NCXs: Balancing calcium and energy homeostasis. Cell Calcium 2020, 86, 102162. [Google Scholar] [CrossRef] [PubMed]
- Tykocki, N.R.; Jackson, W.F.; Watts, S.W. Reverse-mode Na+/Ca2+ exchange is an important mediator of venous contraction. Pharmacol. Res. 2012, 66, 544–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.; Liu, J. Mitochondrial calcium and reactive oxygen species in cardiovascular disease. Cardiovasc. Res. 2022, 2022, cvac134. [Google Scholar] [CrossRef]
- Shankar, T.S.; Ramadurai, D.K.A.; Steinhorst, K.; Sommakia, S.; Badolia, R.; Thodou Krokidi, A.; Calder, D.; Navankasattusas, S.; Sander, P.; Kwon, O.S.; et al. Cardiac-specific deletion of voltage dependent anion channel 2 leads to dilated cardiomyopathy by altering calcium homeostasis. Nat. Commun. 2021, 12, 4583. [Google Scholar] [CrossRef]
- Hong, Z.; Chen, K.-H.; Dasgupta, A.; Potus, F.; Dunham-Snary, K.; Bonnet, S.; Tian, L.; Fu, J.; Breuils-Bonnet, S.; Provencher, S.; et al. MicroRNA-138 and MicroRNA-25 down-regulate mitochondrial calcium uniporter, causing the pulmonary arterial hypertension cancer phenotype. Am. J. Respir. Crit. Care Med. 2017, 195, 515–529. [Google Scholar] [CrossRef] [Green Version]
- Honrath, B.; Matschke, L.; Meyer, T.; Magerhans, L.; Perocchi, F.; Ganjam, G.K.; Zischka, H.; Krasel, C.; Gerding, A.; Bakker, B.M.; et al. SK2 channels regulate mitochondrial respiration and mitochondrial Ca2+ uptake. Cell Death Differ. 2017, 24, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Assali, E.A.; Jones, A.E.; Veliova, M.; Acín-Pérez, R.; Taha, M.; Miller, N.; Shum, M.; Oliveira, M.F.; Las, G.; Liesa, M.; et al. NCLX prevents cell death during adrenergic activation of the brown adipose tissue. Nat. Commun. 2020, 11, 3347. [Google Scholar] [CrossRef]
- Oropeza-Almazán, Y.; Vázquez-Garza, E.; Chapoy-Villanueva, H.; Torre-Amione, G.; García-Rivas, G. Small interfering RNA targeting mitochondrial calcium uniporter improves cardiomyocyte cell viability in hypoxia/reoxygenation injury by reducing calcium overload. Oxid. Med. Cell. Longev. 2017, 2017, 5750897. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.-C.; Lien, C.-F.; Lee, W.-S.; Chang, H.-R.; Hsu, Y.-C.; Luo, Y.-P.; Jeng, J.-R.; Hsieh, J.-C.; Yang, K.-T. Intermittent hypoxia prevents myocardial mitochondrial Ca2+ overload and cell death during ischemia/reperfusion: The role of reactive oxygen species. Cells 2019, 8, 564. [Google Scholar] [CrossRef]
- Zanou, N.; Dridi, H.; Reiken, S.; Imamura de Lima, T.; Donnelly, C.; de Marchi, U.; Ferrini, M.; Vidal, J.; Sittenfeld, L.; Feige, J.N.; et al. Acute RyR1 Ca2+ leak enhances NADH-linked mitochondrial respiratory capacity. Nat. Commun. 2021, 12, 7219. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of ATP production by mitochondrial Ca(2+). Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [Green Version]
- McCormack, J.G.; Denton, R.M. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 1979, 180, 533–544. [Google Scholar] [CrossRef]
- Denton, R.M.; McCormack, J.G.; Edgell, N.J. Role of calcium ions in the regulation of intramitochondrial metabolism. Effects of Na+, Mg2+ and ruthenium red on the Ca2+-stimulated oxidation of oxoglutarate and on pyruvate dehydrogenase activity in intact rat heart mitochondria. Biochem. J. 1980, 190, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatrinet, R.; Leone, G.; de Luise, M.; Girolimetti, G.; Vidone, M.; Gasparre, G.; Porcelli, A.M. The α-ketoglutarate dehydrogenase complex in cancer metabolic plasticity. Cancer Metab. 2017, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheeran, F.L.; Angerosa, J.; Liaw, N.Y.; Cheung, M.M.; Pepe, S. Adaptations in protein expression and regulated activity of pyruvate dehydrogenase multienzyme complex in human systolic heart failure. Oxid. Med. Cell. Longev. 2019, 2019, 4532592. [Google Scholar] [CrossRef]
- Liu, T.; O’Rourke, B. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J. Bioenerg. Biomembr. 2009, 41, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Denton, R.M.; Richards, D.A.; Chin, J.G. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem. J. 1978, 176, 899–906. [Google Scholar] [CrossRef]
- Lukács, G.L.; Kapus, A.; Fonyó, A. Parallel measurement of oxoglutarate dehydrogenase activity and matrix free Ca2+ in fura-2-loaded heart mitochondria. FEBS Lett. 1988, 229, 219–223. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Waldeck-Weiermair, M.; Bourguignon, M.-P.; Villeneuve, N.; Gottschalk, B.; Klec, C.; Stryeck, S.; Radulovic, S.; Parichatikanond, W.; Frank, S.; et al. Enhanced inter-compartmental Ca2+ flux modulates mitochondrial metabolism and apoptotic threshold during aging. Redox Biol. 2019, 20, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.P.; Luongo, T.S.; Tomar, D.; Jadiya, P.; Gao, E.; Zhang, X.; Lucchese, A.M.; Kolmetzky, D.W.; Shah, N.S.; Elrod, J.W. MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation 2019, 140, 1720–1733. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Wang, J.; Qiu, T.; Wu, J.; An, Y.; Shi, X.; Sun, X.; Jiang, L.; Liu, X.; Yang, G.; et al. Perfluorooctane sulfonate induces mitochondrial calcium overload and early hepatic insulin resistance via autophagy/detyrosinated alpha-tubulin-regulated IP3R2-VDAC1-MICU1 interaction. Sci. Total Environ. 2022, 825, 153933. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.B.; Qin, F.; Morgan, R.J.; Chambers, J.M.; Croteau, D.; Siwik, D.A.; Hobai, I.; Panagia, M.; Luptak, I.; Bachschmid, M.; et al. Redox-resistant SERCA sarco(endo)plasmic reticulum calcium ATPase attenuates oxidant-stimulated mitochondrial calcium and apoptosis in cardiac myocytes and pressure overload-induced myocardial failure in mice. Circulation 2020, 142, 2459–2469. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhang, X.; Xing, P.; Zhang, S.; Zhang, F.; Wang, J.; Yu, J.; Zhu, X.; Chang, P. Grpel2 alleviates myocardial ischemia/reperfusion injury by inhibiting MCU-mediated mitochondrial calcium overload. Biochem. Biophys. Res. Commun. 2022, 609, 169–175. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Fu, A.; Robson-Doucette, C.; Allister, E.M.; Wheeler, M.B.; Screaton, R.; Harper, M.-E. Glutathionylation state of uncoupling protein-2 and the control of glucose-stimulated insulin secretion. J. Biol. Chem. 2012, 287, 39673–39685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Bériault, R.; Lemire, J.; Singh, R.; Chénier, D.R.; Hamel, R.D.; Appanna, V.D. The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS ONE 2007, 2, e690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef]
- Kehrer, J.P.; Klotz, L.-O. Free radicals and related reactive species as mediators of tissue injury and disease: Implications for Health. Crit. Rev. Toxicol. 2015, 45, 765–798. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, C.D.; Wu, R.F.; Terada, L.S. ROS signaling and ER stress in cardiovascular disease. Mol. Aspects Med. 2018, 63, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [Green Version]
- Flint, D.H.; Tuminello, J.F.; Emptage, M.H. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 1993, 268, 22369–22376. [Google Scholar] [CrossRef]
- Mailloux, R.J.; McBride, S.L.; Harper, M.-E. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem. Sci. 2013, 38, 592–602. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Xin, Y.; Tian, G.; Zhou, J.; Liu, X. Mitochondrial ROS-modulated mtDNA: A potential target for cardiac aging. Oxid. Med. Cell. Longev. 2020, 2020, 9423593. [Google Scholar] [CrossRef] [Green Version]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, Y.; Jiang, X.; Zhang, H.; Gao, Z.; Li, Y.; Fu, R.; Li, L.; Li, J.; Cui, H.; et al. ROS-mediated activation and mitochondrial translocation of CaMKII contributes to Drp1-dependent mitochondrial fission and apoptosis in triple-negative breast cancer cells by isorhamnetin and chloroquine. J. Exp. Clin. Cancer Res. 2019, 38, 225. [Google Scholar] [CrossRef]
- Su, L.; Zhang, J.; Gomez, H.; Kellum, J.A.; Peng, Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 2022, 1–14. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- D’Alessandro, A.; El Kasmi, K.C.; Plecitá-Hlavatá, L.; Ježek, P.; Li, M.; Zhang, H.; Gupte, S.A.; Stenmark, K.R. Hallmarks of pulmonary hypertension: Mesenchymal and inflammatory cell metabolic reprogramming. Antioxid. Redox Signal. 2018, 28, 230–250. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Janocha, A.J.; Erzurum, S.C. Metabolism in pulmonary hypertension. Annu. Rev. Physiol. 2021, 83, 551–576. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Comhair, S.A.A.; Chen, R.; Hu, B.; Hou, Y.; Zhou, Y.; Mavrakis, L.A.; Janocha, A.J.; Li, L.; Zhang, D.; et al. Integrative proteomics and phosphoproteomics in pulmonary arterial hypertension. Sci. Rep. 2019, 9, 18623. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Peng, J.; Lu, C.; Hsin, M.; Mura, M.; Wu, L.; Chu, L.; Zamel, R.; Machuca, T.; Waddell, T.; et al. Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS ONE 2014, 9, e88727. [Google Scholar] [CrossRef]
- Lee, M.H.; Sanders, L.; Kumar, R.; Hernandez-Saavedra, D.; Yun, X.; Ford, J.A.; Perez, M.J.; Mickael, C.; Gandjeva, A.; Koyanagi, D.E.; et al. Contribution of fatty acid oxidation to the pathogenesis of pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 323, L355–L371. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.B.; Michelakis, E.D. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci. Transl. Med. 2010, 2, 44ra58. [Google Scholar] [CrossRef] [PubMed]
- Can, M.M.; Kaymaz, C.; Tanboga, I.H.; Tokgoz, H.C.; Canpolat, N.; Turkyilmaz, E.; Sonmez, K.; Ozdemir, N. Increased right ventricular glucose metabolism in patients with pulmonary arterial hypertension. Clin. Nucl. Med. 2011, 36, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Brittain, E.L.; Trammell, A.W.; Fessel, J.P.; Austin, E.D.; Penner, N.; Maynard, K.B.; Gleaves, L.; Talati, M.; Absi, T.; et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Talati, M.H.; Brittain, E.L.; Fessel, J.P.; Penner, N.; Atkinson, J.; Funke, M.; Grueter, C.; Jerome, W.G.; Freeman, M.; Newman, J.H.; et al. Mechanisms of lipid accumulation in the bone morphogenetic protein receptor type 2 mutant right ventricle. Am. J. Respir. Crit. Care Med. 2016, 194, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Lu, Q.; Yegambaram, M.; Kumar, S.; Qu, N.; Srivastava, A.; Wang, T.; Fineman, J.R.; Black, S.M. TGF-β1 attenuates mitochondrial bioenergetics in pulmonary arterial endothelial cells via the disruption of carnitine homeostasis. Redox Biol. 2020, 36, 101593. [Google Scholar] [CrossRef]
- Gupte, S.A.; Wolin, M.S. Mitochondrial Calcium Transport: A potentially prominent, therapeutically targetable contributor to pulmonary arterial hypertension progression. Am. J. Respir. Crit. Care Med. 2017, 195, 420–421. [Google Scholar] [CrossRef]
- Parra, V.; Bravo-Sagua, R.; Norambuena-Soto, I.; Hernández-Fuentes, C.P.; Gómez-Contreras, A.G.; Verdejo, H.E.; Mellado, R.; Chiong, M.; Lavandero, S.; Castro, P.F. Inhibition of mitochondrial fission prevents hypoxia-induced metabolic shift and cellular proliferation of pulmonary arterial smooth muscle cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2891–2903. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, J.; Liu, H.; Ma, W.; Yu, L.; Tan, X.; Wang, S.; Ren, F.; Li, X.; Li, X. Cannabidiol attenuates pulmonary arterial hypertension by improving vascular smooth muscle cells mitochondrial function. Theranostics 2021, 11, 5267–5278. [Google Scholar] [CrossRef]
- Tian, L.; Wu, D.; Dasgupta, A.; Chen, K.-H.; Mewburn, J.; Potus, F.; Lima, P.D.A.; Hong, Z.; Zhao, Y.-Y.; Hindmarch, C.C.T.; et al. Epigenetic metabolic reprogramming of right ventricular fibroblasts in pulmonary arterial hypertension: A pyruvate dehydrogenase kinase-dependent shift in mitochondrial metabolism promotes right ventricular fibrosis. Circ. Res. 2020, 126, 1723–1745. [Google Scholar] [CrossRef]
- Laine, M.T.; Laukontaus, S.J.; Sund, R.; Aho, P.S.; Kantonen, I.; Albäck, A.; Venermo, M. A population-based study of abdominal aortic aneurysm treatment in finland 2000 to 2014. Circulation 2017, 136, 1726–1734. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, M.; Wang, M.; Yu, B. Aberrant mitochondrial dynamics: An emerging pathogenic driver of abdominal aortic aneurysm. Cardiovasc. Ther. 2021, 2021, 6615400. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.-Y.; Lyu, Y.-Y.; Zhang, H.-Y.; Shen, Z.; Lin, G.-Q.; Geng, N.; Wang, Y.-L.; Huang, L.; Feng, Z.-H.; Guo, X.; et al. Nuclear receptor NR1D1 regulates abdominal aortic aneurysm development by targeting the mitochondrial tricarboxylic acid cycle enzyme aconitase-2. Circulation 2022, 146, 1591–1609. [Google Scholar] [CrossRef] [PubMed]
- van der Pluijm, I.; Burger, J.; van Heijningen, P.M.; IJpma, A.; van Vliet, N.; Milanese, C.; Schoonderwoerd, K.; Sluiter, W.; Ringuette, L.-J.; Dekkers, D.H.W.; et al. Decreased mitochondrial respiration in aneurysmal aortas of Fibulin-4 mutant mice is linked to PGC1A regulation. Cardiovasc. Res. 2018, 114, 1776–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ren, P.; Dawson, A.; Vasquez, H.G.; Ageedi, W.; Zhang, C.; Luo, W.; Chen, R.; Li, Y.; Kim, S.; et al. Single-cell transcriptome analysis reveals dynamic cell populations and differential gene expression patterns in control and aneurysmal human aortic tissue. Circulation 2020, 142, 1374–1388. [Google Scholar] [CrossRef]
- Oller, J.; Gabandé-Rodríguez, E.; Ruiz-Rodríguez, M.J.; Desdín-Micó, G.; Aranda, J.F.; Rodrigues-Diez, R.; Ballesteros-Martínez, C.; Blanco, E.M.; Roldan-Montero, R.; Acuña, P.; et al. Extracellular tuning of mitochondrial respiration leads to aortic aneurysm. Circulation 2021, 143, 2091–2109. [Google Scholar] [CrossRef]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. the road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, M.; Huang, W.; Chen, W.; Zhao, Y.; Schulte, M.L.; Volberding, P.; Gerbec, Z.; Zimmermann, M.T.; Zeighami, A.; et al. Mitochondrial metabolic reprogramming by CD36 signaling drives macrophage inflammatory responses. Circ. Res. 2019, 125, 1087–1102. [Google Scholar] [CrossRef]
- Pircher, A.; Treps, L.; Bodrug, N.; Carmeliet, P. Endothelial cell metabolism: A novel player in atherosclerosis? Basic principles and therapeutic opportunities. Atherosclerosis 2016, 253, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Xu, R.-H.; Liu, B.; Wu, J.-D.; Yan, Y.-Y.; Wang, J.-N. miR-143 is involved in endothelial cell dysfunction through suppression of glycolysis and correlated with atherosclerotic plaques formation. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4063–4071. [Google Scholar]
- Liu, L.; Lu, Y.; Martinez, J.; Bi, Y.; Lian, G.; Wang, T.; Milasta, S.; Wang, J.; Yang, M.; Liu, G.; et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1α-dependent. Proc. Natl. Acad. Sci. USA 2016, 113, 1564–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawakol, A.; Singh, P.; Mojena, M.; Pimentel-Santillana, M.; Emami, H.; MacNabb, M.; Rudd, J.H.F.; Narula, J.; Enriquez, J.A.; Través, P.G.; et al. HIF-1α and PFKFB3 mediate a tight relationship between proinflammatory activation and anerobic metabolism in atherosclerotic macrophages. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1463–1471. [Google Scholar] [CrossRef] [Green Version]
- Ouimet, M.; Ediriweera, H.N.; Gundra, U.M.; Sheedy, F.J.; Ramkhelawon, B.; Hutchison, S.B.; Rinehold, K.; van Solingen, C.; Fullerton, M.D.; Cecchini, K.; et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J. Clin. Investig. 2015, 125, 4334–4348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinnouchi, H.; Guo, L.; Sakamoto, A.; Torii, S.; Sato, Y.; Cornelissen, A.; Kuntz, S.; Paek, K.H.; Fernandez, R.; Fuller, D.; et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell. Mol. Life Sci. 2020, 77, 1919–1932. [Google Scholar] [CrossRef]
- Kumar, V.; Santhosh Kumar, T.R.; Kartha, C.C. Mitochondrial membrane transporters and metabolic switch in heart failure. Heart Fail. Rev. 2019, 24, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac energy metabolism in heart failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.-H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef]
- O’Rourke, B.; Ashok, D.; Liu, T. Mitochondrial Ca2+ in heart failure: Not enough or too much? J. Mol. Cell. Cardiol. 2021, 151, 126–134. [Google Scholar] [CrossRef]
- Knowlton, A.A.; Liu, T.T. Mitochondrial dynamics and heart failure. Compr. Physiol. 2015, 6, 507–526. [Google Scholar] [PubMed]
- Wu, C.; Zhang, Z.; Zhang, W.; Liu, X. Mitochondrial dysfunction and mitochondrial therapies in heart failure. Pharmacol. Res. 2022, 175, 106038. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Calcium signaling and reactive oxygen species in mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yang, N.; Sidor, A.; O’Rourke, B. MCU overexpression rescues inotropy and reverses heart failure by reducing SR Ca2+ leak. Circ. Res. 2021, 128, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhou, S.; Zhang, X.; Li, C.; Ji, S.; Mao, H.; Jiang, X. Mitochondrion-specific dendritic lipopeptide liposomes for targeted sub-cellular delivery. Nat. Commun. 2021, 12, 2390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-X.; Cheng, Y.; Liu, D.-Z.; Liu, M.; Cui, H.; Zhang, B.; Mei, Q.-B.; Zhou, S.-Y. Mitochondria-targeted cyclosporin A delivery system to treat myocardial ischemia reperfusion injury of rats. J. Nanobiotechn. 2019, 17, 18. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-S.; Chang, J.-C.; Kuo, S.-J.; Liu, K.-H.; Lin, T.-T.; Cheng, W.-L.; Chuang, S.-F. Delivering healthy mitochondria for the therapy of mitochondrial diseases and beyond. Int. J. Biochem. Cell Biol. 2014, 53, 141–146. [Google Scholar] [CrossRef]
- Nakazato, I.; Okuno, M.; Zhou, C.; Itoh, T.; Tsutsumi, N.; Takenaka, M.; Arimura, S.-I. Targeted base editing in the mitochondrial genome of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2022, 119, e2121177119. [Google Scholar] [CrossRef]
- Zinovkin, R.A.; Zamyatnin, A.A. Mitochondria-targeted drugs. Curr. Mol. Pharmacol. 2019, 12, 202–214. [Google Scholar] [CrossRef]
- Shi, H.; Tang, H.; Ai, W.; Zeng, Q.; Yang, H.; Zhu, F.; Wei, Y.; Feng, R.; Wen, L.; Pu, P.; et al. Schisandrin B antagonizes cardiotoxicity induced by pirarubicin by inhibiting mitochondrial permeability transition pore (mPTP) opening and decreasing cardiomyocyte apoptosis. Front. Pharmacol. 2021, 12, 733805. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Wu, S.-P.; Wang, N.; Seto, S.; Chang, D. Hydroxysafflor yellow A alleviates cerebral ischemia reperfusion injury by suppressing apoptosis via mitochondrial permeability transition pore. Phytomedicine 2021, 85, 153532. [Google Scholar] [CrossRef] [PubMed]
- Godoy, J.A.; Rios, J.A.; Picón-Pagès, P.; Herrera-Fernández, V.; Swaby, B.; Crepin, G.; Vicente, R.; Fernández-Fernández, J.M.; Muñoz, F.J. Mitostasis, calcium and free radicals in health, aging and neurodegeneration. Biomolecules 2021, 11, 1012. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-X.; Cui, S.-M.; Zhang, Y.-M.; Ren, J. Mitochondrial Ca2+ regulation in the etiology of heart failure: Physiological and pathophysiological implications. Acta Pharmacol. Sin. 2020, 41, 1301–1309. [Google Scholar] [CrossRef]
- Schriewer, J.M.; Peek, C.B.; Bass, J.; Schumacker, P.T. ROS-mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia-reperfusion. J. Am. Heart Assoc. 2013, 2, e000159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eirin, A.; Li, Z.; Zhang, X.; Krier, J.D.; Woollard, J.R.; Zhu, X.-Y.; Tang, H.; Herrmann, S.M.; Lerman, A.; Textor, S.C.; et al. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension 2012, 60, 1242–1249. [Google Scholar] [CrossRef] [Green Version]
- Bernal-Ramírez, J.; Silva-Platas, C.; Jerjes-Sánchez, C.; Ramos-González, M.R.; Vázquez-Garza, E.; Chapoy-Villanueva, H.; Ramírez-Rivera, A.; Zarain-Herzberg, Á.; García, N.; García-Rivas, G. Resveratrol prevents right ventricle dysfunction, calcium mishandling, and energetic failure via SIRT3 stimulation in pulmonary arterial hypertension. Oxid. Med. Cell. Longev. 2021, 2021, 9912434. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Halestrap, A.P. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J. Mol. Cell. Cardiol. 1993, 25, 1461–1469. [Google Scholar] [CrossRef]
- Ikeda, G.; Matoba, T.; Nakano, Y.; Nagaoka, K.; Ishikita, A.; Nakano, K.; Funamoto, D.; Sunagawa, K.; Egashira, K. Nanoparticle-mediated targeting of cyclosporine A enhances cardioprotection against ischemia-reperfusion injury through inhibition of mitochondrial permeability transition pore opening. Sci. Rep. 2016, 6, 20467. [Google Scholar] [CrossRef] [Green Version]
- Parodi-Rullán, R.M.; Chapa-Dubocq, X.R.; Javadov, S. Acetylation of mitochondrial proteins in the heart: The role of SIRT3. Front. Physiol. 2018, 9, 1094. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Nagasawa, K.; Münch, C.; Xu, Y.; Satterstrom, K.; Jeong, S.; Hayes, S.D.; Jedrychowski, M.P.; Vyas, F.S.; Zaganjor, E.; et al. Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell 2016, 167, 985–1000.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wang, Y.; Shen, L. Mitochondrial proteins in heart failure: The role of deacetylation by SIRT3. Pharmacol. Res. 2021, 172, 105802. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, S.; Zhang, B.; Liu, J. SIRT3 as a potential therapeutic target for heart failure. Pharmacol. Res. 2021, 165, 105432. [Google Scholar] [CrossRef]
- Sun, W.; Liu, C.; Chen, Q.; Liu, N.; Yan, Y.; Liu, B. SIRT3: A new regulator of cardiovascular diseases. Oxid. Med. Cell. Longev. 2018, 2018, 7293861. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Chen, J.; Liu, M.; Chen, Y.; Wu, Y.; Li, Q.; Ma, T.; Gao, J.; Xia, Y.; Fan, M.; et al. Protective effect of HINT2 on mitochondrial function via repressing MCU complex activation attenuates cardiac microvascular ischemia-reperfusion injury. Basic Res. Cardiol. 2021, 116, 65. [Google Scholar] [CrossRef]
- Wu, N.N.; Bi, Y.; Ajoolabady, A.; You, F.; Sowers, J.; Wang, Q.; Ceylan, A.F.; Zhang, Y.; Ren, J. Parkin insufficiency accentuates high-fat diet-induced cardiac remodeling and contractile dysfunction through VDAC1-mediated mitochondrial Ca2+ overload. JACC Basic Transl. Sci. 2022, 7, 779–796. [Google Scholar] [CrossRef]
- Motegi, K.; Tanonaka, K.; Takenaga, Y.; Takagi, N.; Takeo, S. Preservation of mitochondrial function may contribute to cardioprotective effects of Na+/Ca2+ exchanger inhibitors in ischaemic/reperfused rat hearts. Br. J. Pharmacol. 2007, 151, 963–978. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.-Y.; Wei, X.-X.; Zhi, X.-L.; Wang, X.-H.; Meng, D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol. Sin. 2021, 42, 655–664. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, B.; Zhang, J.; He, D.; Zhang, Q.; Pan, C.; Yuan, Q.; Shi, Y.; Tang, H.; Xu, F.; et al. ALDH2 (Aldehyde Dehydrogenase 2) Protects Against Hypoxia-Induced Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2303–2319. [Google Scholar] [CrossRef]
- Huang, G.; Cong, Z.; Wang, X.; Yuan, Y.; Xu, R.; Lu, Z.; Wang, X.; Qi, J. Targeting HSP90 attenuates angiotensin II-induced adventitial remodelling via suppression of mitochondrial fission. Cardiovasc. Res. 2020, 116, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Feng, N.; Tang, D.; Feng, J.; Li, Z.; Jia, M.; Liu, Z.; Gu, X.; Wang, Y.; Fu, F.; et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J. Pineal Res. 2018, 65, e12491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabanal-Ruiz, Y.; Llanos-González, E.; Alcain, F.J. The Use of Coenzyme Q10 in Cardiovascular Diseases. Antioxidants 2021, 10, 755. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Suo, H.; Song, J. Protective role of mitoquinone against impaired mitochondrial homeostasis in metabolic syndrome. Crit. Rev. Food Sci. Nutr. 2021, 61, 3857–3875. [Google Scholar] [CrossRef]
- Masoumi-Ardakani, Y.; Najafipour, H.; Nasri, H.R.; Aminizadeh, S.; Jafari, S.; Moflehi, D. Effect of combined endurance training and MitoQ on cardiac function and serum level of antioxidants, NO, miR-126, and miR-27a in hypertensive individuals. Biomed Res. Int. 2022, 2022, 8720661. [Google Scholar] [CrossRef]
- Goh, K.Y.; He, L.; Song, J.; Jinno, M.; Rogers, A.J.; Sethu, P.; Halade, G.V.; Rajasekaran, N.S.; Liu, X.; Prabhu, S.D.; et al. Mitoquinone ameliorates pressure overload-induced cardiac fibrosis and left ventricular dysfunction in mice. Redox Biol. 2019, 21, 101100. [Google Scholar] [CrossRef]
- Ribeiro Junior, R.F.; Dabkowski, E.R.; Shekar, K.C.; Connell, K.A.O.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef]
- Méndez, D.; Arauna, D.; Fuentes, F.; Araya-Maturana, R.; Palomo, I.; Alarcón, M.; Sebastián, D.; Zorzano, A.; Fuentes, E. Mitoquinone (MitoQ) inhibits platelet activation steps by reducing ROS levels. Int. J. Mol. Sci. 2020, 21, 6192. [Google Scholar] [CrossRef]
- Cui, L.; Zhou, Q.; Zheng, X.; Sun, B.; Zhao, S. Mitoquinone attenuates vascular calcification by suppressing oxidative stress and reducing apoptosis of vascular smooth muscle cells via the Keap1/Nrf2 pathway. Free Radic. Biol. Med. 2020, 161, 23–31. [Google Scholar] [CrossRef]
- Maun, H.R.; Liu, P.S.; Franke, Y.; Eigenbrot, C.; Forrest, W.F.; Schwartz, L.B.; Lazarus, R.A. Dual functionality of β-tryptase protomers as both proteases and cofactors in the active tetramer. J. Biol. Chem. 2018, 293, 9614–9628. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Pekas, E.J.; Headid, R.J.; Son, W.-M.; Wooden, T.K.; Song, J.; Layec, G.; Yadav, S.K.; Mishra, P.K.; Pipinos, I.I. Acute mitochondrial antioxidant intake improves endothelial function, antioxidant enzyme activity, and exercise tolerance in patients with peripheral artery disease. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H456–H467. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Jiang, H.; Bigham, Z.; Zaldumbide, J.; Huetsch, J.C.; Kliment, C.; Acoba, M.G.; Kirsch, B.J.; Claypool, S.M.; et al. Regulation of mitochondrial fragmentation in microvascular endothelial cells isolated from the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L639–L652. [Google Scholar] [CrossRef] [PubMed]
- Pandi-Perumal, S.R.; Trakht, I.; Srinivasan, V.; Spence, D.W.; Maestroni, G.J.M.; Zisapel, N.; Cardinali, D.P. Physiological effects of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol. 2008, 85, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; García, J.A.; Escames, G.; Ortiz, F.; López, A.; Doerrier, C.; García-Corzo, L.; López, L.C.; Reiter, R.J.; Acuña-Castroviejo, D. Extrapineal melatonin: Analysis of its subcellular distribution and daily fluctuations. J. Pineal Res. 2012, 52, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Nduhirabandi, F.; Maarman, G.J. Melatonin in heart failure: A promising therapeutic strategy? Molecules 2018, 23, 1819. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Ma, Q.; Zhu, P.; Ren, J.; Reiter, R.J.; Chen, Y. Protective role of melatonin in cardiac ischemia-reperfusion injury: From pathogenesis to targeted therapy. J. Pineal Res. 2018, 64, e12471. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Sun, C.; Zhu, H.; Zhai, M.; Zhang, L.; Jiang, L.; Hou, P.; Li, J.; Li, K.; Liu, Z.; et al. Melatonin protects against thoracic aortic aneurysm and dissection through SIRT1-dependent regulation of oxidative stress and vascular smooth muscle cell loss. J. Pineal Res. 2020, 69, e12661. [Google Scholar] [CrossRef]
- Duan, L.; Li, S.; Wang, L.; Jing, Y.; Li, G.; Sun, Y.; Sun, W.; Li, Y.; Zhao, L.; Xin, S. Melatonin plays a critical protective role in nicotine-related abdominal aortic aneurysm. Front. Physiol. 2020, 11, 866. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Bi, Y.; McClements, D.J.; Lip, G.Y.H.; Des Richardson, R.; Reiter, R.J.; Klionsky, D.J.; Ren, J. Melatonin-based therapeutics for atherosclerotic lesions and beyond: Focusing on macrophage mitophagy. Pharmacol. Res. 2022, 176, 106072. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, X.; Liu, M.; Fan, H.; Zheng, H.; Zhang, S.; Rahman, N.; Wołczyński, S.; Kretowski, A.; Li, X. Melatonin inhibits inflammasome-associated activation of endothelium and macrophages attenuating pulmonary arterial hypertension. Cardiovasc. Res. 2020, 116, 2156–2169. [Google Scholar] [CrossRef]
- MacLean, M.R. Melatonin: Shining some light on pulmonary hypertension. Cardiovasc. Res. 2020, 116, 2036–2037. [Google Scholar] [CrossRef] [PubMed]
- Arinno, A.; Maneechote, C.; Khuanjing, T.; Ongnok, B.; Prathumsap, N.; Chunchai, T.; Arunsak, B.; Kerdphoo, S.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; et al. Cardioprotective effects of melatonin and metformin against doxorubicin-induced cardiotoxicity in rats are through preserving mitochondrial function and dynamics. Biochem. Pharmacol. 2021, 192, 114743. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ma, Z.; Di, S.; Yang, Y.; Yang, J.; Xu, L.; Reiter, R.J.; Qiao, S.; Yuan, J. AMPK/PGC1α activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic. Biol. Med. 2018, 129, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Singhanat, K.; Apaijai, N.; Sumneang, N.; Maneechote, C.; Arunsak, B.; Chunchai, T.; Chattipakorn, S.C.; Chattipakorn, N. Therapeutic potential of a single-dose melatonin in the attenuation of cardiac ischemia/reperfusion injury in prediabetic obese rats. Cell. Mol. Life Sci. 2022, 79, 300. [Google Scholar] [CrossRef]
- Lan, H.; Su, Y.; Liu, Y.; Deng, C.; Wang, J.; Chen, T.; Jules, K.E.D.; Masau, J.F.; Li, H.; Wei, X. Melatonin protects circulatory death heart from ischemia/reperfusion injury via the JAK2/STAT3 signalling pathway. Life Sci. 2019, 228, 35–46. [Google Scholar] [CrossRef]
- Ma, W.-Y.; Song, R.-J.; Xu, B.-B.; Xu, Y.; Wang, X.-X.; Sun, H.-Y.; Li, S.-N.; Liu, S.-Z.; Yu, M.-X.; Yang, F.; et al. Melatonin promotes cardiomyocyte proliferation and heart repair in mice with myocardial infarction via miR-143-3p/Yap/Ctnnd1 signaling pathway. Acta Pharmacol. Sin. 2021, 42, 921–931. [Google Scholar] [CrossRef]
- Wen, L.; Wang, M.; Luo, P.; Meng, X.; Zhao, M. Melatonin exerts cardioprotective Effects by inhibiting NLRP3 inflammasome-induced pyroptosis in mice following myocardial infarction. Oxid. Med. Cell. Longev. 2021, 2021, 5387799. [Google Scholar] [CrossRef]
- Jiao, L.; Wang, Y.; Zhang, S.; Wang, Y.; Liu, Z.; Liu, Z.; Zhou, Y.; Zhou, H.; Xu, X.; Li, Z.; et al. Melatonin improves cardiac remodeling and brain-heart sympathetic hyperactivation aggravated by light disruption after myocardial infarction. J. Pineal Res. 2022, 73, e12829. [Google Scholar] [CrossRef]
- Xu, C.-N.; Kong, L.-H.; Ding, P.; Liu, Y.; Fan, Z.-G.; Gao, E.-H.; Yang, J.; Yang, L.-F. Melatonin ameliorates pressure overload-induced cardiac hypertrophy by attenuating Atg5-dependent autophagy and activating the Akt/mTOR pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165848. [Google Scholar] [CrossRef]
- Odinokova, I.; Baburina, Y.; Kruglov, A.; Fadeeva, I.; Zvyagina, A.; Sotnikova, L.; Akatov, V.; Krestinina, O. Effect of melatonin on rat heart mitochondria in acute aeart failure in aged rats. Int. J. Mol. Sci. 2018, 19, 1555. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Lin, N.; Sheng, X.; Zhao, Y.; Su, Y.; Xu, L.; Tong, R.; Yan, Y.; Fu, Y.; He, J.; et al. Melatonin stabilizes rupture-prone vulnerable plaques via regulating macrophage polarization in a nuclear circadian receptor RORα-dependent manner. J. Pineal Res. 2019, 67, e12581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, X.; Bai, X.; Lin, Y.; Li, Z.; Fu, J.; Li, M.; Zhao, T.; Yang, H.; Xu, R.; et al. Melatonin prevents endothelial cell pyroptosis via regulation of long noncoding RNA MEG3/miR-223/NLRP3 axis. J. Pineal Res. 2018, 64, e12449. [Google Scholar] [CrossRef]
- Li, H.-Y.; Leu, Y.-L.; Wu, Y.-C.; Wang, S.-H. Melatonin inhibits in vitro smooth muscle cell inflammation and proliferation and atherosclerosis in apolipoprotein E-deficient mice. J. Agric. Food Chem. 2019, 67, 1889–1901. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin ameliorates the progression of atherosclerosis via mitophagy activation and NLRP3 inflammasome inhibition. Oxid. Med. Cell. Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef]
- Xie, Y.; Lou, D.; Zhang, D. Melatonin alleviates age-associated endothelial injury of atherosclerosis via regulating telomere function. J. Inflamm. Res. 2021, 14, 6799–6812. [Google Scholar] [CrossRef] [PubMed]
- Tekin, G.; İsbir, S.; Şener, G.; Çevik, Ö.; Çetinel, Ş.; Dericioğlu, O.; Arsan, S.; Çobanoğlu, A. The preventive and curative effects of melatonin against abdominal aortic aneurysm in rats. J. Vasc. Surg. 2018, 67, 1546–1555. [Google Scholar] [CrossRef]
- Wang, R.; Pan, J.; Han, J.; Gong, M.; Liu, L.; Zhang, Y.; Liu, Y.; Wang, D.; Tang, Q.; Wu, N.; et al. Melatonin attenuates dasatinib-aggravated hypoxic pulmonary hypertension via inhibiting pulmonary vascular remodeling. Front. Cardiovasc. Med. 2022, 9, 790921. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, P.; Li, Y.; Shen, W.; Wang, C.; Zhao, P.; Cui, H.; Xue, J.-Y.; Du, G.-Q. Melatonin activates the Mst1-Nrf2 signaling to alleviate cardiac hypertrophy in pulmonary arterial hypertension. Eur. J. Pharmacol. 2022, 933, 175262. [Google Scholar] [CrossRef]
- Figueroa, E.G.; Gonzaléz-Candia, A.; Villanueva, C.A.; Ebensperger, G.; Reyes, R.V.; Llanos, A.J.; Herrera, E.A. Beneficial effects of melatonin on prostanoids pathways in pulmonary hypertensive neonates. Vascul. Pharmacol. 2021, 138, 106853. [Google Scholar] [CrossRef]
- Raygan, F.; Ostadmohammadi, V.; Bahmani, F.; Reiter, R.J.; Asemi, Z. Melatonin administration lowers biomarkers of oxidative stress and cardio-metabolic risk in type 2 diabetic patients with coronary heart disease: A randomized, double-blind, placebo-controlled trial. Clin. Nutr. 2019, 38, 191–196. [Google Scholar] [CrossRef]
- Cerrato, C.P.; Pirisinu, M.; Vlachos, E.N.; Langel, Ü. Novel cell-penetrating peptide targeting mitochondria. FASEB J. 2015, 29, 4589–4599. [Google Scholar] [CrossRef] [PubMed]
- Whitson, J.A.; Bitto, A.; Zhang, H.; Sweetwyne, M.T.; Coig, R.; Bhayana, S.; Shankland, E.G.; Wang, L.; Bammler, T.K.; Mills, K.F.; et al. SS-31 and NMN: Two paths to improve metabolism and function in aged hearts. Aging Cell 2020, 19, e13213. [Google Scholar] [CrossRef] [PubMed]
- Chiao, Y.A.; Zhang, H.; Sweetwyne, M.; Whitson, J.; Ting, Y.S.; Basisty, N.; Pino, L.K.; Quarles, E.; Nguyen, N.-H.; Campbell, M.D.; et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. Elife 2020, 9, e55513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Alder, N.N.; Wang, W.; Szeto, H.; Marcinek, D.J.; Rabinovitch, P.S. Reduction of elevated proton leak rejuvenates mitochondria in the aged cardiomyocyte. Elife 2020, 9, e60827. [Google Scholar] [CrossRef]
- Whitson, J.A.; Martín-Pérez, M.; Zhang, T.; Gaffrey, M.J.; Merrihew, G.E.; Huang, E.; White, C.C.; Kavanagh, T.J.; Qian, W.-J.; Campbell, M.D.; et al. Elamipretide (SS-31) treatment attenuates age-associated post-translational modifications of heart proteins. Geroscience 2021, 43, 2395–2412. [Google Scholar] [CrossRef]
- Whitson, J.A.; Johnson, R.; Wang, L.; Bammler, T.K.; Imai, S.-I.; Zhang, H.; Fredrickson, J.; Latorre-Esteves, E.; Bitto, A.; MacCoss, M.J.; et al. Age-related disruption of the proteome and acetylome in mouse hearts is associated with loss of function and attenuated by elamipretide (SS-31) and nicotinamide mononucleotide (NMN) treatment. Geroscience 2022, 44, 1621–1639. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.-F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yang, W.; Sun, X.; Xie, L.; Yang, Y.; Sang, M.; Jiao, R. SS31 ameliorates sepsis-induced heart Injury by inhibiting oxidative stress and inflammation. Inflammation 2019, 42, 2170–2180. [Google Scholar] [CrossRef]
- Aimo, A.; Castiglione, V.; Borrelli, C.; Saccaro, L.F.; Franzini, M.; Masi, S.; Emdin, M.; Giannoni, A. Oxidative stress and inflammation in the evolution of heart failure: From pathophysiology to therapeutic strategies. Eur. J. Prev. Cardiol. 2020, 27, 494–510. [Google Scholar] [CrossRef]
- Machiraju, P.; Wang, X.; Sabouny, R.; Huang, J.; Zhao, T.; Iqbal, F.; King, M.; Prasher, D.; Lodha, A.; Jimenez-Tellez, N.; et al. SS-31 peptide reverses the mitochondrial fragmentation present in fibroblasts from patients with DCMA, a mitochondrial cardiomyopathy. Front. Cardiovasc. Med. 2019, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.-Y.; Shao, P.-L.; Wallace, C.G.; Chua, S.; Sung, P.-H.; Ko, S.-F.; Chai, H.-T.; Chung, S.-Y.; Chen, K.-H.; Lu, H.-I.; et al. Combined therapy with SS31 and mitochondria mitigates myocardial ischemia-reperfusion injury in rats. Int. J. Mol. Sci. 2018, 19, 2782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Feng, M.; Wang, X.; Zhang, H.; Ding, J.; Cheng, Z.; Qian, L. Peptide Szeto-Schiller 31 ameliorates doxorubicin-induced cardiotoxicity by inhibiting the activation of the p38 MAPK signaling pathway. Int. J. Mol. Med. 2021, 47, 63. [Google Scholar] [CrossRef]
- Yeh, J.-N.; Sung, P.-H.; Chiang, J.Y.; Sheu, J.-J.; Huang, C.-R.; Chu, Y.-C.; Chua, S.; Yip, H.-K. Early treatment with combination of SS31 and entresto effectively preserved the heart function in doxorubicin-induced dilated cardiomyopathic rat. Biomed. Pharmacother. 2021, 141, 111886. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhao, H.; Cai, J.; Li, H.; Wu, Q.; Qiao, T.; Li, K. Chronic administration of mitochondrion-targeted peptide SS-31 prevents atherosclerotic development in ApoE knockout mice fed Western diet. PLoS ONE 2017, 12, e0185688. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-I.; Huang, T.-H.; Sung, P.-H.; Chen, Y.-L.; Chua, S.; Chai, H.-Y.; Chung, S.-Y.; Liu, C.-F.; Sun, C.-K.; Chang, H.-W.; et al. Administration of antioxidant peptide SS-31 attenuates transverse aortic constriction-induced pulmonary arterial hypertension in mice. Acta Pharmacol. Sin. 2016, 37, 589–603. [Google Scholar] [CrossRef] [Green Version]
- Chatfield, K.C.; Sparagna, G.C.; Chau, S.; Phillips, E.K.; Ambardekar, A.V.; Aftab, M.; Mitchell, M.B.; Sucharov, C.C.; Miyamoto, S.D.; Stauffer, B.L. Elamipretide improves mitochondrial function in the failing human heart. JACC Basic Transl. Sci. 2019, 4, 147–157. [Google Scholar] [CrossRef]
- Hortmann, M.; Robinson, S.; Mohr, M.; Mauler, M.; Stallmann, D.; Reinöhl, J.; Duerschmied, D.; Peter, K.; Carr, J.; Gibson, C.M.; et al. The mitochondria-targeting peptide elamipretide diminishes circulating HtrA2 in ST-segment elevation myocardial infarction. Eur. Heart J. Acute Cardiovasc. Care 2019, 8, 695–702. [Google Scholar] [CrossRef]
- Gibson, C.M.; Giugliano, R.P.; Kloner, R.A.; Bode, C.; Tendera, M.; Jánosi, A.; Merkely, B.; Godlewski, J.; Halaby, R.; Korjian, S.; et al. EMBRACE STEMI study: A Phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur. Heart J. 2016, 37, 1296–1303. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.; Khan, M.S.; Anker, S.D.; Fonarow, G.C.; Kim, R.J.; Nodari, S.; O’Connor, C.M.; Pieske, B.; Pieske-Kraigher, E.; Sabbah, H.N.; et al. Effects of elamipretide on left ventricular function in patients with heart failure with reduced ejection fraction: The PROGRESS-HF phase 2 trial. J. Card. Fail. 2020, 26, 429–437. [Google Scholar] [CrossRef]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, A.; Budin, S.B.; Mohd Fauzi, N.; Ritchie, R.H.; Zainalabidin, S. Targeting mitochondrial reactive oxygen species-mediated oxidative stress attenuates nicotine-induced cardiac remodeling and dysfunction. Sci. Rep. 2021, 11, 13845. [Google Scholar] [CrossRef] [PubMed]
- Vendrov, A.E.; Stevenson, M.D.; Alahari, S.; Pan, H.; Wickline, S.A.; Madamanchi, N.R.; Runge, M.S. Attenuated superoxide dismutase 2 activity induces atherosclerotic plaque instability during aging in hyperlipidemic mice. J. Am. Heart Assoc. 2017, 6, e006775. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, Z.; Dong, Z.; Wang, C.; Cao, Q.; Fan, F.; Zhao, J.; Liu, X.; Yuan, M.; Sun, X.; et al. Aldehyde dehydrogenase 2 deficiency promotes atherosclerotic plaque instability through accelerating mitochondrial ROS-mediated vascular smooth muscle cell senescence. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1782–1792. [Google Scholar] [CrossRef]
- Ma, Y.; Huang, Z.; Zhou, Z.; He, X.; Wang, Y.; Meng, C.; Huang, G.; Fang, N. A novel antioxidant Mito-Tempol inhibits ox-LDL-induced foam cell formation through restoration of autophagy flux. Free Radic. Biol. Med. 2018, 129, 463–472. [Google Scholar] [CrossRef]
- Moncada, S.; Erusalimsky, J.D. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell Biol. 2002, 3, 214–220. [Google Scholar] [CrossRef]
- Dahm, C.C.; Moore, K.; Murphy, M.P. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: Implications for the interaction of nitric oxide with mitochondria. J. Biol. Chem. 2006, 281, 10056–10065. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Hurd, T.R.; Nadtochiy, S.M.; Brookes, P.S.; Fearnley, I.M.; Lilley, K.S.; Smith, R.A.J.; Murphy, M.P. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): Implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem. J. 2010, 430, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Shiva, S.; Sack, M.N.; Greer, J.J.; Duranski, M.; Ringwood, L.A.; Burwell, L.; Wang, X.; MacArthur, P.H.; Shoja, A.; Raghavachari, N.; et al. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J. Exp. Med. 2007, 204, 2089–2102. [Google Scholar] [CrossRef] [Green Version]
- Duranski, M.R.; Greer, J.J.M.; Dejam, A.; Jaganmohan, S.; Hogg, N.; Langston, W.; Patel, R.P.; Yet, S.-F.; Wang, X.; Kevil, C.G.; et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J. Clin. Investig. 2005, 115, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cochemé, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Methner, C.; Chouchani, E.T.; Buonincontri, G.; Pell, V.R.; Sawiak, S.J.; Murphy, M.P.; Krieg, T. Mitochondria selective S-nitrosation by mitochondria-targeted S-nitrosothiol protects against post-infarct heart failure in mouse hearts. Eur. J. Heart Fail. 2014, 16, 712–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uittenbogaard, M.; Brantner, C.A.; Fang, Z.; Wong, L.-J.C.; Gropman, A.; Chiaramello, A. Novel insights into the functional metabolic impact of an apparent de novo m.8993TG variant in the MT-ATP6 gene associated with maternally inherited form of Leigh Syndrome. Mol. Genet. Metab. 2018, 124, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Kauppila, J.H.K.; Pereira, C.V.; Nissanka, N.; Miranda, M.; Pinto, M.; Williams, S.L.; Larsson, N.-G.; Stewart, J.B.; Moraes, C.T. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018, 24, 1696–1700. [Google Scholar] [CrossRef]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef]
- Jo, A.; Ham, S.; Lee, G.H.; Lee, Y.-I.; Kim, S.; Lee, Y.-S.; Shin, J.-H.; Lee, Y. Efficient mitochondrial genome editing by CRISPR/Cas9. Biomed Res. Int. 2015, 2015, 305716. [Google Scholar] [CrossRef] [Green Version]
- Mok, B.Y.; Kotrys, A.V.; Raguram, A.; Huang, T.P.; Mootha, V.K.; Liu, D.R. CRISPR-free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat. Biotechnol. 2022, 40, 1378–1387. [Google Scholar] [CrossRef]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef]
- Manfredi, G.; Fu, J.; Ojaimi, J.; Sadlock, J.E.; Kwong, J.Q.; Guy, J.; Schon, E.A. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat. Genet. 2002, 30, 394–399. [Google Scholar] [CrossRef]
- Tachibana, M.; Sparman, M.; Sritanaudomchai, H.; Ma, H.; Clepper, L.; Woodward, J.; Li, Y.; Ramsey, C.; Kolotushkina, O.; Mitalipov, S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009, 461, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.-Y.; Guo, L.; Chen, L.-N.; Yin, S.; Wen, J.; Li, S.; Ma, J.-Y.; Jing, T.; Jiang, M.-X.; Sun, X.-H.; et al. Reduction of mtDNA heteroplasmy in mitochondrial replacement therapy by inducing forced mitophagy. Nat. Biomed. Eng. 2022, 6, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Kitani, T.; Kami, D.; Kawasaki, T.; Nakata, M.; Matoba, S.; Gojo, S. Direct human mitochondrial transfer: A novel concept based on the endosymbiotic theory. Transplant. Proc. 2014, 46, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Carl, S.M.; Swerdlow, R.H. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2014, 2, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef]
- Wu, T.-H.; Sagullo, E.; Case, D.; Zheng, X.; Li, Y.; Hong, J.S.; TeSlaa, T.; Patananan, A.N.; McCaffery, J.M.; Niazi, K.; et al. Mitochondrial transfer by photothermal nanoblade restores metabolite profile in mammalian cells. Cell Metab. 2016, 23, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zhong, J.; Wang, L.; Chen, Y. Mitochondrial transfer in cardiovascular disease: From mechanisms to therapeutic implications. Front. Cardiovasc. Med. 2021, 8, 1764. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Kami, D.; Maeda, R.; Murata, Y.; Jo, J.-I.; Kitani, T.; Tabata, Y.; Matoba, S.; Gojo, S. TAT-dextran-mediated mitochondrial transfer enhances recovery from models of reperfusion injury in cultured cardiomyocytes. J. Cell. Mol. Med. 2020, 24, 5007–5020. [Google Scholar] [CrossRef] [Green Version]
- Louwagie, E.J.; Larsen, T.D.; Wachal, A.L.; Gandy, T.C.; Baack, M.L. Mitochondrial transfer improves cardiomyocyte bioenergetics and viability in male rats exposed to pregestational diabetes. Int. J. Mol. Sci. 2021, 22, 2382. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Animals Model | Results and/or Possible Mechanism | Reference |
|---|---|---|---|
| Heart diseases | Doxorubicin induced cardiotoxicity in rats | Melatonin exerts cardioprotective efficacy by preserving mitochondrial function and dynamics | [183,184] |
| Cardiac I/R injury in prediabetic obese rats | Melatonin exerts cardioprotective effects against cardiac I/R injury in prediabetic obese rats by activation of modulating melatonin receptor 2 | [185] | |
| Myocardial I/R injury in a DCD heart rat model | Melatonin exerts cardioprotective effects by activation of the JAK2/STAT3 signaling pathway | [186] | |
| MI rat model induced by ligation of the left anterior descending coronary artery | Melatonin exerts cardioprotective effects by promoting cardiomyocyte proliferation and heart regeneration, and by inhibiting pyroptosis and cardiac fibrosis | [187,188,189] | |
| A murine cardiac hypertrophy model by transverse aortic constriction | Melatonin ameliorates pressure overload-induced cardiac hypertrophy by attenuating Atg5-dependent autophagy and activating the Akt/mTOR pathway | [190] | |
| Isoprenaline hydrochloride induced acute heart failure in aged rat | Melatonin prevents the mitochondrial dysfunction by increasing the CNPase level | [191] | |
| Atherosclerosis | High-fat diet treated ApoE knockout mice | Melatonin decreases pro-inflammatory macrophage polarization, vulnerable plaque rupture, endothelial cell injury and atherosclerotic plaque | [192,193,194,195,196] |
| Aneurysm | Rat AAA model | Melatonin prevents AAA formation through anti-inflammatory, antioxidative and anti-autophagy effects | [179,197] |
| TAAD mouse model | Melatonin exerts therapeutic effects against TAAD by reducing oxidative stress and SMC loss via activation of Sirt1 signaling | [178] | |
| PAH | Hypoxia-induced PAH in mice | Melatonin inhibits inflammasome-associated activation of endothelium and macrophages in PAH | [181] |
| Dasatinib aggravated rat experimental model of hypoxic PAH | Melatonin attenuates dasatinib-induced PAH by inhibiting oxidative damage and apoptosis of ECs and inhibiting abnormal proliferation of SMCs | [198] | |
| MCT-induced rat PAH model | Melatonin treatment reduces MCT-induced RV hypertrophy, fibrosis, and remodeling | [199] | |
| A chronically hypoxia-induced PAH of the newborn sheep model | Melatonin improves vasodilatory function by enhancing the vasodilator prostanoid pathway | [200] |
| Disease | Study Title | Melatonin Dose | Outcome | Clinical Trials.gov Identifier |
|---|---|---|---|---|
| Heart failure | Effect of melatonin on cardiovascular and muscle mass and function in patients with heart failure | 10 mg tablets orally every night for 24 weeks | Unknown | NCT03894683 |
| Coronary artery disease | Melatonin impact on the outcomes of myocardial I/R injury during coronary artery bypass grafting surgery | 60 mg/day 5 days before surgery | Ongoing | NCT05552586 |
| Coronary artery disease | Enhanced recovery after surgery in coronary artery bypass graft/off-pump coronary artery bypass | 5 mg in the evening | Unknown | NCT03956420 |
| Coronary artery disease | Melatonin on coronary artery calcification | 3 mg/day for 6 months | Unknown | NCT03966235 |
| Acute coronary syndrome | Effects of melatonin on reperfusion injury | Intravenous 11.61 mg | Unknown | NCT03303378 |
| Essential hypertension | Melatonin and essential arterial hypertension | 1 mg/day for 1 year | Ongoing | NCT05257291 |
| Hypertension | Trial of oral melatonin in elevated blood pressure | 3 mg for three weeks | Ongoing | NCT03764020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Zeng, Z.; Li, S.; Xie, Y.; Tong, X. The Therapeutic Strategies Targeting Mitochondrial Metabolism in Cardiovascular Disease. Pharmaceutics 2022, 14, 2760. https://doi.org/10.3390/pharmaceutics14122760
Huang X, Zeng Z, Li S, Xie Y, Tong X. The Therapeutic Strategies Targeting Mitochondrial Metabolism in Cardiovascular Disease. Pharmaceutics. 2022; 14(12):2760. https://doi.org/10.3390/pharmaceutics14122760
Chicago/Turabian StyleHuang, Xiaoyang, Zhenhua Zeng, Siqi Li, Yufei Xie, and Xiaoyong Tong. 2022. "The Therapeutic Strategies Targeting Mitochondrial Metabolism in Cardiovascular Disease" Pharmaceutics 14, no. 12: 2760. https://doi.org/10.3390/pharmaceutics14122760