
Optimization of the Solvent and In Vivo Administration Route of Auranofin in a Syngeneic Non-Small Cell Lung Cancer and Glioblastoma Mouse Model
 , , , , , , and
, , , , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Murine Cell Lines
2.3. Tumor Kinetics and Survival
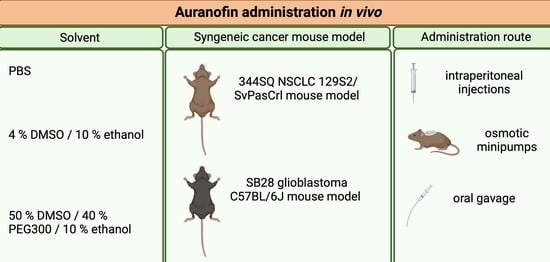
2.4. Auranofin Solvent
2.5. In Vivo Administration of AF
2.6. Thioredoxin Reductase Activity Assay
2.7. Statistics
3. Results
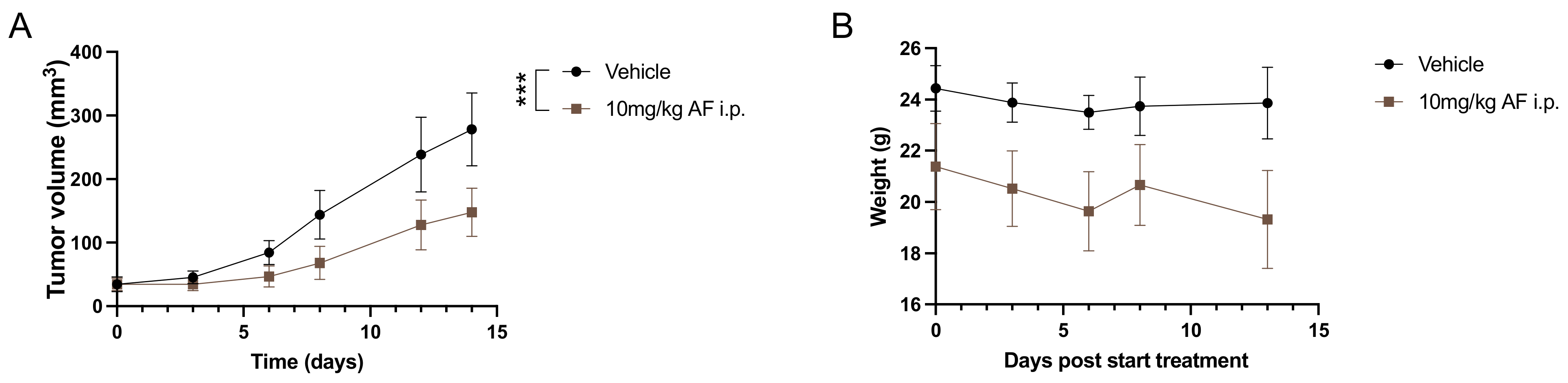
3.1. Daily I.P. Injections with AF Induce Weight Loss and Gut-Related Cytotoxicity in 344SQ 129-Mouse Model
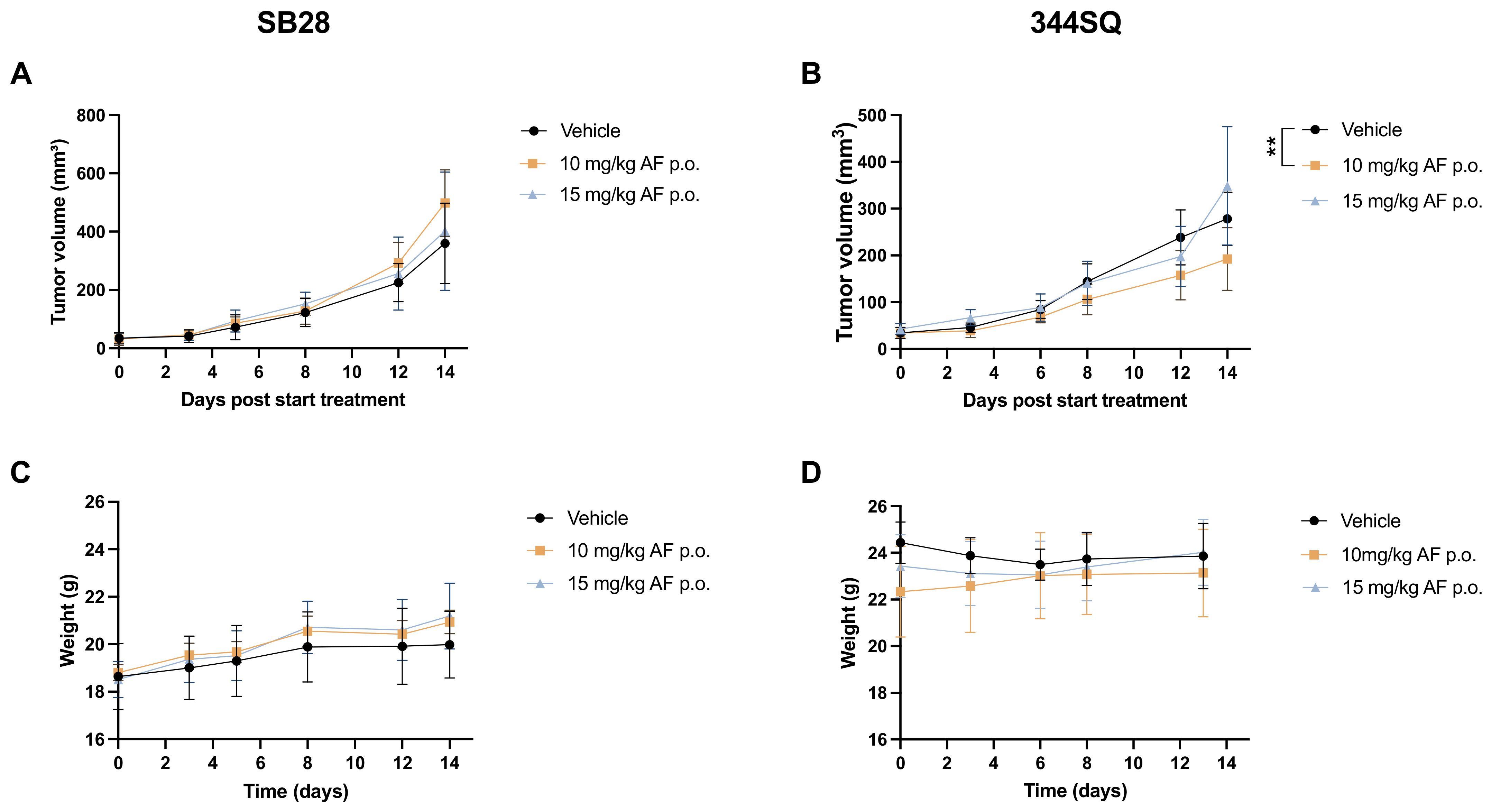
3.2. Continuous Slow Release of AF Treatment in 344SQ- and SB28-Bearing Mice via an Osmotic Minipump
3.3. Oral Administration of AF Treatment in 344SQ- and SB28-Bearing Mice
3.4. AF Inhibits TrxR Activity in SB28 Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kean, W.F.; Hart, L.; Buchanan, W.W. Auranofin. Br. J. Rheumatol. 1997, 36, 560–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooke, S.T.; Snyder, R.M.; Butt, T.R.; Ecker, D.J.; Allaudeen, H.S.; Monia, B.; Mirabelli, C.K. Cellular and molecular pharmacology of auranofin and related gold complexes. Biochem. Pharmacol. 1986, 35, 3423–3431. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, J.; Sadler, P.J.; Tucker, A. A new structural transition of serum albumin dependent on the state of Cys34. Detection by 1H-NMR spectroscopy. Eur. J. Biochem. 1994, 225, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs RD 2015, 15, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiekink, E. Gold compounds in medicine: Potential anti-tumour agents. Gold Bull. 2003, 36, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Mirabelli, C.K.; Johnson, R.K.; Sung, C.M.; Faucette, L.; Muirhead, K.; Crooke, S.T. Evaluation of the in vivo antitumor activity and in vitro cytotoxic properties of auranofin, a coordinated gold compound, in murine tumor models. Cancer Res. 1985, 45, 32–39. [Google Scholar]
- Madeira, J.M.; Gibson, D.L.; Kean, W.F.; Klegeris, A. The biological activity of auranofin: Implications for novel treatment of diseases. Inflammopharmacology 2012, 20, 297–306. [Google Scholar] [CrossRef]
- Gamberi, T.; Chiappetta, G.; Fiaschi, T.; Modesti, A.; Sorbi, F.; Magherini, F. Upgrade of an old drug: Auranofin in innovative cancer therapies to overcome drug resistance and to increase drug effectiveness. Med. Res. Rev. 2022, 42, 1111–1146. [Google Scholar] [CrossRef]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Gupta, S.D.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef] [Green Version]
- Sze, J.H.; Raninga, P.V.; Nakamura, K.; Casey, M.; Khanna, K.K.; Berners-Price, S.J.; Di Trapani, G.; Tonissen, K.F. Anticancer activity of a Gold(I) phosphine thioredoxin reductase inhibitor in multiple myeloma. Redox Biol. 2020, 28, 101310. [Google Scholar] [CrossRef]
- Chen, X.; Shi, X.; Zhao, C.; Li, X.; Lan, X.; Liu, S.; Huang, H.; Liu, N.; Liao, S.; Zang, D.; et al. Anti-rheumatic agent auranofin induced apoptosis in chronic myeloid leukemia cells resistant to imatinib through both Bcr/Abl-dependent and -independent mechanisms. Oncotarget 2014, 5, 9118–9132. [Google Scholar] [CrossRef]
- Fidyt, K.; Pastorczak, A.; Goral, A.; Szczygiel, K.; Fendler, W.; Muchowicz, A.; Bartlomiejczyk, M.A.; Madzio, J.; Cyran, J.; Graczyk-Jarzynka, A.; et al. Targeting the thioredoxin system as a novel strategy against B-cell acute lymphoblastic leukemia. Mol. Oncol. 2019, 13, 1180–1195. [Google Scholar] [CrossRef] [Green Version]
- Celegato, M.; Borghese, C.; Casagrande, N.; Mongiat, M.; Kahle, X.U.; Paulitti, A.; Spina, M.; Colombatti, A.; Aldinucci, D. Preclinical activity of the repurposed drug auranofin in classical Hodgkin lymphoma. Blood 2015, 126, 1394–1397. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, J.; Lopez, E.; Guo, H.; Zhang, H.; Liu, Y.; Chen, Z.; Huang, S.; Zhou, S.; Leeming, A.; et al. Repurposing auranofin to treat TP53-mutated or PTEN-deleted refractory B-cell lymphoma. Blood Cancer J. 2019, 9, 95-95. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Li, X.; Huang, H.; Zhao, C.; Liao, S.; Yang, C.; Liu, S.; Song, W.; Lu, X.; Lan, X.; et al. Clinically used antirheumatic agent auranofin is a proteasomal deubiquitinase inhibitor and inhibits tumor growth. Oncotarget 2014, 5, 5453–5471. [Google Scholar] [CrossRef] [Green Version]
- Nag, D.; Bhanja, P.; Riha, R.; Sanchez-Guerrero, G.; Kimler, B.F.; Tsue, T.T.; Lominska, C.; Saha, S. Auranofin Protects Intestine against Radiation Injury by Modulating p53/p21 Pathway and Radiosensitizes Human Colon Tumor. Clin. Cancer Res. 2019, 25, 4791–4807. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Hu, J.; Wu, S.; Wang, L.; Cao, X.; Zhang, X.; Dai, B.; Cao, M.; Shao, R.; Zhang, R.; et al. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget 2016, 7, 3548–3558. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Zhang, X.; Wang, L.; Zhang, R.; Pu, X.; Wu, S.; Li, L.; Tong, P.; Wang, J.; Meng, Q.H.; et al. Inhibition of Thioredoxin/Thioredoxin Reductase Induces Synthetic Lethality in Lung Cancers with Compromised Glutathione Homeostasis. Cancer Res. 2019, 79, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hill, K.S.; Fields, A.P. PKCι maintains a tumor-initiating cell phenotype that is required for ovarian tumorigenesis. Mol. Cancer Res. 2013, 11, 1624–1635. [Google Scholar] [CrossRef] [Green Version]
- Marzo, T.; Massai, L.; Pratesi, A.; Stefanini, M.; Cirri, D.; Magherini, F.; Becatti, M.; Landini, I.; Nobili, S.; Mini, E.; et al. Replacement of the Thiosugar of Auranofin with Iodide Enhances the Anticancer Potency in a Mouse Model of Ovarian Cancer. ACS Med. Chem. Lett. 2019, 10, 656–660. [Google Scholar] [CrossRef]
- Liu, N.; Guo, Z.; Xia, X.; Liao, Y.; Zhang, F.; Huang, C.; Liu, Y.; Deng, X.; Jiang, L.; Wang, X.; et al. Auranofin lethality to prostate cancer includes inhibition of proteasomal deubiquitinases and disrupted androgen receptor signaling. Eur. J. Pharmacol. 2019, 846, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yang, X.; Deshmukh, D.; Chen, H.; Fang, S.; Qiu, Y. The Role of Crosstalk between AR3 and E2F1 in Drug Resistance in Prostate Cancer Cells. Cells 2020, 9, 1094. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, P.; Zhang, Y.; Lu, W.; Ding, W.; Luo, Y.; Wen, S.; Xu, R.; Liu, P.; Huang, P. Synergy between Auranofin and Celecoxib against Colon Cancer In Vitro and In Vivo through a Novel Redox-Mediated Mechanism. Cancers 2019, 11, 931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, Y.; Lu, W.; Han, Y.; Yang, J.; Jiang, W.; You, X.; Luo, Y.; Wen, S.; Hu, Y.; et al. Mitochondrial TXNRD3 confers drug resistance via redox-mediated mechanism and is a potential therapeutic target in vivo. Redox Biol. 2020, 36, 101652-101652. [Google Scholar] [CrossRef] [PubMed]
- Hatem, E.; Azzi, S.; El Banna, N.; He, T.; Heneman-Masurel, A.; Vernis, L.; Baïlle, D.; Masson, V.; Dingli, F.; Loew, D.; et al. Auranofin/Vitamin C: A Novel Drug Combination Targeting Triple-Negative Breast Cancer. J. Natl. Cancer. Inst. 2018, 111, 597–608. [Google Scholar] [CrossRef]
- Raninga, P.V.; Lee, A.C.; Sinha, D.; Shih, Y.-Y.; Mittal, D.; Makhale, A.; Bain, A.L.; Nanayakarra, D.; Tonissen, K.F.; Kalimutho, M.; et al. Therapeutic cooperation between auranofin, a thioredoxin reductase inhibitor and anti-PD-L1 antibody for treatment of triple-negative breast cancer. Int. J. Cancer 2020, 146, 123–136. [Google Scholar] [CrossRef]
- Rodman, S.N.; Spence, J.M.; Ronnfeldt, T.J.; Zhu, Y.; Solst, S.R.; O’Neill, R.A.; Allen, B.G.; Guan, X.; Spitz, D.R.; Fath, M.A. Enhancement of Radiation Response in Breast Cancer Stem Cells by Inhibition of Thioredoxin- and Glutathione-Dependent Metabolism. Radiat. Res. 2016, 186, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Hrabe, J.E.; O’Leary, B.R.; Fath, M.A.; Rodman, S.N.; Button, A.M.; Domann, F.E.; Spitz, D.R.; Mezhir, J.J. Disruption of thioredoxin metabolism enhances the toxicity of transforming growth factor β-activated kinase 1 (TAK1) inhibition in KRAS-mutated colon cancer cells. Redox Biol. 2015, 5, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhao, Y.; Wang, J.; Yang, Y.; Zhang, Y.; Qu, X.; Peng, S.; Yao, Z.; Zhao, S.; He, B.; et al. FoxO3 reverses 5-fluorouracil resistance in human colorectal cancer cells by inhibiting the Nrf2/TR1 signaling pathway. Cancer Lett. 2020, 470, 29–42. [Google Scholar] [CrossRef]
- Zou, P.; Chen, M.; Ji, J.; Chen, W.; Chen, X.; Ying, S.; Zhang, J.; Zhang, Z.; Liu, Z.; Yang, S.; et al. Auranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget 2015, 6, 36505–36521. [Google Scholar] [CrossRef] [Green Version]
- Fath, M.A.; Ahmad, I.M.; Smith, C.J.; Spence, J.; Spitz, D.R. Enhancement of Carboplatin-Mediated Lung Cancer Cell Killing by Simultaneous Disruption of Glutathione and Thioredoxin Metabolism. Clin. Cancer Res. 2011, 17, 6206. [Google Scholar] [CrossRef]
- Fan, C.; Zheng, W.; Fu, X.; Li, X.; Wong, Y.S.; Chen, T. Enhancement of auranofin-induced lung cancer cell apoptosis by selenocystine, a natural inhibitor of TrxR1 in vitro and in vivo. Cell Death Dis. 2014, 5, e1191. [Google Scholar] [CrossRef] [Green Version]
- Dai, B.; Yoo, S.-Y.; Bartholomeusz, G.; Graham, R.A.; Majidi, M.; Yan, S.; Meng, J.; Ji, L.; Coombes, K.; Minna, J.D.; et al. KEAP1-dependent synthetic lethality induced by AKT and TXNRD1 inhibitors in lung cancer. Cancer Res. 2013, 73, 5532–5543. [Google Scholar] [CrossRef] [Green Version]
- Xiaobo, C.; Majidi, M.; Feng, M.; Shao, R.; Wang, J.; Zhao, Y.; Baladandayuthapani, V.; Song, J.; Fang, B.; Ji, L.; et al. TUSC2(FUS1)-erlotinib Induced Vulnerabilities in Epidermal Growth Factor Receptor(EGFR) Wildtype Non-small Cell Lung Cancer(NSCLC) Targeted by the Repurposed Drug Auranofin. Sci. Rep. 2016, 6, 35741-35741. [Google Scholar] [CrossRef]
- Hou, G.-X.; Liu, P.-P.; Zhang, S.; Yang, M.; Liao, J.; Yang, J.; Hu, Y.; Jiang, W.-Q.; Wen, S.; Huang, P. Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis. Cell Death Dis. 2018, 9, 89. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Zhang, H.; Cao, M.; Wang, L.; Wu, S.; Fang, B. Auranofin Enhances Ibrutinib’s Anticancer Activity in EGFR-Mutant Lung Adenocarcinoma. Mol. Cancer Ther. 2018, 17, 2156–2163. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, W.; Yin, Y.; Li, M.; Li, H.; Xiang, H.; Xu, A.; Mei, X.; Hong, B.; Lin, W. A high-throughput drug screen identifies auranofin as a potential sensitizer of cisplatin in small cell lung cancer. Investig. New Drugs 2019, 37, 1166–1176. [Google Scholar] [CrossRef]
- Huang, H.; Liao, Y.; Liu, N.; Hua, X.; Cai, J.; Yang, C.; Long, H.; Zhao, C.; Chen, X.; Lan, X.; et al. Two clinical drugs deubiquitinase inhibitor auranofin and aldehyde dehydrogenase inhibitor disulfiram trigger synergistic anti-tumor effects in vitro and in vivo. Oncotarget 2016, 7, 2796–2808. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Xu, I.M.-J.; Chiu, D.K.-C.; Leibold, J.; Tse, A.P.-W.; Bao, M.H.-R.; Yuen, V.W.-H.; Chan, C.Y.-K.; Lai, R.K.-H.; Chin, D.W.-C.; et al. Induction of Oxidative Stress Through Inhibition of Thioredoxin Reductase 1 Is an Effective Therapeutic Approach for Hepatocellular Carcinoma. Hepatology 2019, 69, 1768–1786. [Google Scholar] [CrossRef]
- Sobhakumari, A.; Love-Homan, L.; Fletcher, E.V.; Martin, S.M.; Parsons, A.D.; Spitz, D.R.; Knudson, C.M.; Simons, A.L. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS ONE 2012, 7, e48175. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.-L.; Jang, H.; Kim, E.H.; Shin, D. Targeting of the Glutathione, Thioredoxin, and Nrf2 Antioxidant Systems in Head and Neck Cancer. Antioxid. Redox Signal. 2016, 27, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; McDonald, P.; Ottomeyer, M.; Roy, A.; Shoenen, F.J.; Broward, M.; Bruns, T.; Thamm, D.H.; Weir, S.J.; Neville, K.A.; et al. Comparative oncology approach to drug repurposing in osteosarcoma. PLoS ONE 2018, 13, e0194224. [Google Scholar] [CrossRef] [PubMed]
- Pessetto, Z.Y.; Chen, B.; Alturkmani, H.; Hyter, S.; Flynn, C.A.; Baltezor, M.; Ma, Y.; Rosenthal, H.G.; Neville, K.A.; Weir, S.J.; et al. In silico and in vitro drug screening identifies new therapeutic approaches for Ewing sarcoma. Oncotarget 2017, 8, 4079–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashmi, R.; Huang, X.; Floberg, J.M.; Elhammali, A.E.; McCormick, M.L.; Patti, G.J.; Spitz, D.R.; Schwarz, J.K. Radioresistant Cervical Cancers Are Sensitive to Inhibition of Glycolysis and Redox Metabolism. Cancer Res. 2018, 78, 1392–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Loenhout, J.; Freire Boullosa, L.; Quatannens, D.; De Waele, J.; Merlin, C.; Lambrechts, H.; Lau, H.W.; Hermans, C.; Lin, A.; Lardon, F.; et al. Auranofin and Cold Atmospheric Plasma Synergize to Trigger Distinct Cell Death Mechanisms and Immunogenic Responses in Glioblastoma. Cells 2021, 10, 2936. [Google Scholar] [CrossRef]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69–86.e66. [Google Scholar] [CrossRef] [Green Version]
- Alzet. Available online: https://www.alzet.com/formulating-the-solution/ (accessed on 16 January 2021).
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Singmann, H.; Bolker, B.; Westfall, J.; Aust, F. Afex: Analysis of Factorial Experiments, R Package Version 0.25-1; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Lenth, R. Emmeans: Estimated Marginal Means, aka Least-Squares Means, R Package Version 1.4.3.01; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Langford, D.J.; Bailey, A.L.; Chanda, M.L.; Clarke, S.E.; Drummond, T.E.; Echols, S.; Glick, S.; Ingrao, J.; Klassen-Ross, T.; Lacroix-Fralish, M.L.; et al. Coding of facial expressions of pain in the laboratory mouse. Nat. Methods 2010, 7, 447–449. [Google Scholar] [CrossRef]
- Kurdi, A.; De Doncker, M.; Leloup, A.; Neels, H.; Timmermans, J.-P.; Lemmens, K.; Apers, S.; De Meyer, G.R.Y.; Martinet, W. Continuous administration of the mTORC1 inhibitor everolimus induces tolerance and decreases autophagy in mice. Br. J. Pharmacol. 2016, 173, 3359–3371. [Google Scholar] [CrossRef] [Green Version]
- Cameron, A.M.; Adams, D.H.; Greenwood, J.E.; Anderson, P.J.; Cowin, A.J. A Novel Murine Model of Hypertrophic Scarring Using Subcutaneous Infusion of Bleomycin. Plast. Reconstr. Surg. 2014, 133, 69–78. [Google Scholar] [CrossRef]
- Pantziarka, P.; Verbaanderd, C.; Sukhatme, V.; Rica Capistrano, I.; Crispino, S.; Gyawali, B.; Rooman, I.; Van Nuffel, A.M.; Meheus, L.; Sukhatme, V.P.; et al. ReDO_DB: The repurposing drugs in oncology database. Ecancermedicalscience 2018, 12, 886. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.S.; Liu, J.O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef]
- Fogel, D.B. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: A review. Contemp. Clin. Trials Commun. 2018, 11, 156–164. [Google Scholar] [CrossRef]
- Chaffman, M.; Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Auranofin. A preliminary review of its pharmacological properties and therapeutic use in rheumatoid arthritis. Drugs 1984, 27, 378–424. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Abutaleb, N.S.; Seleem, M.N. Auranofin, at clinically achievable dose, protects mice and prevents recurrence from Clostridioides difficile infection. Sci. Rep. 2020, 10, 7701. [Google Scholar] [CrossRef]
- Gandin, V.; Fernandes, A.P.; Rigobello, M.P.; Dani, B.; Sorrentino, F.; Tisato, F.; Björnstedt, M.; Bindoli, A.; Sturaro, A.; Rella, R.; et al. Cancer cell death induced by phosphine gold(I) compounds targeting thioredoxin reductase. Biochem. Pharmacol. 2010, 79, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Selvaraju, K.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Arnér, E.S.J.; Linder, S. Repurposing of auranofin: Thioredoxin reductase remains a primary target of the drug. Biochimie 2019, 162, 46–54. [Google Scholar] [CrossRef]
- Freire Boullosa, L.; Van Loenhout, J.; Flieswasser, T.; De Waele, J.; Hermans, C.; Lambrechts, H.; Cuypers, B.; Laukens, K.; Bartholomeus, E.; Siozopoulou, V.; et al. Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biol. 2021, 42, 101949. [Google Scholar] [CrossRef]
- Blodgett, R.C., Jr.; Pietrusko, R.G. Long-term efficacy and safety of auranofin: A review of clinical experience. Scand. J. Rheumatol. Suppl. 1986, 63, 67–78. [Google Scholar]
- Laxer, R.M. CHAPTER 5—PHARMACOLOGY AND DRUG THERAPY. In Textbook of Pediatric Rheumatology, 5th ed.; Cassidy, J.T., Petty, R.E., Laxer, R.M., Lindsley, C.B., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2005; pp. 76–141. [Google Scholar] [CrossRef]
- Aronson, J.K. (Ed.) Gold and Gold Salts. In Meyler’s Side Effects of Drugs, 16th ed.; Elsevier: Oxford, UK, 2016; pp. 572–583. [Google Scholar] [CrossRef]
- Díez-Martínez, R.; García-Fernández, E.; Manzano, M.; Martínez, Á.; Domenech, M.; Vallet-Regí, M.; García, P. Auranofin-loaded nanoparticles as a new therapeutic tool to fight streptococcal infections. Sci. Rep. 2016, 6, 19525. [Google Scholar] [CrossRef]
- Awasthi, R.; Roseblade, A.; Hansbro, P.M.; Rathbone, M.J.; Dua, K.; Bebawy, M. Nanoparticles in Cancer Treatment: Opportunities and Obstacles. Curr. Drug Targets 2018, 19, 1696–1709. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | In Vivo Cancer Model | Solvent AF Concentration | Administration Route Treatment Schedule | Experimental Outcome | Ref. |
|---|---|---|---|---|---|
| AF MONOTHERAPY | |||||
| AF | CLL cells in TCL-1 transgenic mice (Tcl1-tg:p53−/−) | - AF 10 mg/kg | i.p. 5 times a week, for 2 weeks | Reduction in leukemia cell burden and CLL cells in peritoneal cavity Improvement of survival | [9] |
| AF | RPMI8226 MM xenograft model in NOD/SCID mice | - AF 5 mg/kg | i.p. 5 times a week, for 2 weeks | Inhibition of MM tumor growth Increase in % of apoptotic cells Inhibition of TrxR activity | [10] |
| AF | KBM5 (Bcr-Abl wild-type) and KBM5-T315I (Bcr-Abl-T315I) CML xenografts in nude mice | 10% DMSO, 30% cremophor and 60% NaCl AF 7 mg/kg/day | i.p. 12 days | Inhibition of tumor growth and tumor weight—decrease in proliferative cells (Ki67) Constant body weight | [11] |
| AF | BCP-ALL xenograft model in NSG mice | DMSO AF 10 mg/kg | i.p. 5 times a week, for 3 weeks | Reduction in number of human blasts Prolongation of event-free survival of leukemic mice | [12] |
| AF | Hodgkin lymphoma L-540 gemcitabine-resistant-derived tumor xenograft model in nude mice | - AF 10 mg/kg | i.p. 3 times a week, for 17 days | Reduction in tumor growth | [13] |
| AF | TP53-mutated diffuse large B-cell lymphoma (DLBCL) PDX model | - AF 50 mg/kg | p.o. 21 consecutive days | Reduction in tumor growth No weight loss | [14] |
| AF | HepG2 liver carcinoma and MCF-7 breast cancer xenograft models in nude mice | 10% DMSO, 30% Cremophor EL and 60% NaCl AF 6 mg/kg | i.p. 21 days | Reduction in tumor growth and tumor weight Constant body weight | [15] |
| AF | CT26 colon cancer xenograft model in BALB/c mice | 10% DMSO AF 10 mg/kg | i.p. three times a week for 3 weeks | Reduction in tumor growth Constant body weight | [16] |
| AF | Calu3 NSCLC tumor xenograft in nude mice | 2% DMSO, 10% ethanol and 5% PEG400 AF 10 mg/kg | i.p. - | Significant suppression of tumor growth Constant body weight | [17] |
| AF | NSCLC PDX model in nude mice | 2% DMSO, 10% ethanol, and 5% PEG400 AF 10 mg/kg | i.p. Daily for one week, twice a week for the duration of the experiment | Inhibition of tumor growth Constant body weight | [18] |
| AF | orthotopic ovarian ES2 TIC tumors in immune-deficient nude mice | dissolved in 100% ethanol and diluted in saline (0.9%) AF 12 mg/kg | i.p. 6 times a week for the duration of the experiment | Significant inhibition of tumor growth Decrease mitotic index | [19] |
| AF | A2780 orthotopic and s.c. xenograft model in nude mice | - AF 15 mg/kg | i.p. 3 times a week, for 2 weeks | s.c. model: decrease in tumor volume Orthotopic model: no reduction in tumor volume | [20] |
| AF | 22RV1 prostate cancer xenograft model in nude BALB/c mice | 10% DMSO, 30% Cremophor EL and 60% normal saline AF 6 mg/kg | i.p. 2 weeks | Reduction in tumor volume and tumor weight—increase cleaved caspase-3 and decrease in proliferation (Ki67) Constant body weight | [21] |
| AF | R1-DDR prostate cancer xenograft | - AF 5 mg/kg | - 5 times a week, for undefined period | Decrease in tumor volume Higher number of apoptotic cells Decrease in proliferative cells (Ki67) Constant body weight | [22] |
| COMBINATION THERAPY | |||||
| AF + Celecoxib | DLD-1 colon cancer xenograft model in nude mice | Olive oil AF 10 mg/kg | p.o. 30 days (except Saturday and Sunday) | Constant body weight AF mono: moderate therapeutic effect on tumor volume Combo: significant reduction in tumor volume and tumor weight | [23] |
| AF + sorafenib (+cyclophosphamide) | SK-Hep1OE or SK-Hep1VC liver cancer xenograft model in immune-deficient mice Sorafenib-resistant MV4-11R leukemia xenograft model | - AF 10 mg/kg | i.p. and p.o. 22 days | Reduction tumor growth and tumor weight in both liver and leukemia model Constant body weight (p.o.) | [24] |
| AF + Vitamin C | MDA-MB-231 breast cancer xenograft model in Crl:NU(Ico)-Foxn1nu nude mice | PBS AF 10 mg/kg | i.p. 5 times a week, for 15 days | AF mono: no effect Combo: reduction in tumor volume Constant body weight | [25] |
| AF + aPD-L1 | MDA-MB-231 breast cancer xenograft model in NOD/SCID mice Murine 4T1.2 syngeneic model in immunocompetent mice TNBC PDX model in NOD/SCID mice | - AF mono: 10 mg/kg Combo: 5 mg/kg AF | i.p. 5 times a week, for 2 weeks | AF mono: reduction in tumor growth and tumor weight—upregulation PD-L1 expression—reduction TrxR activity—increase in apoptotic cells Combo in 4T1.2 model: overcomes resistance to PD-L1 | [26] |
| AF + BSO/radiation | MDA-MB-231 breast cancer xenograft model in nude mice | - AF 1.7 mg/kg | i.p. - | Triple combo: significant reduction in tumor volume compared to vehicle, but not to radiation alone—prolongation of survival 5 % loss of body weight No changes in behavior or activity | [27] |
| AF + 5Z-7-oxozeaenol | SW620 colon cancer xenograft model in nude mice | Sterile 2.5% DMSO in vegetable oil AF 1.6 mg/kg | i.p. 11 days | AF mono: no effect Combo: no statistically significant effect | [28] |
| AF + 5-FU | 5-FU-resistant SW620 colorectal cancer xenograft model in nude mice | - AF 6 mg/kg | p.o. Daily for 6 weeks | AF Mono: no effect Combo: reduction in tumor growth, tumor weight and metastatic lung nodules Constant body weight | [29] |
| AF + piperlongumine | SGC-7901 gastric xenograft model in immune-deficient nude mice | - AF 2 mg/kg | i.p. Once per day for 14 days | AF mono: inhibition of tumor growth Combo: more effective inhibition of tumor growth Constant body weight | [30] |
| AF + BSO + carboplatin | A549 and H292 lung cancer xenograft model in nude mice | Absolute ethanol, cremophor EL in normal saline AF 1.6 mg/kg | i.p. 5 times a week, for 2 weeks | Combo AF + BSO: reduction total GSH and TrxR activity Triple combo: most significant decrease in tumor growth rate Constant body weight No behavioral changes Normal blood analysis | [31] |
| AF + selenocystine | A549 lung tumor xenograft model in immuno-deficient nude mice | PBS AF 2 mg/kg | Caudal vein injection Every other day for 16 days | Mono AF: no effect on tumor growth or tumor weight—no changes TrxR expression Combo: inhibition of tumor weight and tumor volume—inhibition TrxR expression—higher cleaved caspase activity—inhibition of proliferation (Ki67) Constant body weight | [32] |
| AF + MK2206 | H1993 lung cancer xenograft model in nude mice | - AF 5 mg/kg | i.p. - | Mono AF: no effect on tumor growth—inhibition TrxR activity Combo: significant reduction tumor growth—prolonged survival—inhibition TrxR activity | [33] |
| AF + erlotinib + TUSC2 gene nanovesicles | wild type EGFR TUSC2-deficient H1299 lung cancer xenograft model in nude mice | PBS AF 10 mg/kg | i.p. 5 times a week, for 2 weeks | Triple combo: inhibits tumor growth and prolongs survival Constant body weight | [34] |
| AF + adriamycin | A549 lung tumor xenograft in nude mice | PBS AF 10 mg/kg | i.p. 5 times a week, for 6 weeks | AF mono: moderate inhibitory effect on tumor growth Combo: strongest effect on tumor growth and tumor weight Constant body weight | [35] |
| AF + ibrutinib | H1975 NSCLC tumor xenograft model in nude mice | 10% DMSO and 10% PEG400 AF 5 mg/kg | Tail vein injection or i.p. - | AF mono: Inhibition of tumor growth—no effect on survival Combo: significant effect on survival—reduction in tumor growth Constant body weight | [36] |
| AF + cisplatin | H69 SCLC xenograft model in nude mice | DMSO AF 10 mg/kg | i.p. Every two days for 4 weeks | Mono AF: no effect on tumor growth or tumor weight Combo: significant effect on tumor growth and tumor weight—decrease proliferation marker Ki67 Constant body weight | [37] |
| AF + disulfiram | HepG2 or SMMC-7721 hepatocarcinoma xenograft models in nude mice | 10% DMSO, 30% Cremophor EL and 60% normal saline AF 3 mg/kg | i.p. 15 days | AF mono: no effect on tumor size or tumor weight Combo: significant effect on tumor size and tumor —increase in cleaved caspase-3 Constant body weight | [38] |
| AF + sorafenib | Tail-vein injection model of CRISPR-Cas9-KO p53 and Pten plasmids in immune-competent C57BL/6N mice Orthotopic tumor model of luciferase-labeled HCC MHCC97L cells implanted in nude mice | 5% PEG400 + 5% Tween-80 in H2O AF 6 mg/kg AF 3 mg/kg | i.p. 14 days 35 days | AF mono: suppression of tumor growth Combo: strongest inhibition of HCC tumor formation and metastases to the lungs Constant body weight | [39] |
| AF + BSO | Cal-27 HNC xenograft model in nude mice | Saline AF 1 mg/kg | i.p. Every day, for 10 days | AF mono: significant reduction in tumor volume Combo: strongest significant reduction in tumor volume Constant body weight | [40] |
| AF + BSO + trigonelline | HN3-cisplatin resistant HNC xenograft model in nude mice | - AF 2 mg/kg | i.p. - | AF mono: significant reduction in tumor volume Triple combination: strongest significant reduction in tumor volume and tumor weight—strong increase in apoptotic cells Constant body weight | [41] |
| AF + vorinostat/rapamycin | KHOS/NP osteosarcoma xenograft model in nude-Foxn1nu mice (canine) Abrams osteosarcoma xenograft model in nude-Foxn1nu mice | DMSO AF 0.1 mg/kg | i.p. 5 times a week, for 3 weeks | AF mono in both models: no effect on tumor size Combo in both models: significant suppression of tumor growth—decrease in proliferative cells (Ki67—increase in apoptotic cells (cleaved caspase-3) | [42] |
| AF + ganetespib | A673 Ewing sarcoma xenograft model in nude mice (injected intramuscularly proximal to tibia) | 0.5% Hydroxypropyl methylcellulose (HPMC, viscosity grade K4M) in 5% dextrose in water AF 12 mg/kg | i.p. 5 times a week | Combo: significant difference in survival rates compared to AF mono and vehicle Constant body weight—no side effects | [43] |
| AF + 2DG + BSO + radiation | SiHa or CaSki cervical cancer xenograft model in nude mice | 0.05% DMSO AF 1.5 mg/kg | i.p. 3 times per week, for period of 35 days | AF + BSO/AF + BSO + 2DG: significant reduction in tumor volume Triple combo + radiation: strong radio-sensitization Constant body weight—no behavioral changes | [44] |
| AF + plasma | s.c. injection of murine SB28 GBM cell lines in syngeneic C57BL/6J mice model | 50% DMSO, 40% PEG300 and 10% ethanol AF 15 mg/kg | p.o. 2 weeks | AF mono: no effect on tumor volume Combo: significant decrease in tumor volume | [45] |
| AF + BSO + 2DG | Vari068 TNBC xenograft model (injected in mammary fat pads) in NOD/SCID mice Luciferase-labeled SUM159 breast cancer xenograft model in NOD/SCID mice (cardiac injection for metastasis formation) | - AF 1.5 mg/kg | i.p. every 2 days, for 7 weeks | AF + BSO/AF + BSO + 2DG: significant reduction in tumor growth—reduction in TrxR activity and rat—of GSH/GSSG—significant inhibition of metastasis formation | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freire Boullosa, L.; Van Loenhout, J.; Hermans, C.; Lau, H.W.; Merlin, C.; Marcq, E.; Takhsha, F.S.; Martinet, W.; De Meyer, G.R.Y.; Lardon, F.; et al. Optimization of the Solvent and In Vivo Administration Route of Auranofin in a Syngeneic Non-Small Cell Lung Cancer and Glioblastoma Mouse Model. Pharmaceutics 2022, 14, 2761. https://doi.org/10.3390/pharmaceutics14122761
Freire Boullosa L, Van Loenhout J, Hermans C, Lau HW, Merlin C, Marcq E, Takhsha FS, Martinet W, De Meyer GRY, Lardon F, et al. Optimization of the Solvent and In Vivo Administration Route of Auranofin in a Syngeneic Non-Small Cell Lung Cancer and Glioblastoma Mouse Model. Pharmaceutics. 2022; 14(12):2761. https://doi.org/10.3390/pharmaceutics14122761
Chicago/Turabian StyleFreire Boullosa, Laurie, Jinthe Van Loenhout, Christophe Hermans, Ho Wa Lau, Céline Merlin, Elly Marcq, Farnaz Sedigheh Takhsha, Wim Martinet, Guido R. Y. De Meyer, Filip Lardon, and et al. 2022. "Optimization of the Solvent and In Vivo Administration Route of Auranofin in a Syngeneic Non-Small Cell Lung Cancer and Glioblastoma Mouse Model" Pharmaceutics 14, no. 12: 2761. https://doi.org/10.3390/pharmaceutics14122761