

Targeting OPA1-Mediated Mitochondrial Fusion Contributed to Celastrol’s Anti-Tumor Angiogenesis Effect
 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Animals and Treatments
2.3. Hematoxylin and Eosin Staining and Immunofluorescence
2.4. Cell Viability Assay
2.5. Matrigel Tube Formation Assay
2.6. Measurement of Mitochondrial Length by High-Content Screening
2.7. Wound-Healing Migration Assay
2.8. Measuring Oxygen Consumption Rate by Seahorse
2.9. Western Blot Assay
2.10. siRNA Transfection in HUVECs
2.11. RT-PCR and qPCR
- hs-OPA1: forward, 5′-CGACCCCAATTAAGGACATCC-3′;
- reverse, 5′-GCGAGGCTGGTAGCCATATTT-3′
- hs-GAPDH: forward, 5′-CTGGGCTACACTGAGCACC-3′;
- reverse, 5′-AAGTGGTCGTTGAGGGCAATG-3′
2.12. Transmission Electron Microscopy (TEM)
2.13. Computational Molecular Docking
2.14. Statistical Analysis
3. Results
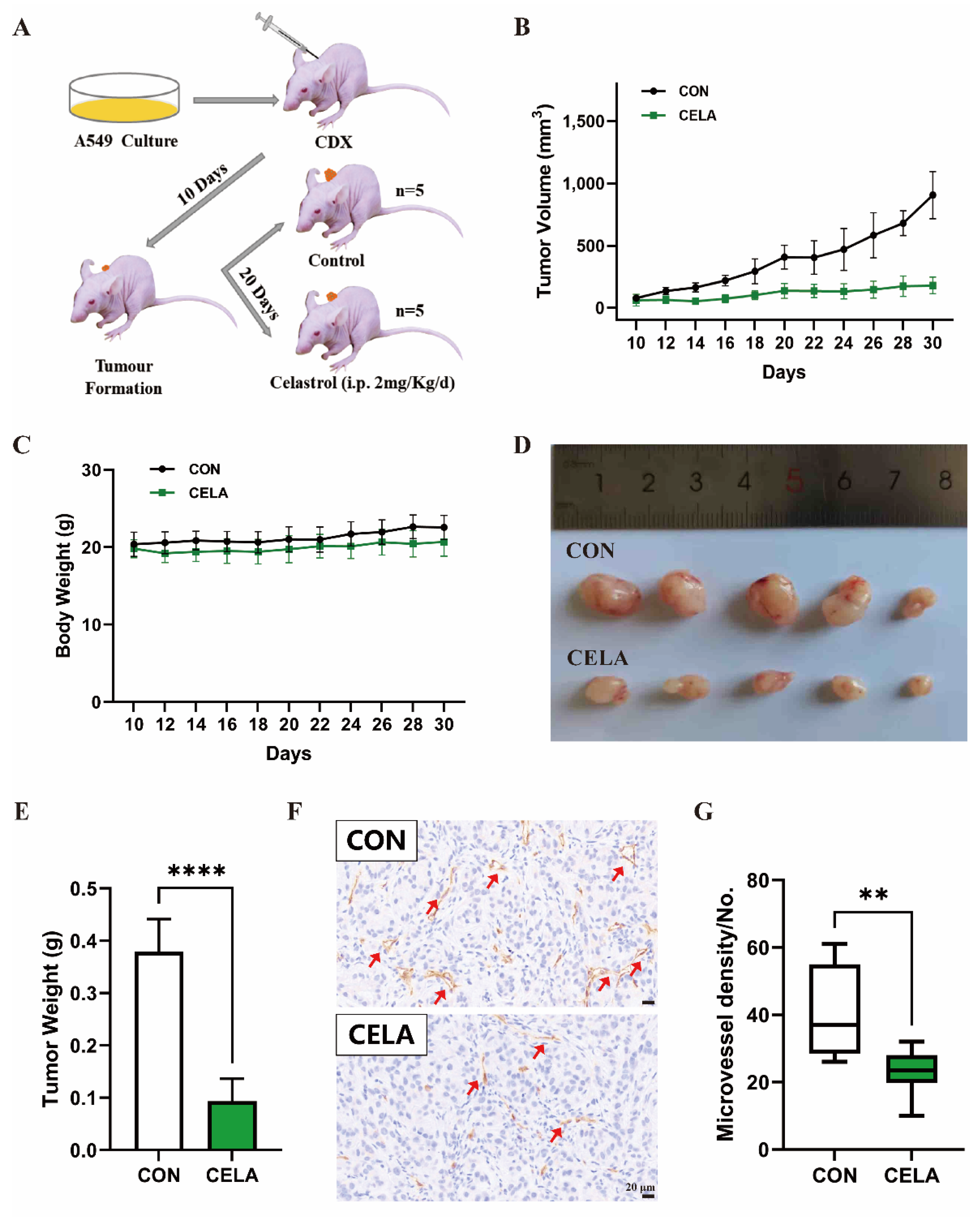
3.1. Celastrol Inhibits Lung-Derived Tumor Growth
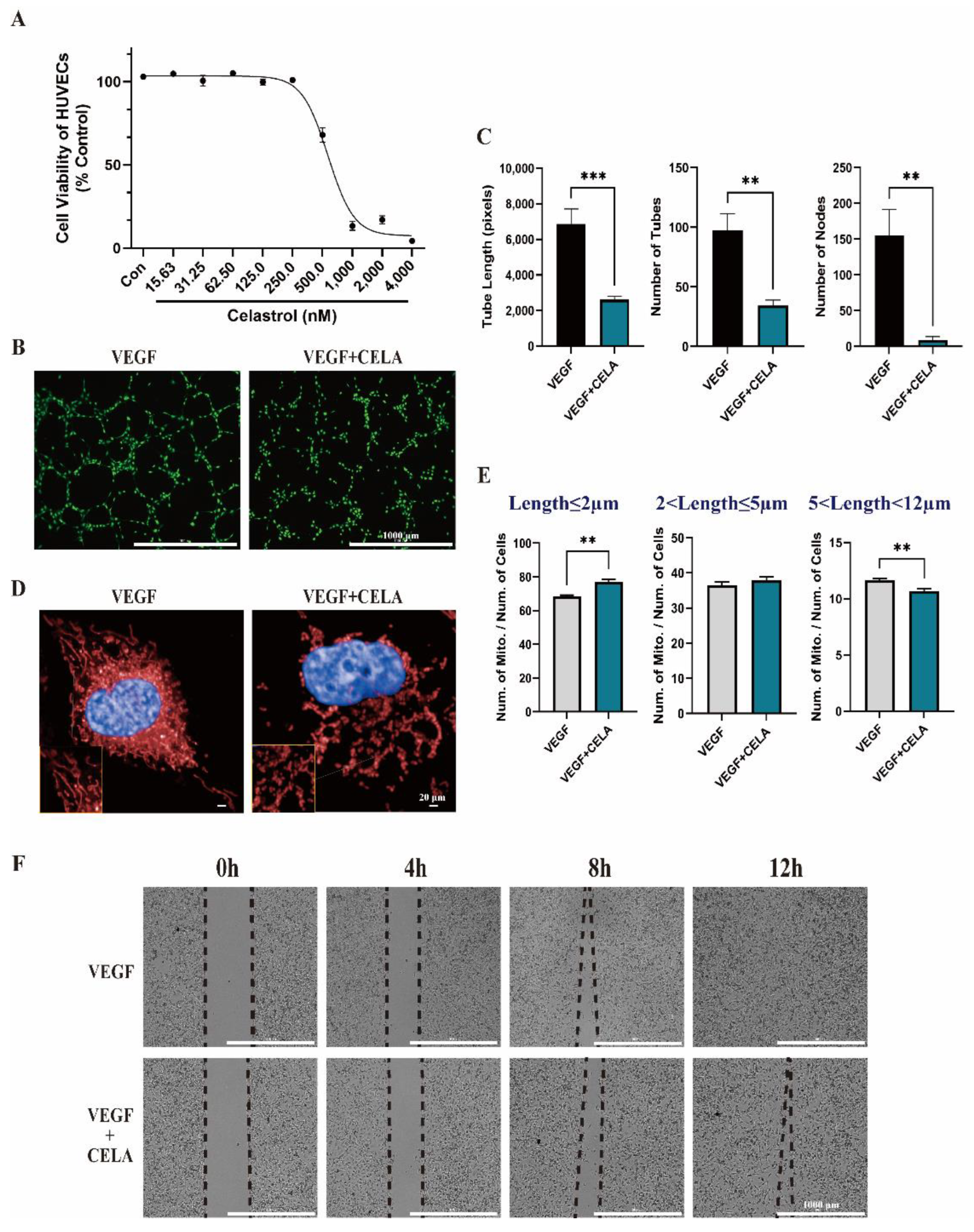
3.2. Celastrol Inhibits HUVEC Tube Formation and Migration In Vitro
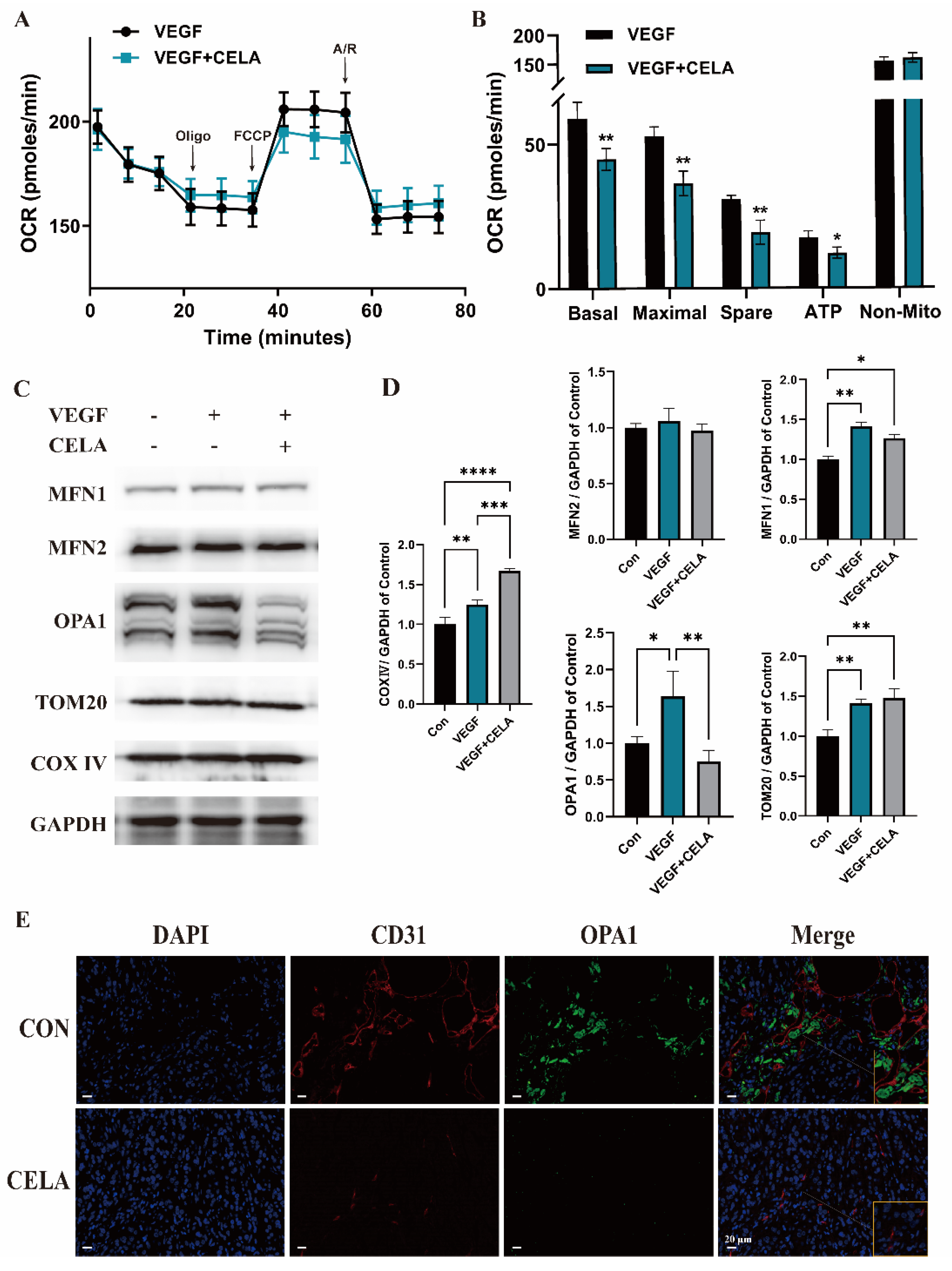
3.3. Celastrol Affects Mitochondrial Function and Morphology
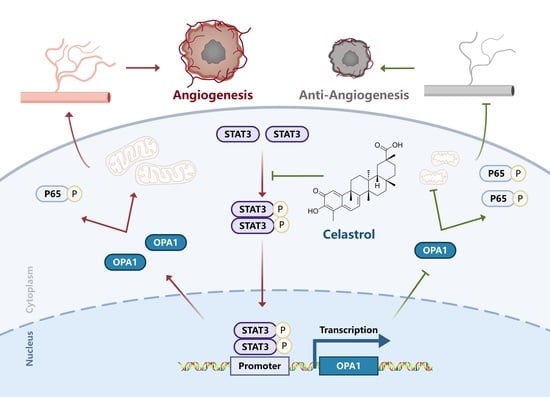
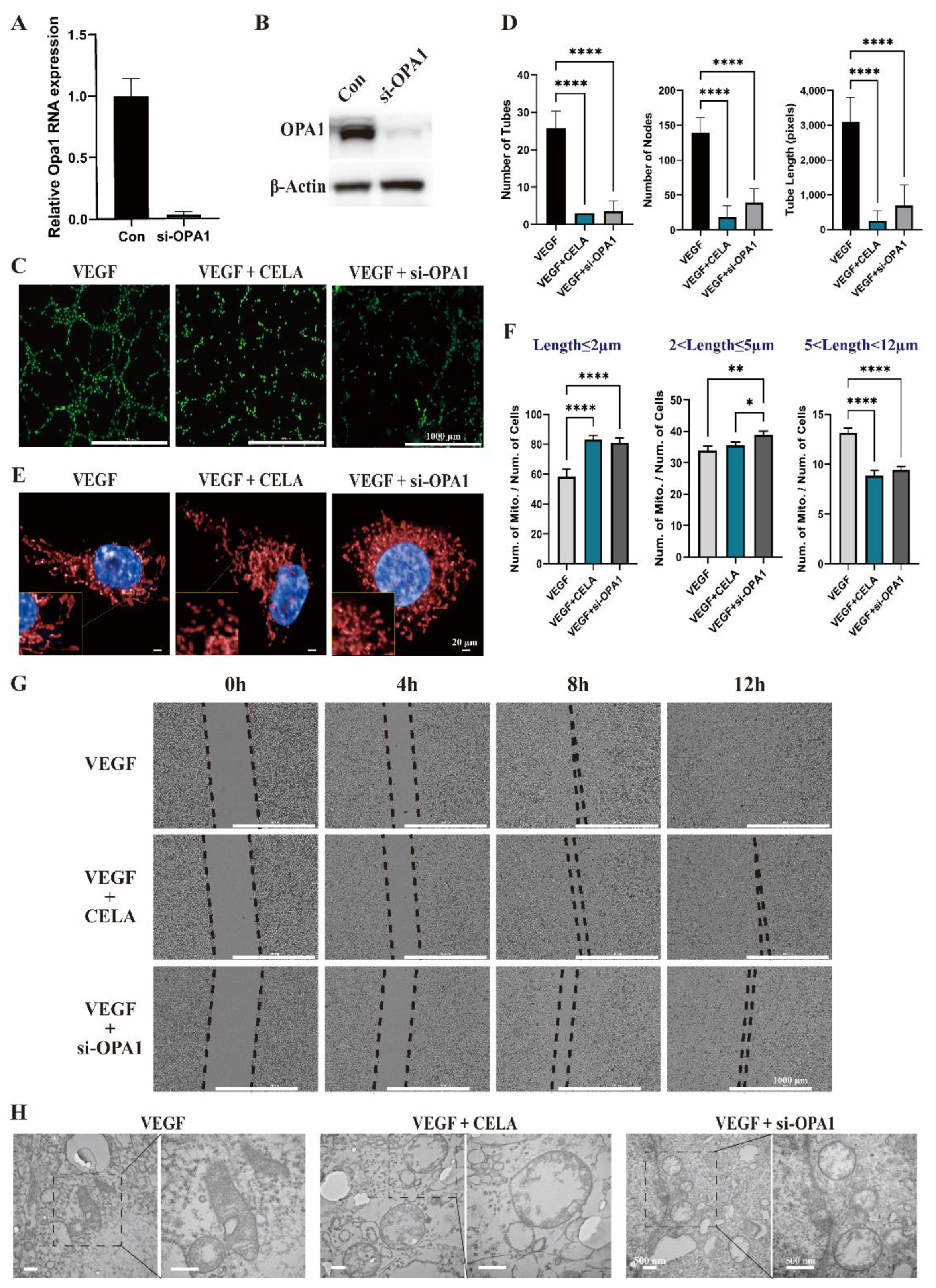
3.4. Celastrol Inhibits Angiogenesis by Downregulating OPA1
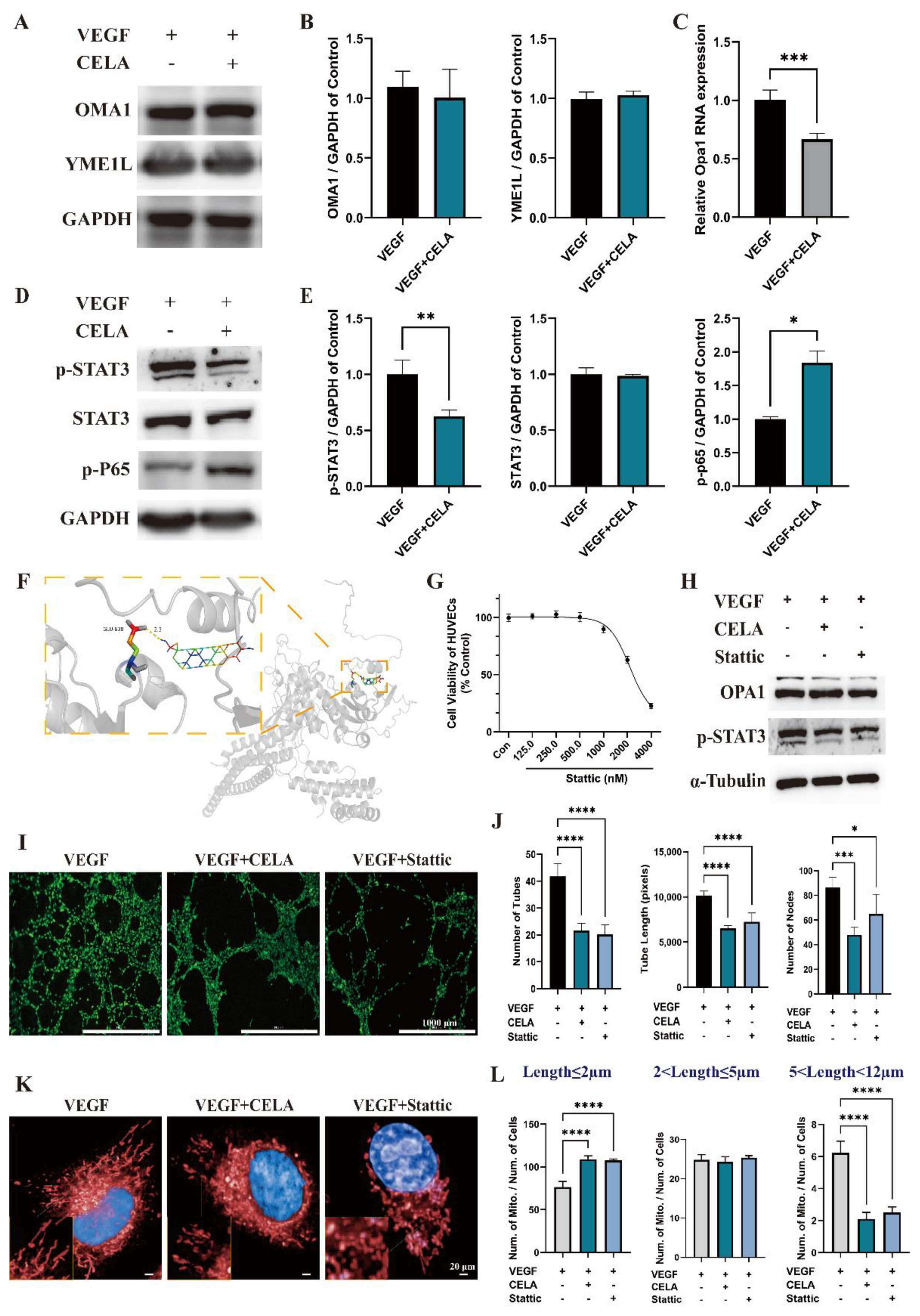
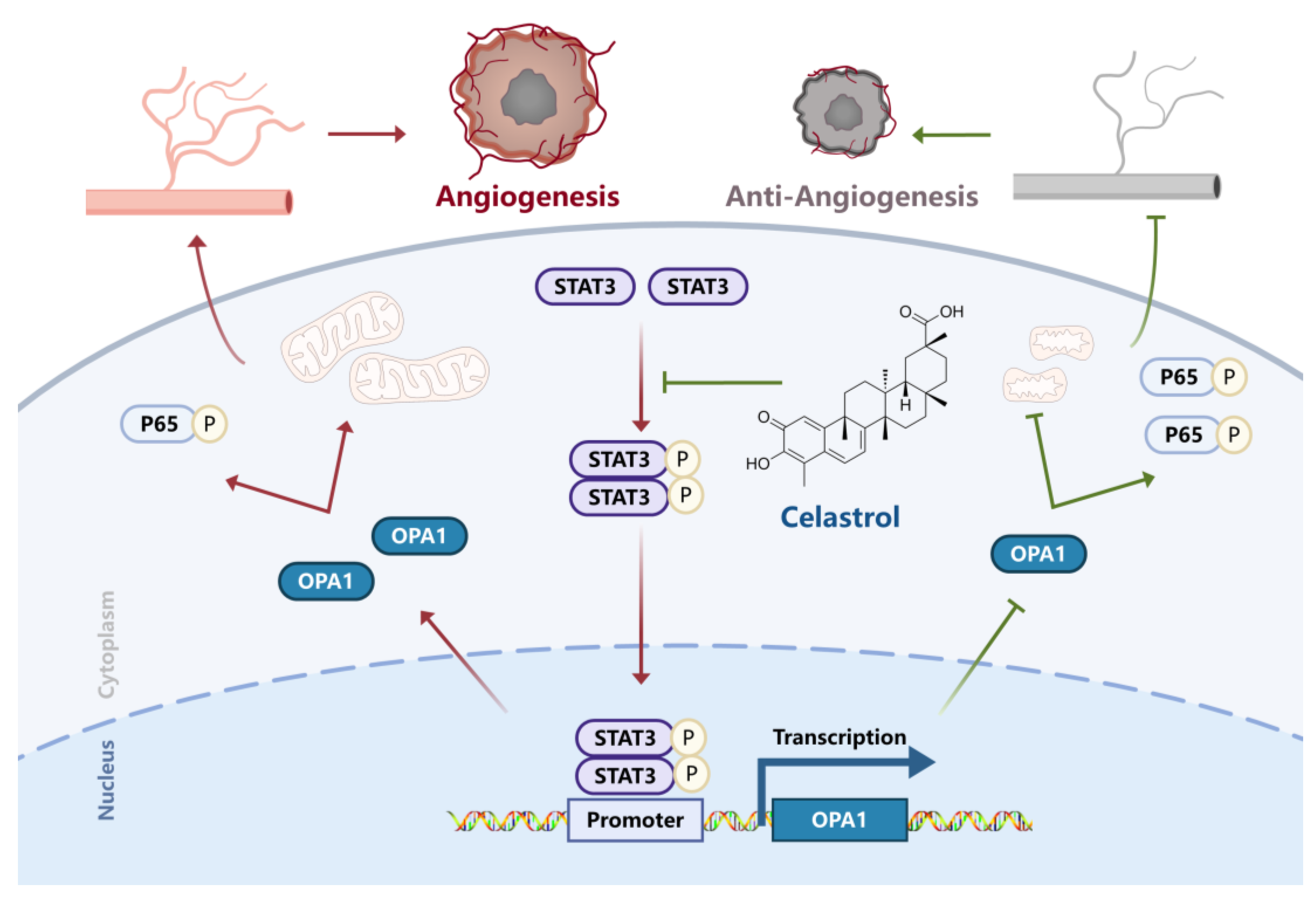
3.5. Celastrol Inhibits Angiogenesis via STAT3/OPA1/p65 Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, M.; Yang, J.; Li, L.; Hu, Y.; Lu, X.; Sun, R.; Wang, Y.; Wang, X.; Zhang, X. Metabolomics reveals that cysteine metabolism plays a role in celastrol-induced mitochondrial apoptosis in hl-60 and nb-4 cells. Sci. Rep. 2020, 10, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Zhu, N.; Long, J.; Wu, H.; Wang, Y.; Liu, B.; Liao, D.; Qin, L. Celastrol induces lipophagy via the lxrα/abca1 pathway in clear cell renal cell carcinoma. Acta Pharmacol. Sin. 2021, 42, 1472–1485. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, Z.; Chu, D.; Feng, H.; Zhang, J.; Zhu, L.; Li, J. Effectively suppressed angiogenesis-mediated retinoblastoma growth using celastrol nanomicelles. Drug Deliv. 2020, 27, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Yi, Z.; Zhang, J.; Lu, B.; Sung, B.; Qu, W.; Aggarwal, B.B.; Liu, M. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of akt/mammalian target of rapamycin pathway. Cancer Res. 2010, 70, 1951–1959. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Wang, H.; Dai, H.; Zhu, Q.; Cui, C.; Sun, X.; Li, Y.; Deng, Z.; Zhou, X.; Ge, Y.; et al. Otulin allies with lubac to govern angiogenesis by editing alk1 linear polyubiquitin. Mol. Cell 2021, 81, 3187–3204. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Tung, K.; Lin, C.; Kuo, C.; Li, L.; Kuo, Y.; Lin, C.; Wu, H. Chc promotes tumor growth and angiogenesis through regulation of hif-1α and vegf signaling. Cancer Lett. 2013, 331, 58–67. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, Z.; Zhang, S.; Ren, J.; Zhang, R.; Zeng, H.; Li, Q.; Wu, G. Inhibitory action of celastrol on hypoxia-mediated angiogenesis and metastasis via the hif-1α pathway. Int. J. Mol. Med. 2011, 27, 407. [Google Scholar]
- Rajendran, P.; Li, F.; Shanmugam, M.K.; Kannaiyan, R.; Goh, J.N.; Wong, K.F.; Wang, W.; Khin, E.; Tergaonkar, V.; Kumar, A.P.; et al. Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of stat3/jak2 signaling cascadein vitro andin vivo. Cancer Prev. Res. 2012, 5, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins mfn1 and mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. Opa1 processing controls mitochondrial fusion and is regulated by mrna splicing, membrane potential, and yme1l. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [Green Version]
- Rainbolt, T.K.; Lebeau, J.; Puchades, C.; Wiseman, R.L. Reciprocal degradation of yme1l and oma1 adapts mitochondrial proteolytic activity during stress. Cell Rep. 2016, 14, 2041–2049. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Shi, X.; Boopathy, S.; Mcdonald, J.; Smith, A.W.; Chao, L.H. Two forms of opa1 cooperate to complete fusion of the mitochondrial inner-membrane. eLife 2020, 9, e50973. [Google Scholar] [CrossRef] [PubMed]
- Herkenne, S.; Ek, O.; Zamberlan, M.; Pellattiero, A.; Chergova, M.; Chivite, I.; Novotná, E.; Rigoni, G.; Fonseca, T.B.; Samardzic, D.; et al. Developmental and tumor angiogenesis requires the mitochondria-shaping protein opa1. Cell Metab. 2020, 31, 987–1003. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Hughes, A.L. Opa1 and angiogenesis: Beyond the fusion function. Cell Metab. 2020, 31, 886–887. [Google Scholar] [CrossRef]
- Montemagno, C.; Pagès, G. Resistance to anti-angiogenic therapies: A mechanism depending on the time of exposure to the drugs. Front. Cell Dev. Biol. 2020, 8, 584. [Google Scholar] [CrossRef]
- Cruys, B.; Wong, B.W.; Kuchnio, A.; Verdegem, D.; Cantelmo, A.R.; Conradi, L.; Vandekeere, S.; Bouché, A.; Cornelissen, I.; Vinckier, S.; et al. Glycolytic regulation of cell rearrangement in angiogenesis. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Zhao, N.; Chen, J.; Ye, P.; Nan, X.; Zhou, H.; Jiang, Q.; Yang, Y.; Huang, J.; Yuan, M.; et al. Celastrol inhibits the growth of ovarian cancer cells in vitro and in vivo. Front. Oncol. 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- De Smet, F.; Segura, I.; De Bock, K.; Hohensinner, P.J.; Carmeliet, P. Mechanisms of vessel branching. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Jou, D.; Wang, Y.; Ma, H.; Liu, T.; Fuchs, J.; Li, P.K.; Lu, J.; Li, C.; Lin, J. Stat3 as a potential therapeutic target in aldh+ and cd44+/cd24+ stem cell-like pancreatic cancer cells. Int. J. Oncol. 2016, 49, 2265–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rini, B.I.; Halabi, S.; Rosenberg, J.E.; Stadler, W.M.; Vaena, D.A.; Ou, S.; Archer, L.; Atkins, J.N.; Picus, J.; Czaykowski, P.; et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: Calgb 90206. J. Clin. Oncol. 2008, 26, 5422–5428. [Google Scholar] [CrossRef] [PubMed]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase iii trial of the cancer and leukemia group b (calgb 80303). J. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef]
- Cao, Y.; Arbiser, J.; D’Amato, R.J.; D’Amore, P.A.; Ingber, D.E.; Kerbel, R.; Klagsbrun, M.; Lim, S.; Moses, M.A.; Zetter, B.; et al. Forty-year journey of angiogenesis translational research. Sci. Transl. Med. 2011, 3, 114rv3. [Google Scholar] [CrossRef]
- Yao, J.C.; Guthrie, K.A.; Moran, C.; Strosberg, J.R.; Kulke, M.H.; Chan, J.A.; Loconte, N.; Mcwilliams, R.R.; Wolin, E.M.; Mattar, B.; et al. Phase iii prospective randomized comparison trial of depot octreotide plus interferon alfa-2b versus depot octreotide plus bevacizumab in patients with advanced carcinoid tumors: Swog s0518. J. Clin. Oncol. 2017, 35, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and bevacizumab in progressive glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Cremolini, C.; Marmorino, F.; Bergamo, F.; Aprile, G.; Salvatore, L.; Masi, G.; Dell Aquila, E.; Antoniotti, C.; Murgioni, S.; Allegrini, G.; et al. Phase ii randomised study of maintenance treatment with bevacizumab or bevacizumab plus metronomic chemotherapy after first-line induction with folfoxiri plus bevacizumab for metastatic colorectal cancer patients: The moma trial. Eur. J. Cancer (1990) 2019, 109, 175–182. [Google Scholar] [CrossRef]
- Lim, H.Y.; Ong, P.S.; Wang, L.; Goel, A.; Ding, L.; Li-Ann Wong, A.; Ho, P.C.; Sethi, G.; Xiang, X.; Goh, B.C. Celastrol in cancer therapy: Recent developments, challenges and prospects. Cancer Lett. 2021, 521, 252–267. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, L.; Deng, C.; Zhao, T.; Shi, Z.; Yan, J.; Gong, Y.; Liao, D.; Qin, L. Celastrol ameliorates vascular neointimal hyperplasia through wnt5a-involved autophagy. Int. J. Biol. Sci. 2021, 17, 2561–2575. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Won, Y.; Park, K.; Lee, M.; Tachibana, H.; Yamada, K.; Seo, K. Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ros-dependent mitochondrial pathway and the suppression of pi3k/akt signaling. Apoptosis 2012, 17, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, X.; Zhao, M.; Wang, Y.; Cheng, X.; Wang, D.; Xu, Y.; Du, Z.; Yu, X. Celastrol targets mitochondrial respiratory chain complex i to induce reactive oxygen species-dependent cytotoxicity in tumor cells. BMC Cancer 2011, 11, 170. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Zhou, L.; Deng, H.; Huang, C.; Wang, N.; Yue, L.; Zhang, P.; Zhou, Y.; Zhou, W.; Gao, Y. Targeting OPA1-Mediated Mitochondrial Fusion Contributed to Celastrol’s Anti-Tumor Angiogenesis Effect. Pharmaceutics 2023, 15, 48. https://doi.org/10.3390/pharmaceutics15010048
Li G, Zhou L, Deng H, Huang C, Wang N, Yue L, Zhang P, Zhou Y, Zhou W, Gao Y. Targeting OPA1-Mediated Mitochondrial Fusion Contributed to Celastrol’s Anti-Tumor Angiogenesis Effect. Pharmaceutics. 2023; 15(1):48. https://doi.org/10.3390/pharmaceutics15010048
Chicago/Turabian StyleLi, Gaofu, Lei Zhou, Huifang Deng, Congshu Huang, Ningning Wang, Lanxin Yue, Pengfei Zhang, Yongqiang Zhou, Wei Zhou, and Yue Gao. 2023. "Targeting OPA1-Mediated Mitochondrial Fusion Contributed to Celastrol’s Anti-Tumor Angiogenesis Effect" Pharmaceutics 15, no. 1: 48. https://doi.org/10.3390/pharmaceutics15010048