
PPAR Gamma Agonist Leriglitazone Recovers Alterations Due to Pank2-Deficiency in hiPS-Derived Astrocytes
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Human Astrocytes from NPC
2.2. Astrocytes Treatments
2.3. Immunofluorescence
2.4. Cell Viability Assay
2.5. Determination of Respiratory Activity
2.6. Iron Staining
2.7. Statistical Analysis
3. Results
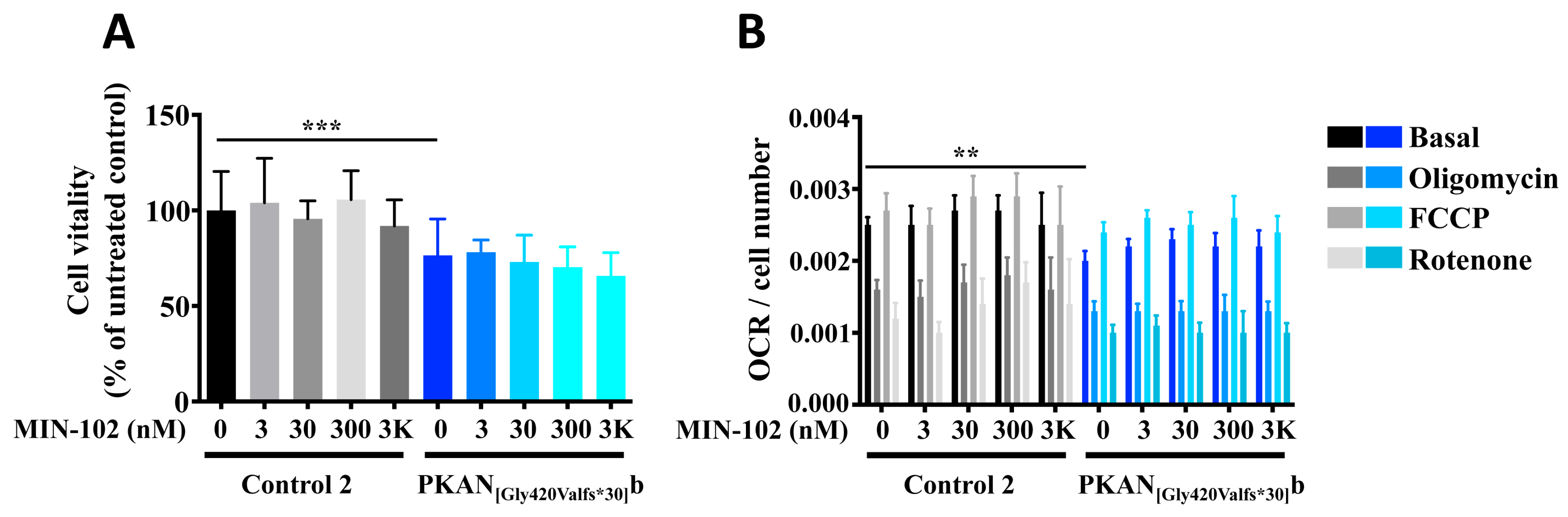
3.1. A Short Treatment with Leriglitazone Does Not Interfere with the Viability and Respiration of Astrocytes
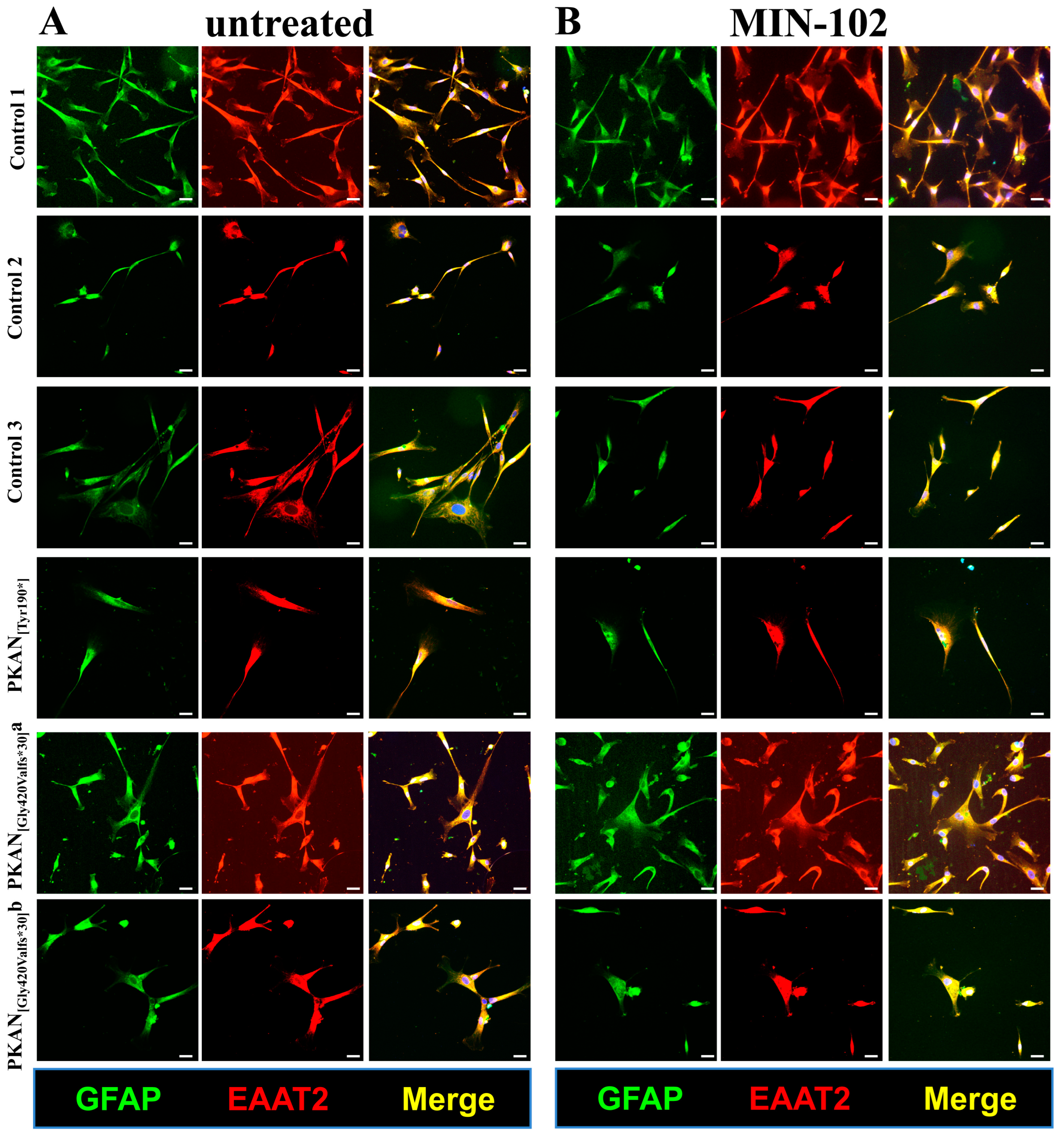
3.2. An Extended Treatment with Leriglitazone Does Not Interfere with the Astrocyte Differentiation
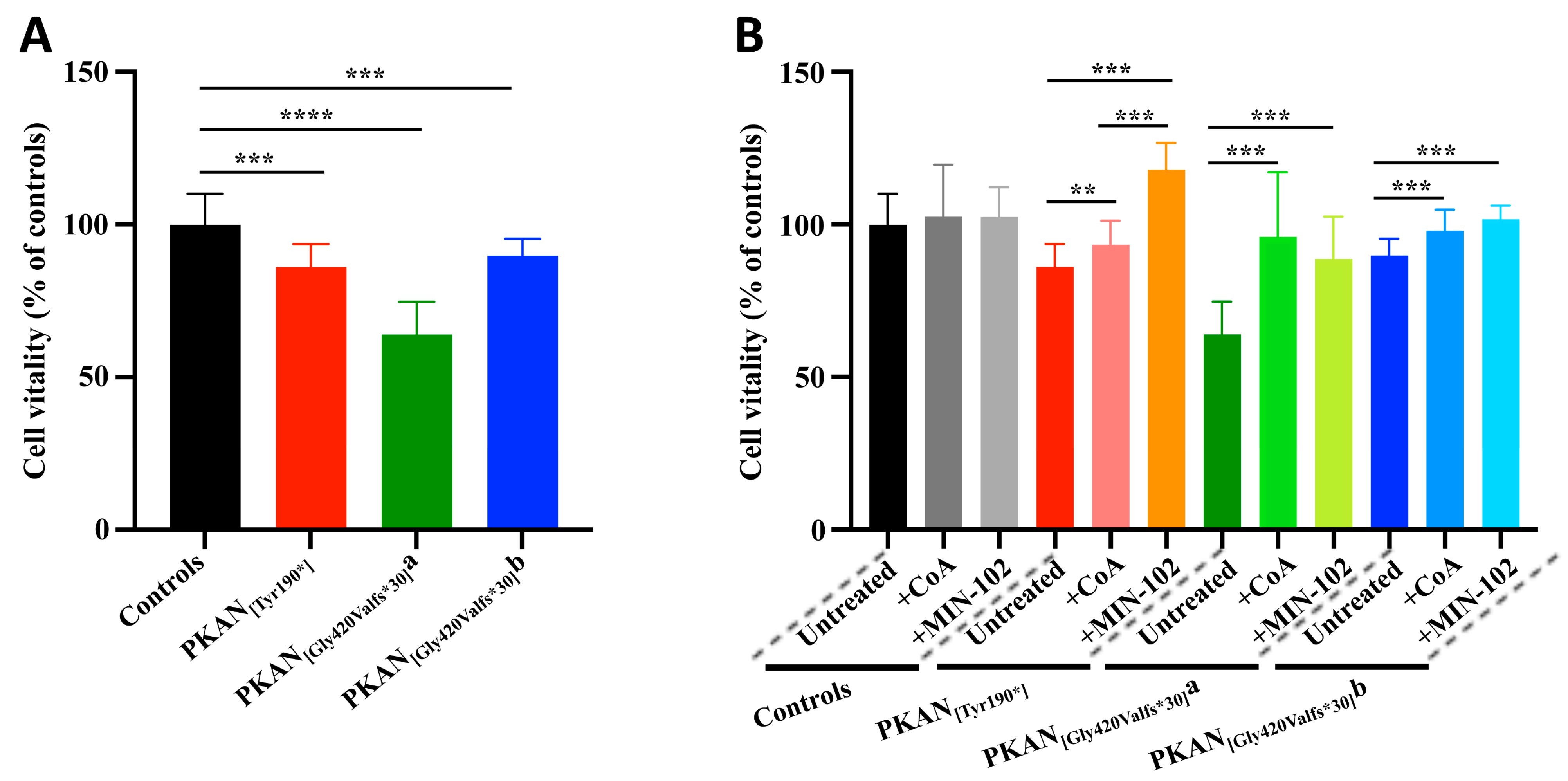
3.3. Treatment with Leriglitazone Improves PKAN d-Astrocytes Vitality
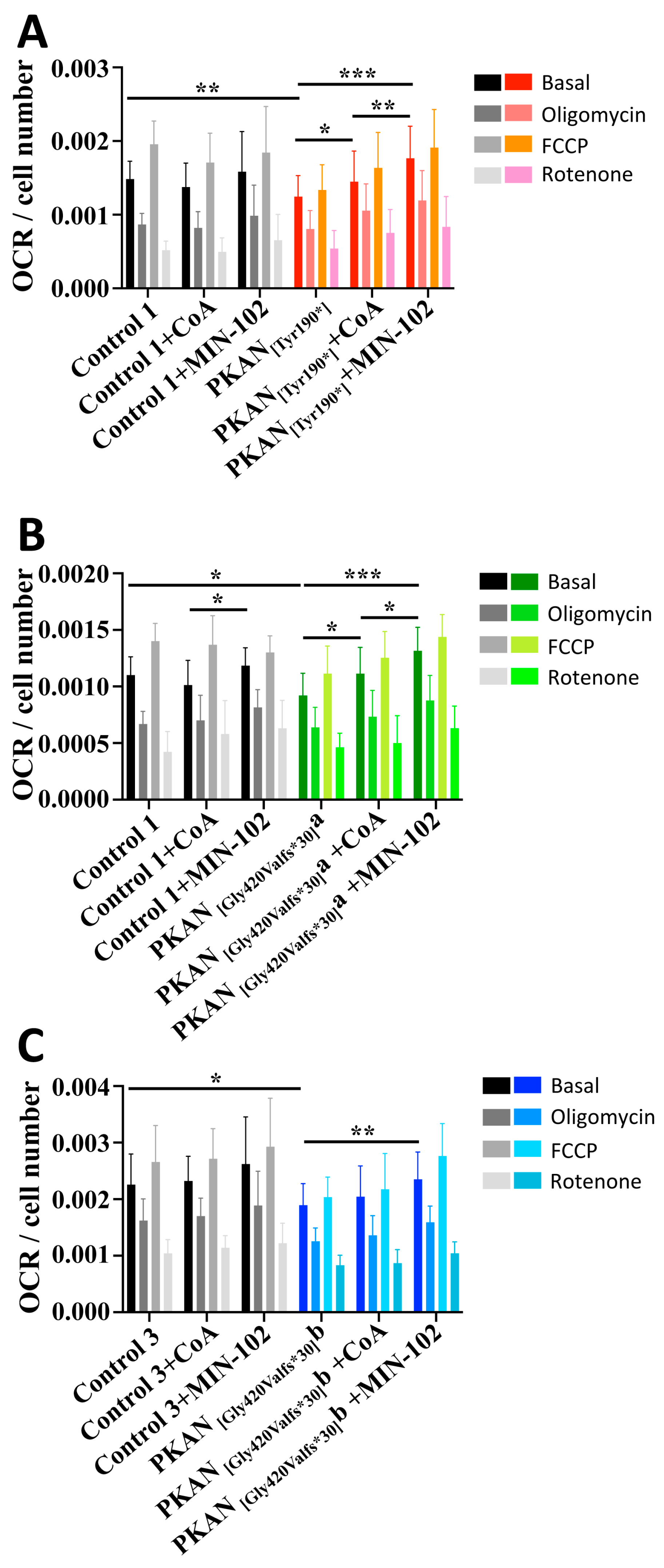
3.4. Treatment with Leriglitazone Improves PKAN d-Astrocytes Respiratory Activity
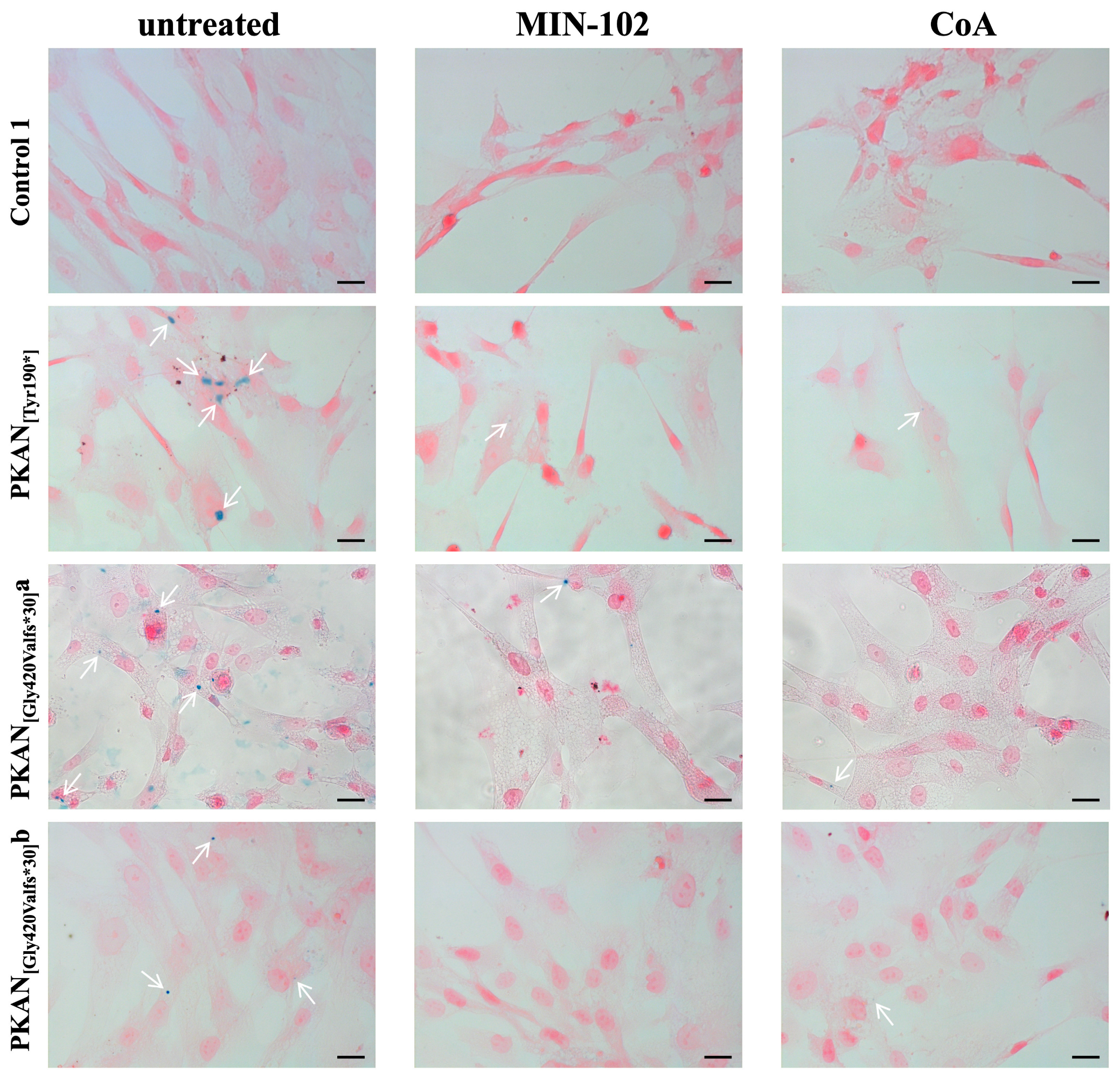
3.5. Treatment with Leriglitazone Strongly Reduces Iron Deposition in PKAN d-Astrocytes
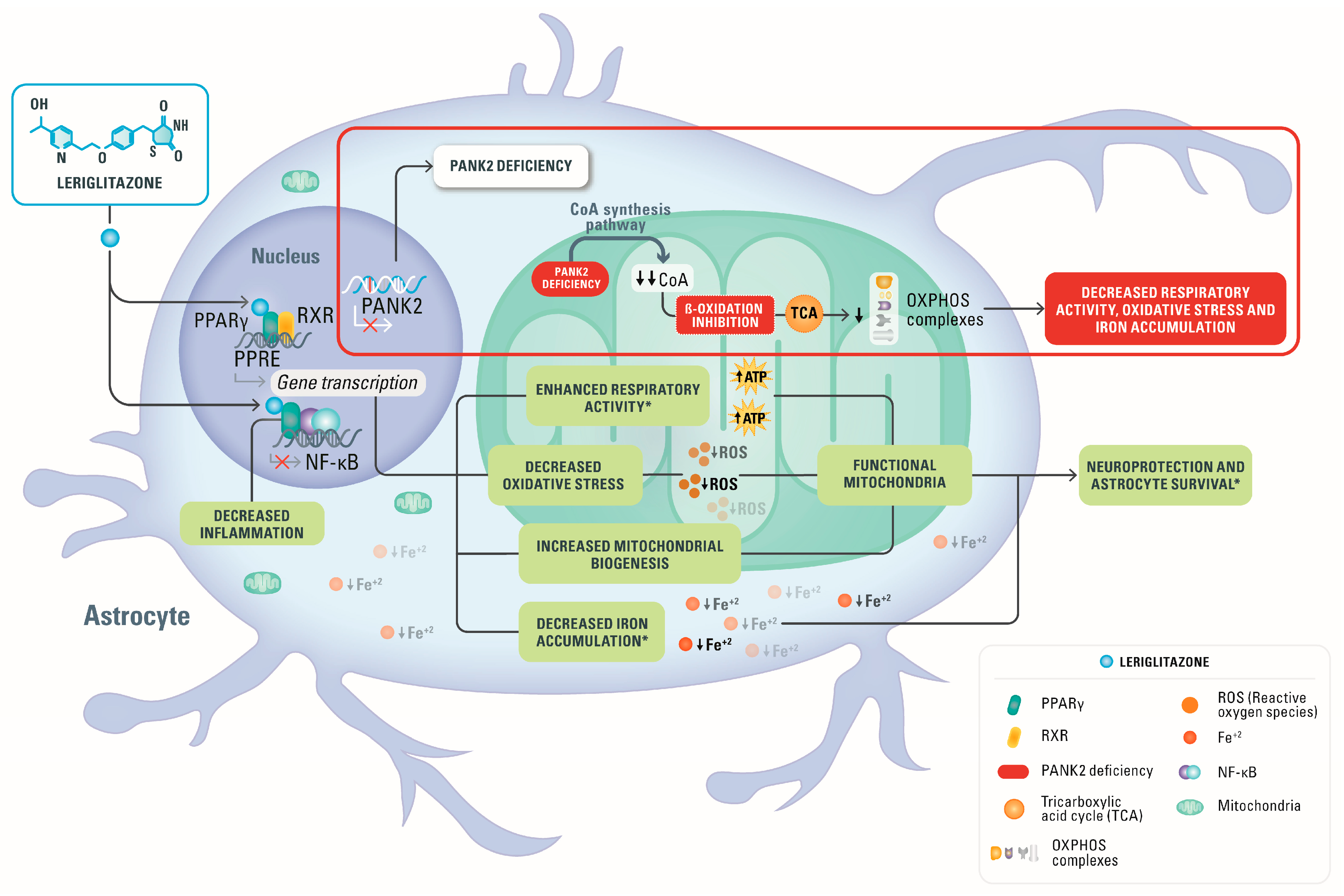
4. Discussion
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levi, S.; Tiranti, V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals 2019, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Kurian, M.A.; Hayflick, S.J. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): Review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int. Rev. Neurobiol. 2013, 110, 49–71. [Google Scholar]
- Schneider, S.A.; Bhatia, K.P. Excess Iron Harms the Brain: The Syndromes of Neurodegeneration with Brain Iron Accumulation (NBIA). J. Neural. Transm. 2013, 120, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, G.; Zibordi, F.; Chiapparini, L.; Bertini, E.; Russo, L.; Piga, A.; Longo, F.; Garavaglia, B.; Aquino, D.; Savoiardo, M.; et al. Iron-related MRI images in patients with pantothenate kinase-associated neurodegeneration (PKAN) treated with deferiprone: Results of a phase II pilot trial. Mov. Disord. 2011, 26, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.; Polster, B.J.; Hayflick, S.J. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J. Med. Genet. 2009, 46, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Westaway, S.K.; Levinson, B.; Zhou, B.; Johnson, M.A.; Ching, K.H.L.; Gitschier, J. Genetic, Clinical, and Radiographic Delineation of Hallervorden-Spatz Syndrome. N. Engl. J. Med. 2003, 348, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Thakur, N.; Klopstock, T.; Jackowski, S.; Kuscer, E.; Tricta, F.; Videnovic, A.; Jinnah, H.A. Rational Design of Novel Therapies for Pantothenate Kinase–Associated Neurodegeneration. Mov. Disord. 2021, 36, 2005–2016. [Google Scholar] [CrossRef]
- Zhou, B.; Westaway, S.K.; Levinson, B.; Johnson, M.A.; Gitschier, J.; Hayflick, S.J. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef]
- Johnson, M.A.; Kuo, Y.M.; Westaway, S.K.; Parker, S.M.; Ching, K.H.L.; Gitschier, J.; Hayflick, S.J. Mitochondrial Localization of Human PANK2 and Hypotheses of Secondary Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration. Ann. N. Y. Acad. Sci. 2004, 1012, 282–298. [Google Scholar] [CrossRef]
- Brunetti, D.; Dusi, S.; Morbin, M.; Uggetti, A.; Moda, F.; D’Amato, I.; Giordano, C.; D’Amati, G.; Cozzi, A.; Levi, S.; et al. Pantothenate kinase-associated neurodegeneration: Altered mitochondria membrane potential and defective respiration in Pank2 knock-out mouse model. Hum. Mol. Genet. 2012, 21, 5294–5305. [Google Scholar] [CrossRef] [Green Version]
- Hörtnagel, K.; Prokisch, H.; Meitinger, T. An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Hum. Mol. Genet. 2003, 12, 321–327. [Google Scholar] [CrossRef]
- Leonardi, R.; Zhang, Y.; Rock, C.; Jackowski, S. Coenzyme A: Back in Action. Prog. Lipid. Res. 2005, 44, 125–153. [Google Scholar] [CrossRef]
- Garcia, M.; Leonardi, R.; Zhang, Y.-M.; Rehg, J.E.; Jackowski, S. Germline deletion of pantothenate kinases 1 and 2 reveals the key roles for CoA in postnatal metabolism. PLoS ONE 2012, 7, e40871. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; van der Zwaag, M.; Wedman, J.J.; Permentier, H.; Plomp, N.; Jia, X.; Kanon, B.; Eggens-Meijer, E.; Buist, G.; Harmsen, H.; et al. Coenzyme A precursors Flow from Mother to Zygote and from Microbiome to Host. Mol. Cell 2022, 82, 2650–2665.e12. [Google Scholar] [CrossRef]
- Meo, I.D.; Carecchio, M.; Tiranti, V. Inborn errors of coenzyme a metabolism and neurodegeneration. J. Inherit. Metab. Dis. 2019, 42, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti, D.; Dusi, S.; Giordano, C.; Lamperti, C.; Morbin, M.; Fugnanesi, V.; Marchet, S.; Fagiolari, G.; Sibon, O.; Moggio, M.; et al. Pantethine treatment is effective in recovering the disease phenotype induced by ketogenic diet in a pantothenate kinase-associated neurodegeneration mouse model. Brain 2014, 137, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Wu, Z.; Kuo, Y.M.; Zhou, B. Dietary rescue of fumble—A Drosophila model for pantothenate-kinase-associated neurodegeneration. J. Inherit. Metab. Dis. 2005, 28, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Zizioli, D.; Tiso, N.; Guglielmi, A.; Saraceno, C.; Busolin, G.; Giuliani, R.; Khatri, D.; Monti, E.; Borsani, G.; Argenton, F.; et al. Knock-down of pantothenate kinase 2 severely affects the development of the nervous and vascular system in zebrafish, providing new insights into PKAN disease. Neurobiol. Dis. 2016, 85, 35–48. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Hogarth, P.; Placzek, A.; Gregory, A.M.; Fox, R.; Zhen, D.; Hamada, J.; Van Der Zwaag, M.; Lambrechts, R.; Jin, H.; et al. 4′-Phosphopantetheine corrects CoA, iron, and dopamine metabolic defects in mammalian models of PKAN. EMBO Mol. Med. 2019, 11, e10489. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, R.A.; Schepers, H.; Yu, Y.; van der Zwaag, M.; Autio, K.J.; Vieira-Lara, M.A.; Bakker, B.M.; Tijssen, M.A.; Hayflick, S.J.; Vieira-Lara, M.A.; et al. CoA-dependent activation of mitochondrial acyl carrier protein links four neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10488. [Google Scholar] [CrossRef] [PubMed]
- Drecourt, A.; Babdor, J.; Dussiot, M.; Petit, F.; Goudin, N.; Garfa-Traoré, M.; Habarou, F.; Bole-Feysot, C.; Nitschké, P.; Ottolenghi, C.; et al. Impaired Transferrin Receptor Palmitoylation and Recycling in Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2018, 102, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme A corrects pathological defects in human neurons of PANK 2-associated neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Angelova, P.R.; Wiethoff, S.; Tsuchiya, Y.; Mazzacuva, F.; Preza, E.; Bhatia, K.P.; Mills, K.; Gout, I.; Abramov, A.Y.; et al. iPSC-derived neuronal models of PANK2-associated neurodegeneration reveal mitochondrial dysfunction contributing to early disease. PLoS ONE 2017, 12, e0184104. [Google Scholar] [CrossRef] [Green Version]
- Santambrogio, P.; Ripamonti, M.; Paolizzi, C.; Panteghini, C.; Carecchio, M.; Chiapparini, L.; Raimondi, M.; Rubio, A.; Di Meo, I.; Cozzi, A.; et al. Harmful Iron-Calcium Relationship in Pantothenate kinase Associated Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 3664. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Hiken, M.; Gregory, A.; Malandrini, A.; Clark, D.; Hogarth, P.; Grafe, M.; Hayflick, S.J.; Woltjer, R.L. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 2011, 134, 947–958. [Google Scholar] [CrossRef]
- Santambrogio, P.; Ripamonti, M.; Cozzi, A.; Raimondi, M.; Cavestro, C.; Di Meo, I.; Rubio, A.; Taverna, S.; Tiranti, V.; Levi, S. Massive iron accumulation in PKAN-derived neurons and astrocytes: Light on the human pathological phenotype. Cell Death Dis. 2022, 13, 1–12. [Google Scholar] [CrossRef]
- Rodríguez-Pascau, L.; Vilalta, A.; Cerrada, M.; Traver, E.; Forss-Petter, S.; Weinhofer, I.; Bauer, J.; Kemp, S.; Pina, G.; Pascual, S.; et al. The brain penetrant PPARγ agonist leriglitazone restores multiple altered pathways in models of X-linked adrenoleukodystrophy. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in Health and Disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α (PGC-1α): Transcriptional Coactivator and Metabolic Regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Pascau, L.; Britti, E.; Calap-Quintana, P.; Na Dong, Y.; Vergara, C.; Delaspre, F.; Medina-Carbonero, M.; Tamarit, J.; Pallardó, F.V.; Gonzalez-Cabo, P.; et al. PPAR Gamma Agonist Leriglitazone Improves Frataxin-Loss Impairments in Cellular and Animal Models of Friedreich Ataxia. Neurobiol. Dis. 2020, 148, 105162. [Google Scholar] [CrossRef]
- Cozzi, A.; Santambrogio, P.; Privitera, D.; Broccoli, V.; Rotundo, L.I.; Garavaglia, B.; Benz, R.; Altamura, S.; Goede, J.S.; Muckenthaler, M.U.; et al. Human L-ferritin deficiency is characterized by idiopathic generalized seizures and atypical restless leg syndrome. J. Exp. Med. 2013, 210, 1779–1791. [Google Scholar] [CrossRef]
- Ripamonti, M.; Santambrogio, P.; Racchetti, G.; Cozzi, A.; Di Meo, I.; Tiranti, V.; Levi, S. PKAN hiPS-Derived Astrocytes Show Impairment of Endosomal Trafficking: A Potential Mechanism Underlying Iron Accumulation. Front. Cell Neurosci. 2022, 16, 281. [Google Scholar] [CrossRef]
- Pandolfo, M.; Reetz, K.; Darling, A.; De Rivera, F.J.; Henry, P.G.; Joers, J.; Lenglet, C.; Adanyeguh, I.; Deelchand, D.; Mochel, F.; et al. E ffi cacy and Safety of Leriglitazone in Patients with Friedreich Ataxia. Neurol. Genet. 2022, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mignani, L.; Gnutti, B.; Zizioli, D.; Finazzi, D. Coenzyme a Biochemistry: From Neurodevelopment to Neurodegeneration. Brain Sci. 2021, 11, 1031. [Google Scholar] [CrossRef] [PubMed]
- Dansie, L.E.; Reeves, S.; Miller, K.; Zano, S.P.; Frank, M.; Pate, C.; Wang, J.; Jackowski, S. Physiological roles of the pantothenate kinases. Biochem. Soc. Trans. 2014, 42, 1033–1036. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Zhang, D.-L.; Ghosh, M.C.; Jain, A.; SantaMaria, A.M.; Rouault, T.A. Mechanisms of cellular iron sensing, regulation of erythropoiesis and mitochondrial iron utilization. Semin. Hematol. 2021, 58, 161–174. [Google Scholar] [CrossRef]
- Lee, W.-S.; Kim, J. Peroxisome Proliferator-Activated Receptors and the Heart: Lessons from the Past and Future Directions. PPAR Res. 2015, 2015, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piloni, N.E.; Fermandez, V.; Videla, L.A.; Puntarulo, S. Acute Iron Overload and Oxidative Stress in Brain. Toxicology 2013, 314, 174–182. [Google Scholar] [CrossRef]
- Cossu, G.; Abbruzzese, G.; Matta, G.; Murgia, D.; Melis, M.; Ricchi, V.; Galanello, R.; Barella, S.; Origa, R.; Balocco, M.; et al. Efficacy and safety of deferiprone for the treatment of pantothenate kinase-associated neurodegeneration (PKAN) and neurodegeneration with brain iron accumulation (NBIA): Results from a four years follow-up. Park. Relat. Disord. 2014, 20, 651–654. [Google Scholar] [CrossRef]
- Rohani, M.; Razmeh, S.; Shahidi, G.A.; Orooji, M. A Pilot Trial of Deferiprone in Pantothenate Kinase-Associated Neurodegeneration Patients. Neurol. Int. 2017, 9, 79–81. [Google Scholar] [CrossRef]
- Klopstock, T.; Tricta, F.; Neumayr, L.; Karin, I.; Zorzi, G.; Fradette, C.; Kmieć, T.; Büchner, B.; Steele, H.E.; Horvath, R.; et al. Safety and efficacy of deferiprone for pantothenate kinase-associated neurodegeneration: A randomised, double-blind, controlled trial and an open-label extension study. Lancet Neurol. 2019, 18, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A. Neurodegeneration with Brain Iron Accumulation. Curr. Neurol. Neurosci. Rep. 2016, 16, 1–9. [Google Scholar] [CrossRef]
- Karin, I.; Büchner, B.; Gauzy, F.; Klucken, A.; Klopstock, T. Treat Iron-Related Childhood-Onset Neurodegeneration (TIRCON)—An International Network on Care and Research for Patients With Neurodegeneration With Brain Iron Accumulation (NBIA). Front. Neurol. 2021, 12, 185. [Google Scholar] [CrossRef]
- Iankova, V.; Karin, I.; Klopstock, T.; Schneider, S.A. Emerging Disease-Modifying Therapies in Neurodegeneration With Brain Iron Accumulation (NBIA) Disorders. Front. Neurol. 2021, 12, 629414. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Subramanian, C.; Yun, M.-K.; Frank, M.W.; White, S.W.; Rock, C.O.; Lee, R.E.; Jackowski, S. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject (Code) | DNA Mutation | Protein Mutation |
|---|---|---|
| NeoL (Control 1) | --- | --- |
| CB (Control 2) | --- | --- |
| DIGI (Control 3) | --- | --- |
| Patient 1 (PKAN[Tyr190*]) | c. [569_570insA] | p. [Tyr190*] |
| Patient 2 (PKAN[Gly420Valfs*30]a) | c. [1259delG] | p. [Gly420Valfs*30]a |
| Patient 3 (PKAN[Gly420Valfs*30]b) | c. [1259delG] | p. [Gly420Valfs*30]b |
| Sample | Counted Fields | Total Cells | Fe Positive Cells | Fe Positive Cells (%) | Reduction of Fe % |
|---|---|---|---|---|---|
| Control 1 ut | 10 | 962 | 45 | 4.7 | --- |
| Control 1 MIN-102 | 10 | 308 | 4 | 1.3 | 74.5 |
| Control 1 CoA | 9 | 323 | 7 | 2.2 | 73.5 |
| Control 2 ut | 10 | 71 | 2 | 2.8 | --- |
| Control 2 MIN-102 | 10 | 31 | 0 | 0 | 100 |
| Control 2 CoA | 3 | 22 | 0 | 0 | 100 |
| Control 3 ut | 3 | 77 | 1 | 1.3 | --- |
| Control 3 MIN-102 | 3 | 43 | 0 | 0 | 100 |
| Control 3 CoA | 3 | 44 | 0 | 0 | 100 |
| PKAN[Tyr190*] ut | 6 | 104 | 20 | 19.2 | --- |
| PKAN[Tyr190*] MIN-102 | 6 | 111 | 5 | 4.5 | 78.4 |
| PKAN[Tyr190*] CoA | 6 | 98 | 6 | 6.1 | 70.7 |
| PKAN[Gly420Valfs*30]a ut | 10 | 556 | 127 | 22.8 | --- |
| PKAN[Gly420Valfs*30]a MIN-102 | 10 | 708 | 18 | 2.5 | 89 |
| PKAN[Gly420Valfs*30]a CoA | 10 | 152 | 10 | 6.6 | 71 |
| PKAN[Gly420Valfs*30]b ut | 10 | 148 | 39 | 26.4 | --- |
| PKAN[Gly420Valfs*30]b MIN-102 | 10 | 163 | 4 | 2.4 | 90.9 |
| PKAN[Gly420Valfs*30]b CoA | 7 | 109 | 5 | 4.6 | 82.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santambrogio, P.; Cozzi, A.; Di Meo, I.; Cavestro, C.; Vergara, C.; Rodríguez-Pascau, L.; Martinell, M.; Pizcueta, P.; Tiranti, V.; Levi, S. PPAR Gamma Agonist Leriglitazone Recovers Alterations Due to Pank2-Deficiency in hiPS-Derived Astrocytes. Pharmaceutics 2023, 15, 202. https://doi.org/10.3390/pharmaceutics15010202
Santambrogio P, Cozzi A, Di Meo I, Cavestro C, Vergara C, Rodríguez-Pascau L, Martinell M, Pizcueta P, Tiranti V, Levi S. PPAR Gamma Agonist Leriglitazone Recovers Alterations Due to Pank2-Deficiency in hiPS-Derived Astrocytes. Pharmaceutics. 2023; 15(1):202. https://doi.org/10.3390/pharmaceutics15010202
Chicago/Turabian StyleSantambrogio, Paolo, Anna Cozzi, Ivano Di Meo, Chiara Cavestro, Cristina Vergara, Laura Rodríguez-Pascau, Marc Martinell, Pilar Pizcueta, Valeria Tiranti, and Sonia Levi. 2023. "PPAR Gamma Agonist Leriglitazone Recovers Alterations Due to Pank2-Deficiency in hiPS-Derived Astrocytes" Pharmaceutics 15, no. 1: 202. https://doi.org/10.3390/pharmaceutics15010202