

Mitochondrial Dysfunction: Pathophysiology and Mitochondria-Targeted Drug Delivery Approaches
 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Physiological Importance of Mitochondria
2.1. Mitochondria and Oxidative Phosphorylation
2.2. Mitochondria and Reactive Oxygen Species (ROS)
2.3. Mitochondria and Calcium Homeostasis
2.4. Mitochondria and Apoptosis
2.5. Mitochondria and Fe/S Clusters
3. Role of Mitochondrial Dysfunction in Pathophysiology
3.1. Mitochondrial Dysfunction and Cancer
3.1.1. Metabolic Alterations Associated with Cancer
3.1.2. Structural Differences Associated with Cancer
3.1.3. OXPHOS Pathway Differences Associated with Cancer
3.1.4. Physiological Differences Associated with Cancer
3.1.5. Necrosis Associated with Cancer
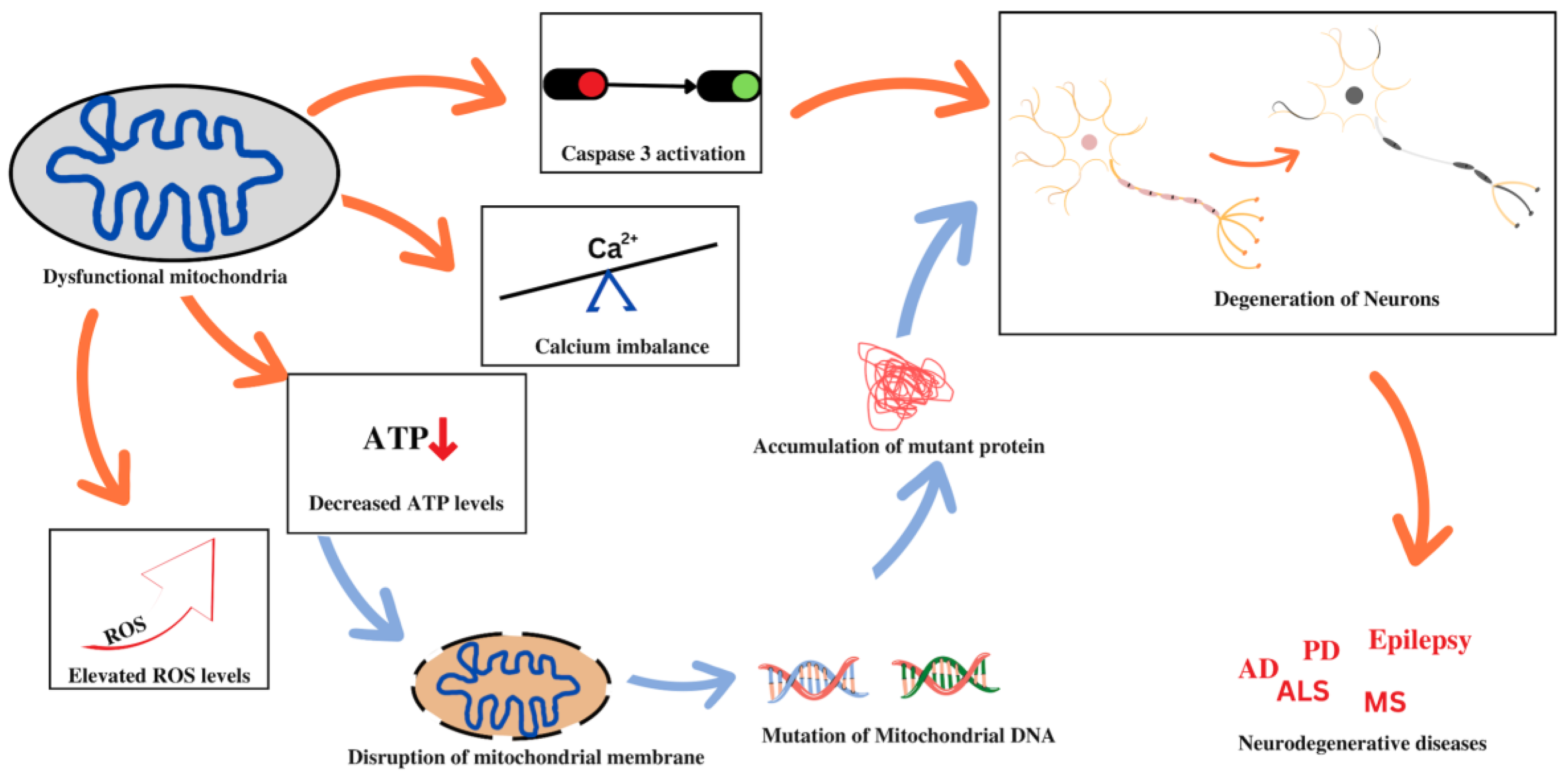
3.2. Mitochondrial Dysfunction and Neurodegenerative Diseases
3.2.1. Alzheimer’s Disease
3.2.2. Parkinson’s Disease
3.2.3. Multiple Sclerosis
3.2.4. Amyotrophic Lateral Sclerosis
3.3. Mitochondrial Dysfunction and Cardiovascular Diseases (CVD)
3.3.1. Atherosclerosis
3.3.2. Ischemic Stroke
4. Strategies for Mitochondria-Targeted Therapy
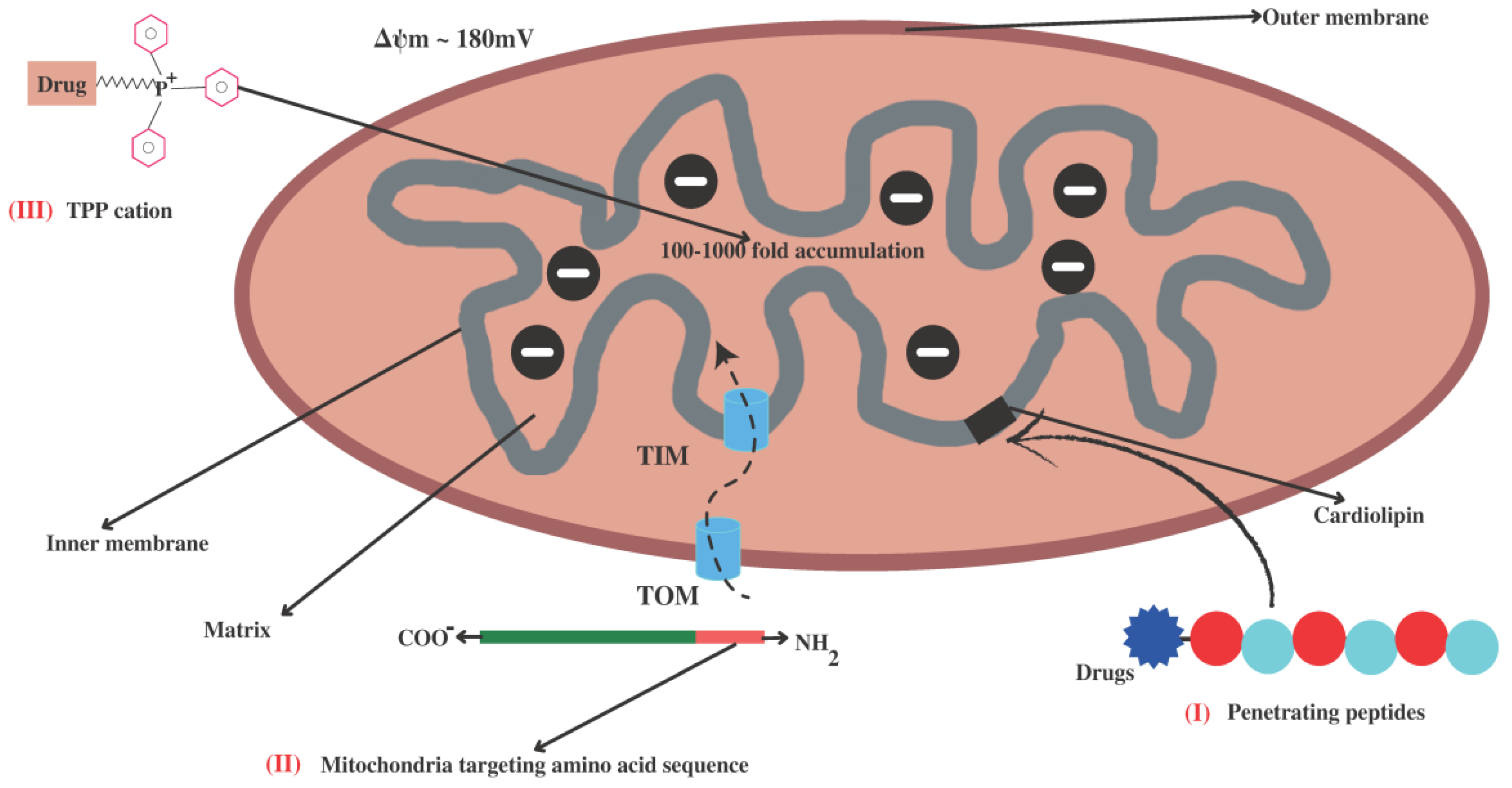
4.1. Small Lipophilic Cationic Molecules Targeting Mitochondria
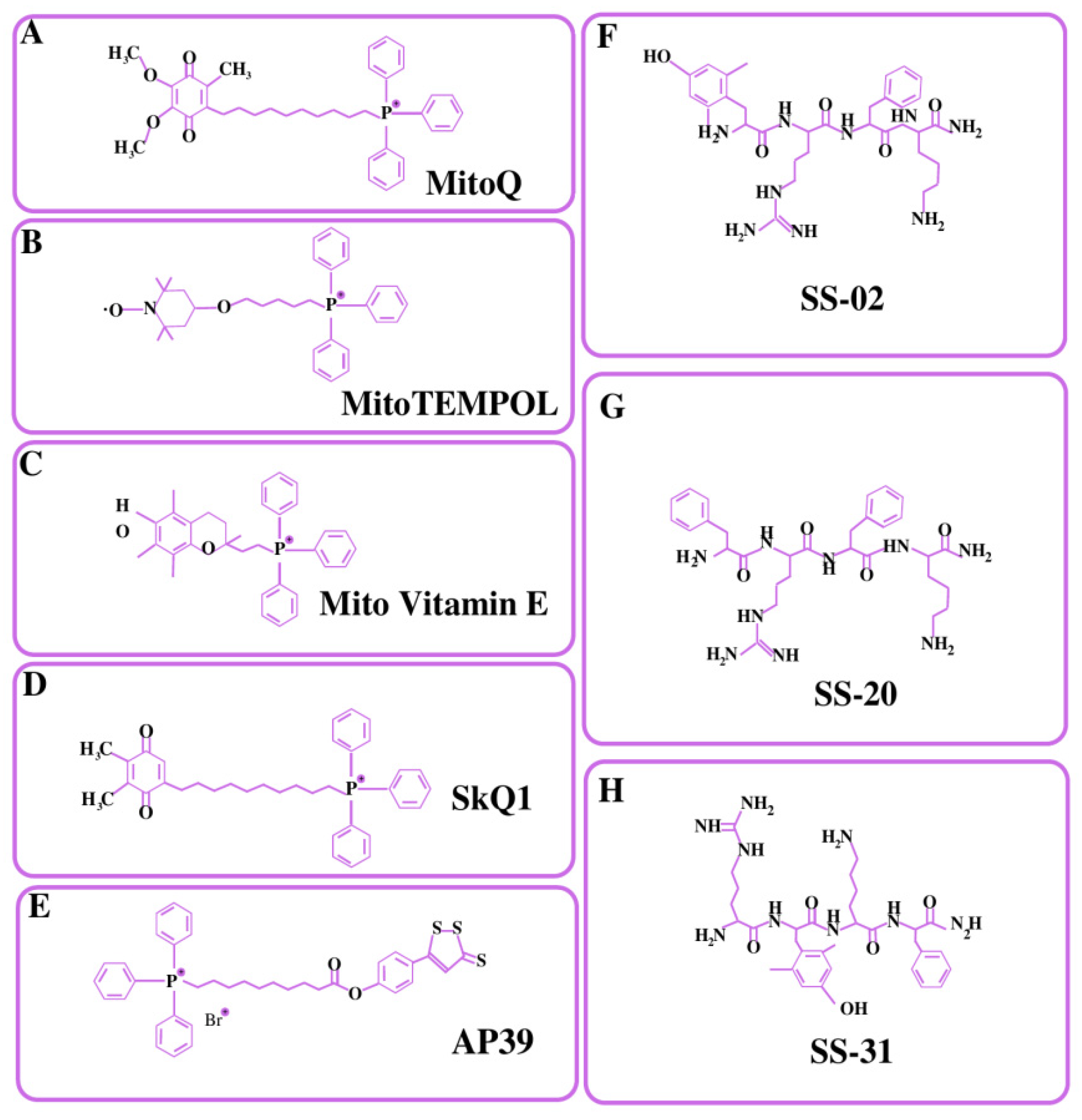
4.1.1. Triphenylphosphonium Cation (TPP+)
4.1.2. Rhodamine
4.1.3. Pyridinium Salts
4.2. Mitochondria-Targeting Signal Peptides
4.3. Cardiolipin Targeting (Penetrating Peptides)
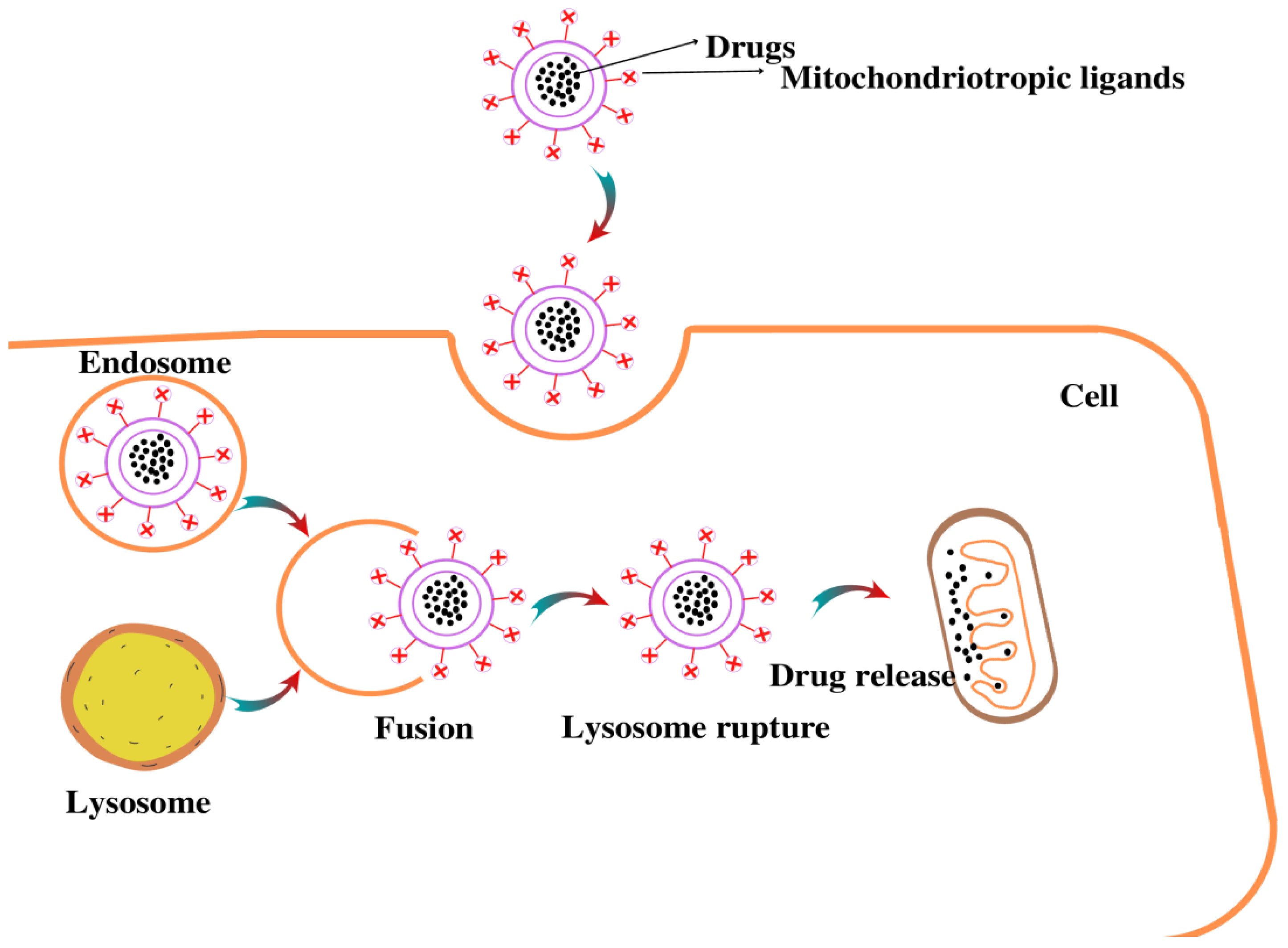
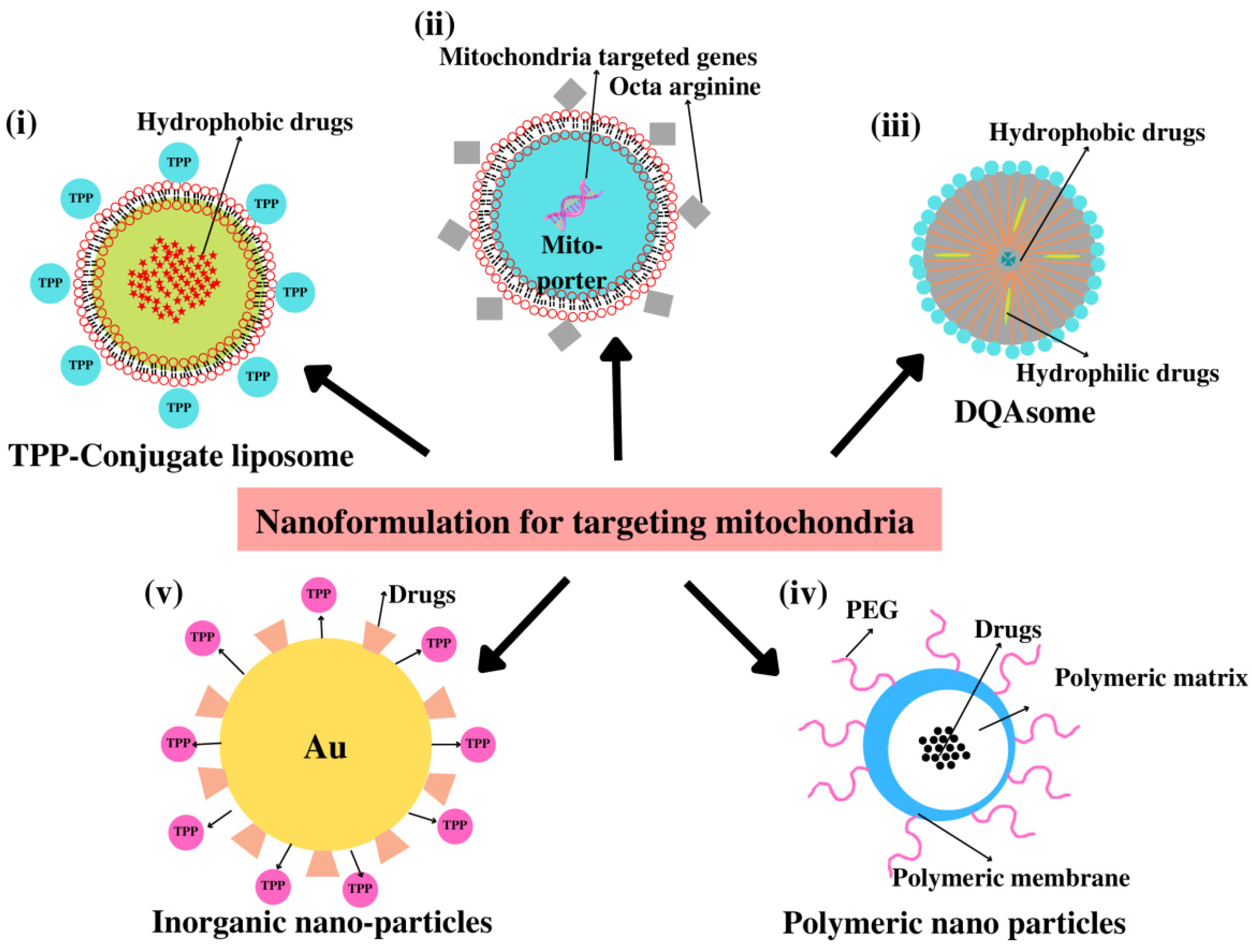
4.4. Nanoparticle (NPs)-Based Drug Delivery
4.4.1. Dequalinium (DQA)
4.4.2. Liposomes
4.4.3. Polymeric Nanoparticles
4.4.4. Inorganic Nanoparticles
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IMM | Inner mitochondrial membrane |
| OMM | Outer mitochondrial membrane |
| IMS | Intermembrane space |
| TPP | Triphenylphosphonium |
| Dmt | Dimethyltyrosine |
| DQA | Dequalinium |
| MitoE | TPP-vitamin E |
| Mito-Q | TPP-ubiquinone |
| STPP | Stearyl conjugated TPP liposomes |
| PEG | Polyethylene glycol |
| PTX | Paclitaxel |
| PLGA | Poly (lactic-co-glycolic acid) |
| BCL-2 | B-cell lymphoma |
| ALS | Amyotrophic lateral sclerosis |
| PD | Parkinson’s disease |
| AD | Alzheimer’s disease |
| MS | Multiple sclerosis |
References
- Javadov, S.; Kuznetsov, A.V. Mitochondria: The Cell Powerhouse and Nexus of Stress; Frontiers Media SA: Lausanne, Switzerland, 2013; Volume 4, p. 207. [Google Scholar]
- Pathak, R.K.; Kolishetti, N.; Dhar, S. Targeted nanoparticles in mitochondrial medicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 315–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, M.F.; Kelso, G.; Blaikie, F.H.; James, A.M.; Cocheme, H.M.; Filipovska, A.; Da Ros, T.; Hurd, T.; Smith, R.A.; Murphy, M.P. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry 2005, 70, 222–230. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.R.; Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waseem, R.; Shamsi, A.; Kazim, S.N.; Islam, A. An insight into mitochondrial dysfunction and its implications in neurological diseases. Curr. Drug Targets 2021, 22, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial disease. Lancet 2006, 368, 70–82. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R. Apoptotic pathways: The roads to ruin. Cell 1998, 94, 695–698. [Google Scholar] [CrossRef] [Green Version]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-f. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, R. Coenzyme Q10: The essential nutrient. J. Pharm. Bioallied Sci. 2011, 3, 466–467. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; McAllister, D.M.; Mackinnon, A.C.; Joseph, J.; Dwinell, M.B.; Kalyanaraman, B. Mitochondria-targeted vitamin E analogs inhibit breast cancer cell energy metabolism and promote cell death. BMC Cancer 2013, 13, 285. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1282–1294. [Google Scholar] [CrossRef] [Green Version]
- Kelso, G.F.; Maroz, A.; Cochemé, H.M.; Logan, A.; Prime, T.A.; Peskin, A.V.; Winterbourn, C.C.; James, A.M.; Ross, M.F.; Brooker, S. A mitochondria-targeted macrocyclic Mn (II) superoxide dismutase mimetic. Chem. Biol. 2012, 19, 1237–1246. [Google Scholar] [CrossRef] [Green Version]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ sensing: Its role in calcium homeostasis and signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer. Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [Green Version]
- Ryan, K.C.; Ashkavand, Z.; Norman, K.R. The role of mitochondrial calcium homeostasis in Alzheimer’s and related diseases. Int. J. Mol. Sci. 2020, 21, 9153. [Google Scholar] [CrossRef] [PubMed]
- David, E.C. Calcium signaling. Cell 1995, 47, 423–431. [Google Scholar]
- Pradelli, L.A.; Bénéteau, M.; Ricci, J.-E. Mitochondrial control of caspase-dependent and-independent cell death. Cell. Mol. Life Sci. 2010, 67, 1589–1597. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beinert, H.; Holm, R.H.; Munck, E. Iron-sulfur clusters: Nature’s modular, multipurpose structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.W.; Broderick, J.B. Emerging paradigms for complex iron-sulfur cofactor assembly and insertion. Annu. Rev. Biochem. 2012, 81, 429–450. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Biogenesis of iron-sulfur clusters in mammalian cells: New insights and relevance to human disease. Dis. Model. Mech. 2012, 5, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Lill, R. Function and biogenesis of iron–sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef]
- Navarro-Sastre, A.; Tort, F.; Stehling, O.; Uzarska, M.A.; Arranz, J.A.; Del Toro, M.; Labayru, M.T.; Landa, J.; Font, A.; Garcia-Villoria, J. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am. J. Hum. Genet. 2011, 89, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Sheftel, A.; Stehling, O.; Lill, R. Iron–sulfur proteins in health and disease. Trends Endocrinol. Metab. 2010, 21, 302–314. [Google Scholar] [CrossRef]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.-C.; Tseng, L.-M.; Lee, H.-C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]
- Fu, X.; Shi, Y.; Qi, T.; Qiu, S.; Huang, Y.; Zhao, X.; Sun, Q.; Lin, G. Precise design strategies of nanomedicine for improving cancer therapeutic efficacy using subcellular targeting. Signal Transduct. Target. Ther. 2020, 5, 262. [Google Scholar] [CrossRef]
- Forrest, M.D. Why cancer cells have a more hyperpolarised mitochondrial membrane potential and emergent prospects for therapy. BioRxiv 2015, 025197. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Asp. Med. 2010, 31, 1–20. [Google Scholar] [CrossRef]
- Mazurek, S.; Boschek, C.; Eigenbrodt, E. The role of phosphometabolites in cell proliferation, energy metabolism, and tumor therapy. J. Bioenerg. Biomembr. 1997, 29, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Fisher, B.; Costantino, J.P.; Wickerham, D.L.; Redmond, C.K.; Kavanah, M.; Cronin, W.M.; Vogel, V.; Robidoux, A.; Dimitrov, N.; Atkins, J. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. JNCI J. Natl. Cancer Inst. 1998, 90, 1371–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundholm, K.; Edström, S.; Karlberg, I.; Ekman, L.; Schersten, T. Glucose turnover, gluconeogenesis from glycerol, and estimation of net glucose cycling in cancer patients. Cancer 1982, 50, 1142–1150. [Google Scholar] [CrossRef]
- Ockner, R.K.; Kaikaus, R.M.; Bass, N.M. Fatty-acid metabolism and the pathogenesis of hepatocellular carcinoma: Review and hypothesis. Hepatology 1993, 18, 669–676. [Google Scholar] [CrossRef]
- Pedersen, P.L.; Greenawalt, J.W.; Reynafarje, B.; Hullihen, J.; Decker, G.L.; Soper, J.W.; Bustamente, E. Preparation and characterization of mitochondria and submitochondrial particles of rat liver and liver-derived tissues. Methods Cell Biol. 1978, 20, 411–481. [Google Scholar] [PubMed]
- Garcea, R.L.; Alberts, B. Comparative studies of histone acetylation in nucleosomes, nuclei, and intact cells. Evidence for special factors which modify acetylase action. J. Biol. Chem. 1980, 255, 11454–11463. [Google Scholar] [CrossRef] [PubMed]
- Capuano, F.; Guerrieri, F.; Papa, S. Oxidative phosphorylation enzymes in normal and neoplastic cell growth. J. Bioenerg. Biomembr. 1997, 29, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Cuezva, J.M.; Ostronoff, L.K.; Ricart, J.; de Heredia, M.L.; Di Liegro, C.M.; Izquierdo, J.M. Mitochondrial biogenesis in the liver during development and oncogenesis. J. Bioenerg. Biomembr. 1997, 29, 365–377. [Google Scholar] [CrossRef]
- Rampino, N.; Yamamoto, H.; Ionov, Y.; Li, Y.; Sawai, H.; Reed, J.C.; Perucho, M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275, 967–969. [Google Scholar] [CrossRef]
- Brimmell, M.; Mendiola, R.; Mangion, J.; Packham, G. BAX frameshift mutations in cell lines derived from human haemopoietic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene 1998, 16, 1803–1812. [Google Scholar] [CrossRef] [Green Version]
- Shirmanova, M.V.; Druzhkova, I.N.; Lukina, M.M.; Matlashov, M.E.; Belousov, V.V.; Snopova, L.B.; Prodanetz, N.N.; Dudenkova, V.V.; Lukyanov, S.A.; Zagaynova, E.V. Intracellular pH imaging in cancer cells in vitro and tumors in vivo using the new genetically encoded sensor SypHer2. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 1905–1911. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Cho, Y.-Y.; Shim, M.S.; Lee, J.Y.; Lee, H.S.; Kang, H.C. Mitochondria-targeted drug delivery in cancers. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165808. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, V. Targeting mitochondria in fighting cancer. Curr. Pharm. Des. 2011, 17, 4034–4046. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.; Bouchier-Hayes, L.; Green, D. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef] [Green Version]
- Kristián, T.; Siesjö, B.K. Calcium in ischemic cell death. Stroke 1998, 29, 705–718. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Qian, T.; Bradham, C.A.; Brenner, D.A.; Cascio, W.E.; Trost, L.C.; Nishimura, Y.; Nieminen, A.-L.; Herman, B. Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J. Bioenerg. Biomembr. 1999, 31, 305–319. [Google Scholar] [CrossRef]
- Moll, U.; Schramm, L. p53--an acrobat in tumorigenesis. Crit. Rev. Oral Biol. Med. 1998, 9, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Calió, M.L.; Henriques, E.; Siena, A.; Bertoncini, C.R.A.; Gil-Mohapel, J.; Rosenstock, T.R. Mitochondrial dysfunction, neurogenesis, and epigenetics: Putative implications for amyotrophic lateral sclerosis neurodegeneration and treatment. Front. Neurosci. 2020, 14, 679. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: From mechanism to therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Mahad, D.J.; Lassmann, H.; van Horssen, J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med. 2014, 20, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Muyderman, H.; Chen, T. Mitochondrial dysfunction in amyotrophic lateral sclerosis–a valid pharmacological target? Br. J. Pharmacol. 2014, 171, 2191–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folbergrová, J.; Kunz, W.S. Mitochondrial dysfunction in epilepsy. Mitochondrion 2012, 12, 35–40. [Google Scholar] [CrossRef]
- Sulaiman, S.A.; Mohd Rani, Z.; Mohd Radin, F.Z.; Abdul Murad, N.A. Advancement in the diagnosis of mitochondrial diseases. J. Transl. Genet. Genom. 2020, 4, 159–187. [Google Scholar] [CrossRef]
- Jurcau, A. Insights into the pathogenesis of neurodegenerative diseases: Focus on mitochondrial dysfunction and oxidative stress. Int. J. Mol. Sci. 2021, 22, 11847. [Google Scholar] [CrossRef]
- Larosa, V.; Remacle, C. Insights into the respiratory chain and oxidative stress. Biosci. Rep. 2018, 38, BSR20171492. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003. [Google Scholar]
- Selvaraji, S.; Poh, L.; Natarajan, V.; Mallilankaraman, K.; Arumugam, T.V. Negative conditioning of mitochondrial dysfunction in age-related neurodegenerative diseases. Cond. Med. 2019, 2, 30. [Google Scholar]
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Waseem, R.; Shamsi, A.; Mohammad, T.; Alhumaydhi, F.A.; Kazim, S.N.; Hassan, M.I.; Ahmad, F.; Islam, A. Multispectroscopic and molecular docking insight into elucidating the interaction of irisin with Rivastigmine tartrate: A combinational therapy approach to fight Alzheimer’s disease. ACS Omega 2021, 6, 7910–7921. [Google Scholar] [CrossRef] [PubMed]
- Waseem, R.; Shamsi, A.; Mohammad, T.; Hassan, M.I.; Kazim, S.N.; Chaudhary, A.A.; Rudayni, H.A.; Al-Zharani, M.; Ahmad, F.; Islam, A. FNDC5/Irisin: Physiology and Pathophysiology. Molecules 2022, 27, 1118. [Google Scholar] [CrossRef] [PubMed]
- Förstl, H.; Kurz, A. Clinical features of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef]
- Waseem, R.; Anwar, S.; Khan, S.; Shamsi, A.; Hassan, M.I.; Anjum, F.; Shafie, A.; Islam, A.; Yadav, D.K. MAP/Microtubule Affinity Regulating Kinase 4 Inhibitory Potential of Irisin: A New Therapeutic Strategy to Combat Cancer and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 10986. [Google Scholar] [CrossRef]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; Lezi, E.; Cardoso, S.M.; Swerdlow, R.H. Mitochondrial abnormalities in Alzheimer’s disease: Possible targets for therapeutic intervention. Adv. Pharmacol. 2012, 64, 83–126. [Google Scholar]
- Lee, J. Mitochondrial drug targets in neurodegenerative diseases. Bioorg. Med. Chem. Lett. 2016, 26, 714–720. [Google Scholar] [CrossRef]
- Blagov, A.V.; Grechko, A.V.; Nikiforov, N.G.; Borisov, E.E.; Sadykhov, N.K.; Orekhov, A.N. Role of impaired mitochondrial dynamics processes in the pathogenesis of Alzheimer’s disease. Int. J. Mol. Sci. 2022, 23, 6954. [Google Scholar] [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial dysfunction in Alzheimer’s disease and progress in mitochondria-targeted therapeutics. Curr. Behav. Neurosci. Rep. 2019, 6, 88–102. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-g.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Xu, H.; Yu, H.; Zhang, N.; Ye, Y.; Estevez, A.G.; Deng, H.; Gibson, G.E. Inactivation and reactivation of the mitochondrial α-ketoglutarate dehydrogenase complex. J. Biol. Chem. 2011, 286, 17640–17648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenini, G.; Voos, W. Mitochondria as potential targets in Alzheimer disease therapy: An update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, I.; Jha, S. Mitochondrial dysfunction and Alzheimer’s disease: Role of microglia. Front. Aging Neurosci. 2020, 252. [Google Scholar] [CrossRef] [PubMed]
- Feeney, C.J.; Frantseva, M.V.; Carlen, P.L.; Pennefather, P.S.; Shulyakova, N.; Shniffer, C.; Mills, L.R. Vulnerability of glial cells to hydrogen peroxide in cultured hippocampal slices. Brain Res. 2008, 1198, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.-S.; Geng, W.-S.; Jia, J.-J.; Chen, L.; Zhang, P.-P. Cellular and molecular basis of neurodegeneration in Parkinson disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Marsili, L.; Rizzo, G.; Colosimo, C. Diagnostic criteria for Parkinson’s disease: From James Parkinson to the concept of prodromal disease. Front. Neurol. 2018, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef] [Green Version]
- Greggio, E.; Cookson, M.R. Leucine-rich repeat kinase 2 mutations and Parkinson’s disease: Three questions. ASN Neuro 2009, 1, AN20090007. [Google Scholar] [CrossRef] [Green Version]
- Wauters, F.; Cornelissen, T.; Imberechts, D.; Martin, S.; Koentjoro, B.; Sue, C.; Vangheluwe, P.; Vandenberghe, W. LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 2020, 16, 203–222. [Google Scholar] [CrossRef]
- Cha, M.-Y.; Kim, D.K.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef] [Green Version]
- Momb, J.; Lewandowski, J.P.; Bryant, J.D.; Fitch, R.; Surman, D.R.; Vokes, S.A.; Appling, D.R. Deletion of Mthfd1l causes embryonic lethality and neural tube and craniofacial defects in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 549–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco-Iborra, S.; Cuadros, T.; Parent, A.; Romero-Gimenez, J.; Vila, M.; Perier, C. Defective mitochondrial protein import contributes to complex I-induced mitochondrial dysfunction and neurodegeneration in Parkinson’s disease. Cell Death Dis. 2018, 9, 1122. [Google Scholar] [CrossRef] [Green Version]
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2, a009258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Reyes, P.F.; Golden, G.T.; Woltjer, R.L.; Hulette, C.; Montine, T.J.; Zhang, J. Mitochondrial DNA deletions/rearrangements in parkinson disease and related neurodegenerative disorders. J. Neuropathol. Exp. Neurol. 2002, 61, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Luoma, P.; Melberg, A.; Rinne, J.O.; Kaukonen, J.A.; Nupponen, N.N.; Chalmers, R.M.; Oldfors, A.; Rautakorpi, I.; Peltonen, L.; Majamaa, K. Parkinsonism, premature menopause, and mitochondrial DNA polymerase γ mutations: Clinical and molecular genetic study. Lancet 2004, 364, 875–882. [Google Scholar] [CrossRef]
- Su, K.; Bourdette, D.; Forte, M. Mitochondrial dysfunction and neurodegeneration in multiple sclerosis. Front. Physiol. 2013, 4, 169. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.-Y.; Lu, M.-H.; Yuan, D.-J.; Xu, D.-E.; Yao, P.-P.; Ji, W.-L.; Chen, H.; Liu, W.-L.; Yan, C.-X.; Xia, Y.-Y. Mitochondrial dysfunction in neural injury. Front. Neurosci. 2019, 13, 30. [Google Scholar] [CrossRef] [Green Version]
- Sadeghian, M.; Mastrolia, V.; Rezaei Haddad, A.; Mosley, A.; Mullali, G.; Schiza, D.; Sajic, M.; Hargreaves, I.; Heales, S.; Duchen, M.R. Mitochondrial dysfunction is an important cause of neurological deficits in an inflammatory model of multiple sclerosis. Sci. Rep. 2016, 6, 33249. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, L.; De Aguilar, J.-l.G.; Oudart, H.; De Tapia, M.; Barbeito, L.; Loeffler, J.-P. Mitochondria in amyotrophic lateral sclerosis: A trigger and a target. Neurodegener. Dis. 2004, 1, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Keon, M.; Musrie, B.; Dinger, M.; Brennan, S.E.; Santos, J.; Saksena, N.K. Destination amyotrophic lateral sclerosis. Front. Neurol. 2021, 12, 392. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Akiyama, T.; Warita, H.; Aoki, M. Omics approach to axonal dysfunction of motor neurons in amyotrophic lateral sclerosis (ALS). Front. Neurosci. 2020, 14, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, M.A.; Villavicencio-Tejo, F.; Quintanilla, R.A. The use of fibroblasts as a valuable strategy for studying mitochondrial impairment in neurological disorders. Transl. Neurodegener. 2022, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, X.; Huo, Z.; Chen, Y.; Liu, J.; Zhao, Z.; Meng, F.; Su, Q.; Bao, W.; Zhang, L. The Impact of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2049. [Google Scholar] [CrossRef]
- Collaborators, G.; Ärnlöv, J. Global age-sex-specific fertility, mortality, healthy life expectancy (HALE), and population estimates in 204 countries and territories, 1950–2019: A comprehensive demographic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1160–1203. [Google Scholar]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is mitochondrial dysfunction a common root of noncommunicable chronic diseases? Endocr. Rev. 2020, 41, bnaa005. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K. Mitochondria and cardiovascular diseases—From pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Lesèche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef] [Green Version]
- Camara, A.K.; Lesnefsky, E.J.; Stowe, D.F. Potential therapeutic benefits of strategies directed to mitochondria. Antioxid. Redox Signal. 2010, 13, 279–347. [Google Scholar] [CrossRef] [Green Version]
- Mantzarlis, K.; Tsolaki, V.; Zakynthinos, E. Role of oxidative stress and mitochondrial dysfunction in sepsis and potential therapies. Oxidative Med. Cell. Longev. 2017, 2017, 5985209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadlec, A.O.; Chabowski, D.S.; Ait-Aissa, K.; Gutterman, D.D. Role of PGC-1α in vascular regulation: Implications for atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1467–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, F.-I.; Lien, L.-M.; Chen, S.-T.; Bai, C.-H.; Sun, M.-C.; Tseng, H.-P.; Chen, Y.-W.; Chen, C.-H.; Jeng, J.-S.; Tsai, S.-Y. Get with the guidelines-stroke performance indicators: Surveillance of stroke care in the Taiwan stroke registry: Get with the guidelines-stroke in Taiwan. Circulation 2010, 122, 1116–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwamm, L.H.; Fonarow, G.C.; Reeves, M.J.; Pan, W.; Frankel, M.R.; Smith, E.E.; Ellrodt, G.; Cannon, C.P.; Liang, L.; Peterson, E. Get With the Guidelines–Stroke is associated with sustained improvement in care for patients hospitalized with acute stroke or transient ischemic attack. Circulation 2009, 119, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-M.; Zipfel, G.J.; Choi, D.W. The changing landscape of ischaemic brain injury mechanisms. Nature 1999, 399, A7–A14. [Google Scholar] [CrossRef]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef]
- Chen, S.D.; Lin, T.K.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Protective effects of peroxisome proliferator-activated receptors γ coactivator-1α against neuronal cell death in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res. 2010, 88, 605–613. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, J.; Schapira, A.H. Mitochondrial respiratory chain disorders II: Neurodegenerative disorders and nuclear gene defects. Lancet 2000, 355, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. Mitochondrial dysfunction and oxidative stress: Cause and consequence of epileptic seizures. Free Radic. Biol. Med. 2004, 37, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Chao, D.T.; Korsmeyer, S.J. BCL-2 family: Regulators of cell death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef]
- Kitayama, T.; Ogita, K.; Yoneda, Y. Sustained potentiation of AP1 DNA binding is not always associated with neuronal death following systemic administration of kainic acid in murine hippocampus. Neurochem. Int. 1999, 35, 453–462. [Google Scholar] [CrossRef]
- Liu, D.; Lu, C.; Wan, R.; Auyeung, W.W.; Mattson, M.P. Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of Bax translocation and cytochrome c release. J. Cereb. Blood Flow Metab. 2002, 22, 431–443. [Google Scholar] [CrossRef]
- Rami, A.; Bechmann, I.; Stehle, J.H. Exploiting endogenous anti-apoptotic proteins for novel therapeutic strategies in cerebral ischemia. Prog. Neurobiol. 2008, 85, 273–296. [Google Scholar] [CrossRef]
- Song, Y.S.; Lee, Y.-S.; Narasimhan, P.; Chan, P.H. Reduced oxidative stress promotes NF-κB-mediated neuroprotective gene expression after transient focal cerebral ischemia: Lymphocytotrophic cytokines and antiapoptotic factors. J. Cereb. Blood Flow Metab. 2007, 27, 764–775. [Google Scholar] [CrossRef]
- Zhao, H.; Yenari, M.A.; Cheng, D.; Sapolsky, R.M.; Steinberg, G.K. Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. J. Neurochem. 2003, 85, 1026–1036. [Google Scholar] [CrossRef]
- Ferrer, I.; Friguls, B.; Dalfo, E.; Justicia, C.; Planas, A. Caspase-dependent and caspase-independent signalling of apoptosis in the penumbra following middle cerebral artery occlusion in the adult rat. Neuropathol. Appl. Neurobiol. 2003, 29, 472–481. [Google Scholar] [CrossRef]
- Saito, A.; Hayashi, T.; Okuno, S.; Ferrand-Drake, M.; Chan, P.H. Interaction between XIAP and Smac/DIABLO in the mouse brain after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 1010–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Culmsee, C.; Zhu, C.; Landshamer, S.; Becattini, B.; Wagner, E.; Pellecchia, M.; Blomgren, K.; Plesnila, N. Apoptosis-inducing factor triggered by poly (ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J. Neurosci. 2005, 25, 10262–10272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plesnila, N.; Zhu, C.; Culmsee, C.; Gröger, M.; Moskowitz, M.A.; Blomgren, K. Nuclear translocation of apoptosis-inducing factor after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2004, 24, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Jameson, V.J.; Cochemé, H.M.; Logan, A.; Hanton, L.R.; Smith, R.A.; Murphy, M.P. Synthesis of triphenylphosphonium vitamin E derivatives as mitochondria-targeted antioxidants. Tetrahedron 2015, 71, 8444–8453. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Koilkonda, R.D.; Chou, T.-H.; Porciatti, V.; Ozdemir, S.S.; Chiodo, V.; Boye, S.L.; Boye, S.E.; Hauswirth, W.W.; Lewin, A.S. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, E1238–E1247. [Google Scholar] [CrossRef]
- Szeto, H.; Birk, A. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin. Pharmacol. Ther. 2014, 96, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef]
- Kagan, V.E.; Borisenko, G.G.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Potapovich, A.I.; Kini, V.; Amoscato, A.A.; Fujii, Y. Oxidative lipidomics of apoptosis: Redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic. Biol. Med. 2004, 37, 1963–1985. [Google Scholar] [CrossRef]
- Pfanner, N.; Geissler, A. Versatility of the mitochondrial protein import machinery. Nat. Rev. Mol. Cell Biol. 2001, 2, 339–349. [Google Scholar] [CrossRef]
- Galley, H.F. Bench-to-bedside review: Targeting antioxidants to mitochondria in sepsis. Crit. Care 2010, 14, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, R.J.; Smith, R.A.; Murphy, M.P. Synthesis and characterization of thiobutyltriphenylphosphonium bromide, a novel thiol reagent targeted to the mitochondrial matrix. Arch. Biochem. Biophys. 1995, 322, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Módis, K.; Yanagi, K.; Coletta, C.; Le Trionnaire, S.; Perry, A.; Wood, M.E.; Whiteman, M.; Szabo, C. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 2014, 41, 120–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [Green Version]
- Antonenko, Y.N.; Avetisyan, A.; Bakeeva, L.; Chernyak, B.; Chertkov, V.; Domnina, L.; Ivanova, O.Y.; Izyumov, D.; Khailova, L.; Klishin, S. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef]
- Trnka, J.; Blaikie, F.H.; Logan, A.; Smith, R.A.; Murphy, M.P. Antioxidant properties of MitoTEMPOL and its hydroxylamine. Free Radic. Res. 2009, 43, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Elrod, J.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, A.; Olah, G.; Szczesny, B.; Wood, M.E.; Whiteman, M.; Szabo, C. AP39, a mitochondrially-targeted hydrogen sulfide donor, exerts protective effects in renal epithelial cells subjected to oxidative stress in vitro and in acute renal injury in vivo. Shock 2016, 45, 88. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.-l.; Fang, F.; Qiao, P.-f.; Yan, N.; Gao, D.; Yan, Y. AP39, a mitochondria-targeted hydrogen sulfide donor, supports cellular bioenergetics and protects against Alzheimer’s disease by preserving mitochondrial function in APP/PS1 mice and neurons. Oxidative Med. Cell. Longev. 2016, 2016, 8360738. [Google Scholar] [CrossRef] [Green Version]
- Iomdina, E.N.; Khoroshilova-Maslova, I.P.; Robustova, O.V.; Averina, O.A.; Kovaleva, N.A.; Aliev, G.; Reddy, V.P.; Zamyatnin, A.A., Jr.; Skulachev, M.V.; Senin, I.I. Mitochondria-targeted antioxidant SkQ1 reverses glaucomatous lesions in rabbits. Front. Biosci.-Landmark 2015, 20, 892–901. [Google Scholar]
- Markovets, A.M.; Fursova, A.Z.; Kolosova, N.G. Therapeutic action of the mitochondria-targeted antioxidant SkQ1 on retinopathy in OXYS rats linked with improvement of VEGF and PEDF gene expression. PLoS ONE 2011, 6, e21682. [Google Scholar] [CrossRef] [PubMed]
- Baracca, A.; Sgarbi, G.; Solaini, G.; Lenaz, G. Rhodamine 123 as a probe of mitochondrial membrane potential: Evaluation of proton flux through F0 during ATP synthesis. Biochim. Biophys. Acta (BBA)-Bioenerg. 2003, 1606, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Lampidis, T.; Hasin, Y.; Weiss, M.a.; Chen, L.B. Selective killing of carcinoma cells" in vitro" by lipophilic-cationic compounds: A cellular basis. Biomed. Pharmacother. = Biomed. Pharmacother. 1985, 39, 220–226. [Google Scholar] [PubMed]
- Antonenko, Y.N.; Avetisyan, A.V.; Cherepanov, D.A.; Knorre, D.A.; Korshunova, G.A.; Markova, O.V.; Ojovan, S.M.; Perevoshchikova, I.V.; Pustovidko, A.V.; Rokitskaya, T.I. Derivatives of rhodamine 19 as mild mitochondria-targeted cationic uncouplers. J. Biol. Chem. 2011, 286, 17831–17840. [Google Scholar] [CrossRef] [Green Version]
- Qian, K.; Chen, H.; Qu, C.; Qi, J.; Du, B.; Ko, T.; Xiang, Z.; Kandawa-Schulz, M.; Wang, Y.; Cheng, Z. Mitochondria-targeted delocalized lipophilic cation complexed with human serum albumin for tumor cell imaging and treatment. Nanomed. Nanotechnol. Biol. Med. 2020, 23, 102087. [Google Scholar] [CrossRef]
- von Heijne, G. Mitochondrial targeting sequences may form amphiphilic helices. EMBO J. 1986, 5, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Neupert, W.; Herrmann, J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef] [Green Version]
- Flierl, A.; Jackson, C.; Cottrell, B.; Murdock, D.; Seibel, P.; Wallace, D. Targeted delivery of DNA to the mitochondrial compartment via import sequence-conjugated peptide nucleic acid. Mol. Ther. 2003, 7, 550–557. [Google Scholar] [CrossRef]
- Cha, M.-Y.; Han, S.-H.; Son, S.M.; Hong, H.-S.; Choi, Y.-J.; Byun, J.; Mook-Jung, I. Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef]
- Horton, K.L.; Stewart, K.M.; Fonseca, S.B.; Guo, Q.; Kelley, S.O. Mitochondria-penetrating peptides. Chem. Biol. 2008, 15, 375–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.C.; Son, M.; Kang, S.; Im, S.; Piao, Y.; Lim, K.S.; Song, M.-Y.; Park, K.-S.; Kim, Y.-H.; Pak, Y.K. Cell-penetrating artificial mitochondria-targeting peptide-conjugated metallothionein 1A alleviates mitochondrial damage in Parkinson’s disease models. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeto, H.H.; Schiller, P.W. Novel therapies targeting inner mitochondrial membrane—From discovery to clinical development. Pharm. Res. 2011, 28, 2669–2679. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.R.; Oh, K.T.; Oh, Y.T.; Baik, H.J.; Park, S.Y.; Youn, Y.S.; Lee, E.S. A novel pH-responsive polysaccharidic ionic complex for proapoptotic D-(KLAKLAK) 2 peptide delivery. Chem. Commun. 2011, 47, 3852–3854. [Google Scholar] [CrossRef]
- Jiang, L.; Li, L.; He, X.; Yi, Q.; He, B.; Cao, J.; Pan, W.; Gu, Z. Overcoming drug-resistant lung cancer by paclitaxel loaded dual-functional liposomes with mitochondria targeting and pH-response. Biomaterials 2015, 52, 126–139. [Google Scholar] [CrossRef]
- Li, L.; Geisler, I.; Chmielewski, J.; Cheng, J.-X. Cationic amphiphilic polyproline helix P11LRR targets intracellular mitochondria. J. Controll. Release 2010, 142, 259–266. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-penetrating peptides: From basic research to clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Szabó, I.; Yousef, M.a.; Soltész, D.; Bató, C.; Mező, G.; Bánóczi, Z. Redesigning of Cell-Penetrating Peptides to Improve Their Efficacy as a Drug Delivery System. Pharmaceutics 2022, 14, 907. [Google Scholar] [CrossRef]
- Weissig, V. DQAsomes as the prototype of mitochondria-targeted pharmaceutical nanocarriers: Preparation, characterization, and use. In Mitochondrial Medicine; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–11. [Google Scholar]
- Bae, Y.; Jung, M.K.; Lee, S.; Song, S.J.; Mun, J.Y.; Green, E.S.; Han, J.; Ko, K.S.; Choi, J.S. Dequalinium-based functional nanosomes show increased mitochondria targeting and anticancer effect. Eur. J. Pharm. Biopharm. 2018, 124, 104–115. [Google Scholar] [CrossRef]
- D’Souza, G.G.; Rammohan, R.; Cheng, S.-M.; Torchilin, V.P.; Weissig, V. DQAsome-mediated delivery of plasmid DNA toward mitochondria in living cells. J. Controll. Release 2003, 92, 189–197. [Google Scholar] [CrossRef]
- Zupancic, S.; Kocbek, P.; Zariwala, M.G.; Renshaw, D.; Gul, M.O.; Elsaid, Z.; Taylor, K.M.; Somavarapu, S. Design and development of novel mitochondrial targeted nanocarriers, DQAsomes for curcumin inhalation. Mol. Pharm. 2014, 11, 2334–2345. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.G.; Cheng, S.-M.; Boddapati, S.V.; Horobin, R.W.; Weissig, V. Nanocarrier-assisted sub-cellular targeting to the site of mitochondria improves the pro-apoptotic activity of paclitaxel. J. Drug Target. 2008, 16, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-I.; Yeh, M.-K. Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2012, 7, 49. [Google Scholar]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCain, C.; Grytnes, J. Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Chichester, UK, 2010. [Google Scholar]
- Guo, F.; Yu, M.; Wang, J.; Tan, F.; Li, N. The mitochondria-targeted and IR780-regulated theranosomes for imaging and enhanced photodynamic/photothermal therapy. RSC Adv. 2016, 6, 11070–11076. [Google Scholar] [CrossRef]
- Rin Jean, S.; Tulumello, D.V.; Wisnovsky, S.P.; Lei, E.K.; Pereira, M.P.; Kelley, S.O. Molecular vehicles for mitochondrial chemical biology and drug delivery. ACS Chem. Biol. 2014, 9, 323–333. [Google Scholar] [CrossRef]
- Boddapati, S.V.; D’Souza, G.G.; Erdogan, S.; Torchilin, V.P.; Weissig, V. Organelle-targeted nanocarriers: Specific delivery of liposomal ceramide to mitochondria enhances its cytotoxicity in vitro and in vivo. Nano Lett. 2008, 8, 2559–2563. [Google Scholar] [CrossRef]
- Biswas, S.; Dodwadkar, N.S.; Deshpande, P.P.; Torchilin, V.P. Liposomes loaded with paclitaxel and modified with novel triphenylphosphonium-PEG-PE conjugate possess low toxicity, target mitochondria and demonstrate enhanced antitumor effects in vitro and in vivo. J. Controll. Release 2012, 159, 393–402. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, B.; Liao, H.; Song, X.; Wu, H.; Wang, H.; Shen, H.; Ma, X.; Tan, M. Liposomal nanohybrid cerasomes for mitochondria-targeted drug delivery. J. Mater. Chem. B 2015, 3, 7291–7299. [Google Scholar] [CrossRef]
- Yasuzaki, Y.; Yamada, Y.; Harashima, H. Mitochondrial matrix delivery using MITO-Porter, a liposome-based carrier that specifies fusion with mitochondrial membranes. Biochem. Biophys. Res. Commun. 2010, 397, 181–186. [Google Scholar] [CrossRef]
- Yamada, Y.; Furukawa, R.; Yasuzaki, Y.; Harashima, H. Dual function MITO-Porter, a nano carrier integrating both efficient cytoplasmic delivery and mitochondrial macromolecule delivery. Mol. Ther. 2011, 19, 1449–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Akita, H.; Kamiya, H.; Kogure, K.; Yamamoto, T.; Shinohara, Y.; Yamashita, K.; Kobayashi, H.; Kikuchi, H.; Harashima, H. MITO-Porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim. Biophys. Acta (BBA)-Biomembr. 2008, 1778, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Hirata, E.; Ushijima, N.; Harashima, H.; Yamada, Y. An effective in vivo mitochondria-targeting nanocarrier combined with a π-extended porphyrin-type photosensitizer. Nanoscale Adv. 2021, 3, 5919–5927. [Google Scholar]
- Kawamura, E.; Maruyama, M.; Abe, J.; Sudo, A.; Takeda, A.; Takada, S.; Yokota, T.; Kinugawa, S.; Harashima, H.; Yamada, Y. Validation of gene therapy for mutant mitochondria by delivering mitochondrial RNA using a MITO-porter. Mol. Ther.-Nucleic Acids 2020, 20, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Maruyama, M.; Kita, T.; Usami, S.-i.; Kitajiri, S.-i.; Harashima, H. The use of a MITO-Porter to deliver exogenous therapeutic RNA to a mitochondrial disease’s cell with a A1555G mutation in the mitochondrial 12S rRNA gene results in an increase in mitochondrial respiratory activity. Mitochondrion 2020, 55, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Tutar, Y. Editorial (Thematic Issue: Controlled Release and Drug Delivery in Diseases). Protein Pept. Lett. 2014, 21, 1085–1086. [Google Scholar] [CrossRef]
- Zhang, L.; Gu, F.; Chan, J.; Wang, A.; Langer, R.; Farokhzad, O. Nanoparticles in medicine: Therapeutic applications and developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [Google Scholar] [CrossRef]
- Marrache, S.; Dhar, S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc. Natl. Acad. Sci. USA 2012, 109, 16288–16293. [Google Scholar] [CrossRef] [Green Version]
- Giljohann, D.A.; Seferos, D.S.; Daniel, W.L.; Massich, M.D.; Patel, P.C.; Mirkin, C.A. Gold nanoparticles for biology and medicine. Spherical Nucleic Acids 2020, 55–90. [Google Scholar]
- Muhammad, N.; Zhao, H.; Song, W.; Gu, M.; Li, Q.; Liu, Y.; Li, C.; Wang, J.; Zhan, H. Silver nanoparticles functionalized Paclitaxel nanocrystals enhance overall anti-cancer effect on human cancer cells. Nanotechnology 2020, 32, 085105. [Google Scholar] [CrossRef]
- Oladimeji, O.; Akinyelu, J.; Daniels, A.; Singh, M. Modified gold nanoparticles for efficient delivery of betulinic acid to cancer cell mitochondria. Int. J. Mol. Sci. 2021, 22, 5072. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Yang, Y.; Gao, Z.; Jiang, H.; Ye, L.; Lai, Y.; Shen, Z.; Wu, Z.-S. Endogenous Stimuli-Responsive Autonomous Separation of Dual-Targeting DNA Guided Missile from Nanospacecraft for Intelligent Targeted Cancer Therapy. ACS Appl. Mater. Interfaces 2022, 14, 45201–45216. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fang, Z.; Song, M.; Liu, K. Mitochondrial targeted hierarchical drug delivery system based on ha-modified liposomes for cancer therapy. Eur. J. Med. Chem. 2022, 241, 114648. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Nakamura, K.; Abe, J.; Hyodo, M.; Haga, S.; Ozaki, M.; Harashima, H. Mitochondrial delivery of Coenzyme Q10 via systemic administration using a MITO-Porter prevents ischemia/reperfusion injury in the mouse liver. J. Controll. Release 2015, 213, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Munechika, R.; Kubota, F.; Sato, Y.; Sakurai, Y.; Harashima, H. Mitochondrial delivery of an anticancer drug via systemic administration using a mitochondrial delivery system that inhibits the growth of drug-resistant cancer engrafted on mice. J. Pharm. Sci. 2020, 109, 2493–2500. [Google Scholar] [CrossRef]
- Wang, X.-X.; Li, Y.-B.; Yao, H.-J.; Ju, R.-J.; Zhang, Y.; Li, R.-J.; Yu, Y.; Zhang, L.; Lu, W.-L. The use of mitochondrial targeting resveratrol liposomes modified with a dequalinium polyethylene glycol-distearoylphosphatidyl ethanolamine conjugate to induce apoptosis in resistant lung cancer cells. Biomaterials 2011, 32, 5673–5687. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, Z.-H.; Zhang, L.; Yao, H.-J.; Zhang, Y.; Li, R.-J.; Ju, R.-J.; Wang, X.-X.; Zhou, J.; Li, N. Mitochondrial targeting topotecan-loaded liposomes for treating drug-resistant breast cancer and inhibiting invasive metastases of melanoma. Biomaterials 2012, 33, 1808–1820. [Google Scholar] [CrossRef]
- Biswas, S.; Dodwadkar, N.S.; Sawant, R.R.; Koshkaryev, A.; Torchilin, V.P. Surface modification of liposomes with rhodamine-123-conjugated polymer results in enhanced mitochondrial targeting. J. Drug Target. 2011, 19, 552–561. [Google Scholar] [CrossRef]
- Zhan, H.; Song, W.; Gu, M.; Zhao, H.; Liu, Y.; Liu, B.; Wang, J. A New Gold Nanoparticles and Paclitaxel Co-Delivery System for Enhanced Anti-Cancer Effect Through Chemo-Photothermal Combination. J. Biomed. Nanotechnol. 2022, 18, 957–975. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, H.; Song, W.; Gu, M.; Liu, Y.; Liu, B.; Zhan, H. Gold Nanoparticle-Decorated Drug Nanocrystals for Enhancing Anticancer Efficacy and Reversing Drug Resistance Through Chemo-/Photothermal Therapy. Mol. Pharm. 2022, 19, 2518–2534. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, X.; Shen, Q.; Xing, D. Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nat. Commun. 2019, 10, 1704. [Google Scholar] [CrossRef] [Green Version]
- Qu, Q.; Ma, X.; Zhao, Y. Anticancer effect of α-tocopheryl succinate delivered by mitochondria-targeted mesoporous silica nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 34261–34269. [Google Scholar] [CrossRef]
- Phillip, J.M.; Lin, R.; Cheetham, A.; Stern, D.; Li, Y.; Wang, Y.; Wang, H.; Rini, D.; Cui, H.; Walston, J.D. Nature-inspired delivery of mitochondria-targeted angiotensin receptor blocker. Proc. Natl. Acad. Sci. Nexus 2022, 1, pgac147. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vector | Mitochondriotropic Moiety | Drug/Cargo | Model | Effect | Ref. |
|---|---|---|---|---|---|
| Liposomes | TPP+ | Paclitaxel, Doxorubicin | Hela and 4T1 cancer cell line | Shows enchanced cytotoxicity to cancer cells as compared to free drug, antitumor activity, and high cell uptake efficiency | [181,196] |
| Liposomes | MITO-Porter | Coenzyme Q10, Doxorubicin | Mouse liver ischemia/reperfusion injury (I/R injury) model, OS-RC-2 cells | Decreases the level of alanine amino-transferase, antitumor activity | [197,198] |
| Liposomes | DQA | Resveratrol | Human lung adenocarcinoma A549 cells and resistant A549/cDDP cells | Induced apoptosis in both cell lines via mitochondria | [199] |
| Liposomes | DQA | Topotecan | Breast cancer MCF-7 and resistant MCF-7/adr cells | Shows enhanced accumulation in mitochondria and anti-cancer effect | [200] |
| Liposomes | Rh123 | Paclitaxel | Hela and B16-F10 cancer cell line | Induced apoptosis and high toxicity to cancer cells | [201] |
| Liposomes | STPP+ | Ceramide | 4T1 mammalian carcinoma cells and animal model | Enhanced specific drug delivery, show anti-tumor effect | [180] |
| Cerasomes | TPP+ | Doxorubicin | Hela cells | Sustainable drug release, high biocompatibility, show antitumor effect | [182] |
| AuNP (Polydopamine) | - | Paclitaxel | Cancer cell line | Downregulates anti-apoptotic gene, enhanced anti-cancer efficacy | [202] |
| AuNP (Hyaluronic acid) | - | Camptothecin | Cancer cell line | Upregulates proapoptotic genes, sensitizes drug-resistant cancer cells, enhanced anti-cancer efficacy | [203] |
| PLGA-PEG NPs | TPP+ | Curcumin, 2,4-dinitrophenol, lonidamine, α-tocopheryl succinate | HeLa cells | Enhanced specific drug delivery, enhanced cytotoxicity to cancer cells | [191,204] |
| Mesoporous silica NPs | TPP+ | α-tocopheryl succinate | HeLa and HepG2 cancerous cell line | Enhanced cytotoxicity, disrupt mitochondrial membrane potential | [205] |
| AgNPs (Polydopamine) | RGDARF peptide (Tumor targeting peptide) | Paclitaxel | Cancer cell line | Strong apoptotic-inducing potency, activation of pro-apoptotic factor P53 and caspase 3 | [193,206] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, T.; Waseem, R.; Zehra, Z.; Aiman, A.; Bhardwaj, P.; Ansari, J.; Hassan, M.I.; Islam, A. Mitochondrial Dysfunction: Pathophysiology and Mitochondria-Targeted Drug Delivery Approaches. Pharmaceutics 2022, 14, 2657. https://doi.org/10.3390/pharmaceutics14122657
Khan T, Waseem R, Zehra Z, Aiman A, Bhardwaj P, Ansari J, Hassan MI, Islam A. Mitochondrial Dysfunction: Pathophysiology and Mitochondria-Targeted Drug Delivery Approaches. Pharmaceutics. 2022; 14(12):2657. https://doi.org/10.3390/pharmaceutics14122657
Chicago/Turabian StyleKhan, Tanzeel, Rashid Waseem, Zainy Zehra, Ayesha Aiman, Priyanka Bhardwaj, Jaoud Ansari, Md. Imtaiyaz Hassan, and Asimul Islam. 2022. "Mitochondrial Dysfunction: Pathophysiology and Mitochondria-Targeted Drug Delivery Approaches" Pharmaceutics 14, no. 12: 2657. https://doi.org/10.3390/pharmaceutics14122657