1. Introduction
One silver lining of the COVID-19 pandemic is the unprecedented, global sharing of clinical and scientific data. These shared databases have revealed many insights into novel coronaviruses, and SARS-CoV-2 in particular, including the astounding number and speed of protein mutations. At the same time, many open questions have been exposed in the cell biology of respiratory viral infections. One particular open question centers on the mechanisms affected by the SARS-CoV-2 protein mutations and their impact on onset and progression of infection, including whether the impacts are uniform versus heterogeneous in the population. This causal, mechanistic link between viral RNA modifications and human respiratory infection outcomes is extremely cloudy as there are many complex processes that lie between the molecular and organ scales. In this article and study, we focus on one remarkable aspect of COVID-19 clinical data. Namely, nasal swab titers collected from individual, non-hospitalized, positive tests have varied by six orders of magnitude [
1,
2,
3,
4,
5,
6,
7,
8,
9]. This dramatic heterogeneity has persisted throughout the pandemic and therefore within and between variants, in all countries reporting data, and prior to and after previous SARS-CoV-2 exposure, infection, or vaccination. Due to the unprecedented global sharing of clinical data for COVID-19, it remains unclear whether this dramatic population heterogeneity in nasal infection is unique to SARS-CoV-2 or potentially generic to respiratory viruses. We turn to computational modeling to seek insights into the possible drivers of such dramatic heterogeneity in nasal infection tests between individuals.
In January of 2020, our group began development of a within-host, agent-based, computational model of human respiratory tract (RT) exposure to and infection by a novel virus. Like all models, choices must be made as to what features to include or not, and the efficacy of model predictions comes with the caveats of the choices made. For biologists and clinicians, as well as the practitioners of computational modeling, one should bear in mind the famous quote from 1976 by statistician George Box: “all models are wrong, some are useful”. In January of 2020, a physiologically faithful, spatially resolved model of the inhalation of a virus onto the air-mucus or air-alveolar liquid interface, and the subsequent diverse outcomes, did not exist. We built such a model, choosing to incorporate the kinetic processes of viral diffusion, virus-cell encounters, and, once a cell is infected, the processes of cellular uptake of the virus and viral hijacking of cellular machinery to make viral RNA copies, followed by cellular shedding of viral RNA copies into the airway surface liquid until cell death. Our group was well-positioned to build such a model because of (1) 25 years of research on lung physiology and biology, in particular the lung branching structure with generation-dependent mucus layer thickness and clearance velocity toward the trachea due to propulsion by beating cilia and (2) 10 years of research on sexually transmitted viral infections in the female cervicovaginal tract, which is also coated with a mucus layer that drains gravitationally. The baseline model [
10] incorporates the complex anatomy and physiology of the human RT, as well as the kinetic processes of virion diffusivity
in the mucosal barrier, the probability
of cell infection per virion encounter, the latency time
of an infected cell prior to shedding viral RNA, and, once shedding starts, the shedding rate
of infectious viral RNA copies until cell death. The latency time
spans the moment of cell infection until the onset of extracellular shedding of viral RNA.
We note that these “model features and mechanisms” are examples of the choices that one must make in order to capture, in an approximate manner, sufficient key impacts on outcomes. For example, (1) we assume the mucus layer is uniformly thick in each generation and moves like an escalator with the same velocity at each height of the layer; (2) we assume a virus, when it diffuses through the airway surface liquid to encounter an infectable cell (assumed to be 50% of epithelial cells), infects or not according to a flip of a biased coin (e.g., infection 1 out of 5 encounters) based on best available experimental data; and (3) once a cell is infected, we impose a latency time (a prescribed delay phase) after which the cell begins shedding viral RNA copies at some prescribed rate, but we do not resolve the processes and timescales for cellular uptake of the virus and hijacking of cellular machinery to produce viral RNA copies. There is strong cell culture evidence linking protein mutations and the pathway and speed for cellular uptake of the virus. Since infected cells typically live longer than 2 days, cell death does not enter the present study. All of the above mechanistic parameters and physiological features were estimated at mean population values in [
10], providing a framework to simulate outcomes of human respiratory infection that is physiologically faithful and incorporates the diffusive mobility of viruses in airway surface liquids and the kinetics of virus–cell infection, replication, and shedding. Below, we summarize the mathematical structure of this model. Additional extensions of the model to include innate [
11] and adaptive [
12] immunity have been developed, but they are not included in this study motivated by the overwhelming evidence of immune escape over the 48 h or longer post-infection period [
9,
13,
14,
15,
16,
17,
18,
19,
20,
21], independent of vaccination status and prior infection.
One important advantage of computational modeling in biology is that, despite the assumptions that render the model only an approximation of in vivo behavior, the model is able to provide predictions of outcomes and test whether features or mechanisms within the model are sufficient to replicate clinical or experimental observations and thus pose candidates for experimental or clinical confirmation. Indeed, the model may shed insights into the relative importance of physiological and in vivo conditions underlying clinical data, as well as the relative importance of ex vivo experimental controls underlying experimental data. We note two such illustrations in our previous work, which further motivate the present study. In [
10], in the nasal passage, trachea, and the first few upper branches of the human RT, mucus layer advection is strong and dominates diffusion of viruses while in the mucus layer. In vivo, strong mucus advection creates, from each initial infected cell, “thin streaks” of infected epithelial cells and shed viruses within mucus. Further, mucus advection accelerates growth in viral load and infected cells relative to a stationary mucus layer of the same thickness that might exist in an ex vivo culture experiment. The upshot is that ex vivo cultures with identical mucus layers produce extreme underestimates of in vivo viral load and infected cells. In [
22], using the same model and code from [
10], we performed a limited parameter sensitivity study of viral load and infected cells in the nasal passage, e.g., by varying the kinetic parameters governing cell infection, replication, and shedding over ranges guided by the literature. The study was limited in that only kinetic parameters and ranges were considered, not physiological parameters, and further limited by the parameter search. Namely, each parameter variation was studied by fixing all other parameters at mean population estimates, and not sampling in all directions of the full parameter space. One can think of this sampling of parameter space as extremely sparse, with each search starting from the mean of all parameters and exploring one parameter direction at a time from the global parameter mean. In addition to an extremely limited sampling of parameter space, moving only one parameter at a time rather than the freedom to move along any direction in the parameter space, the search is blind to correlations between parameters and the physiology or mechanisms they represent. Nonetheless, the following results in [
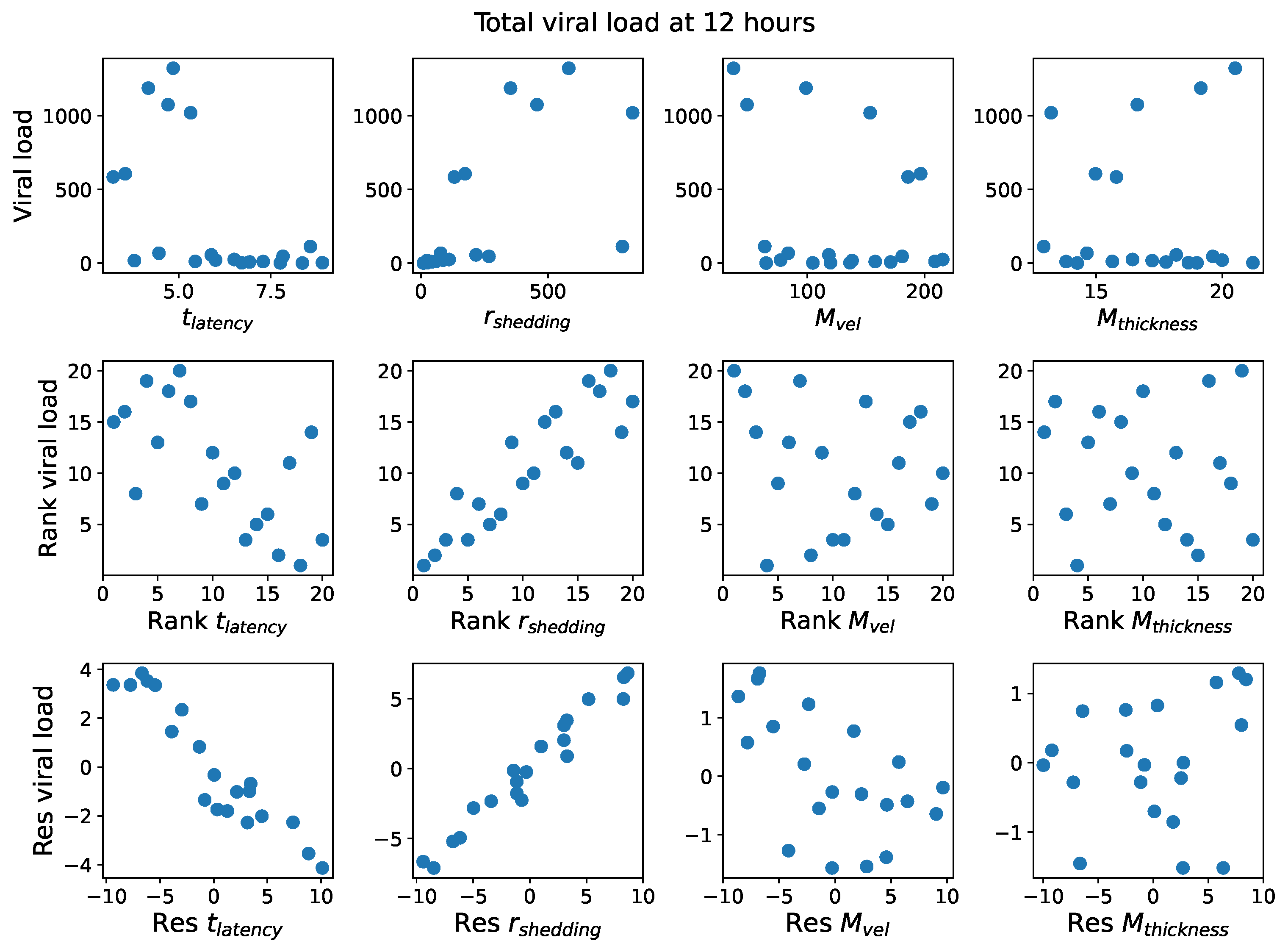
22] are suggestive and guide the present study.
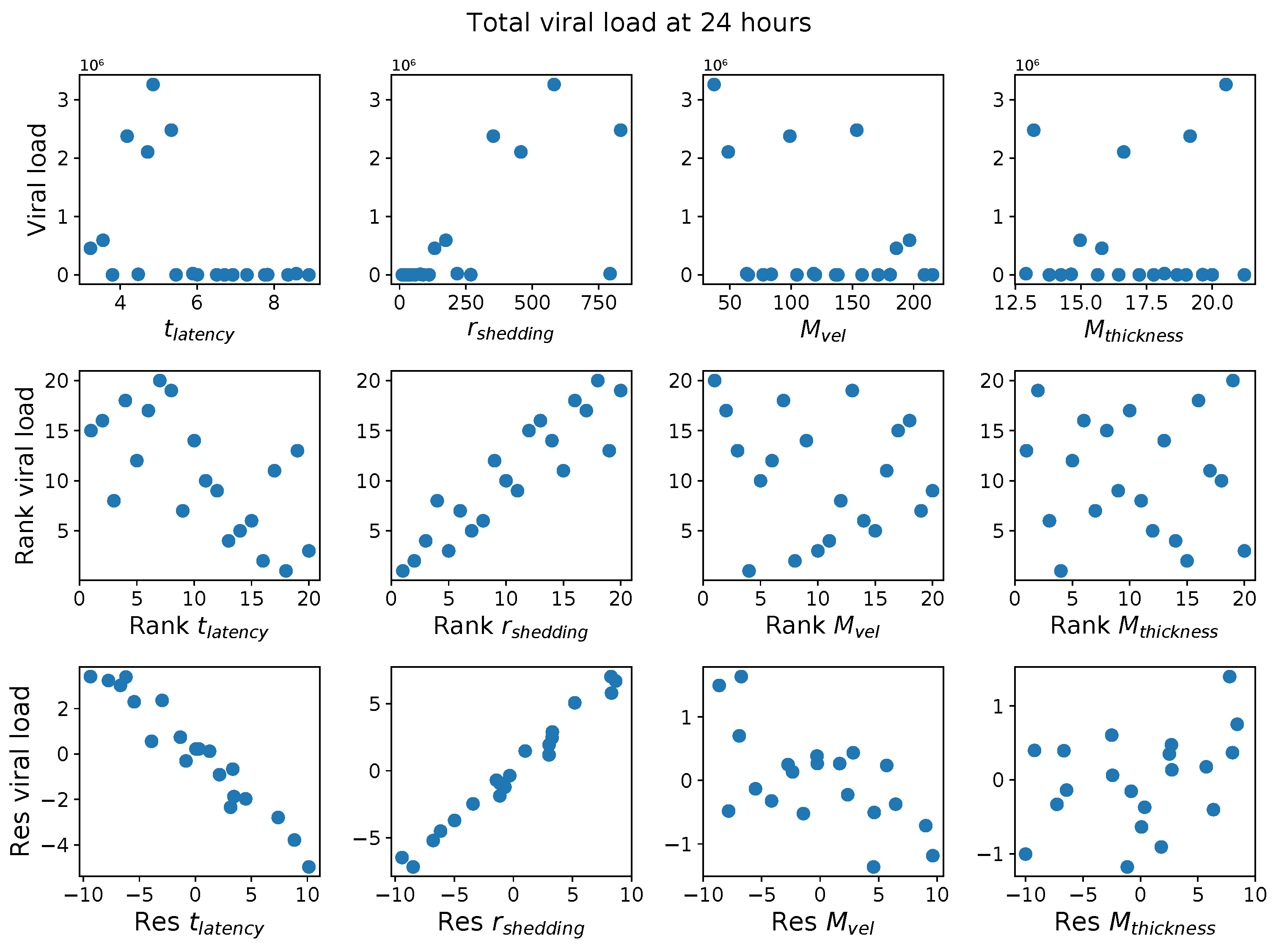
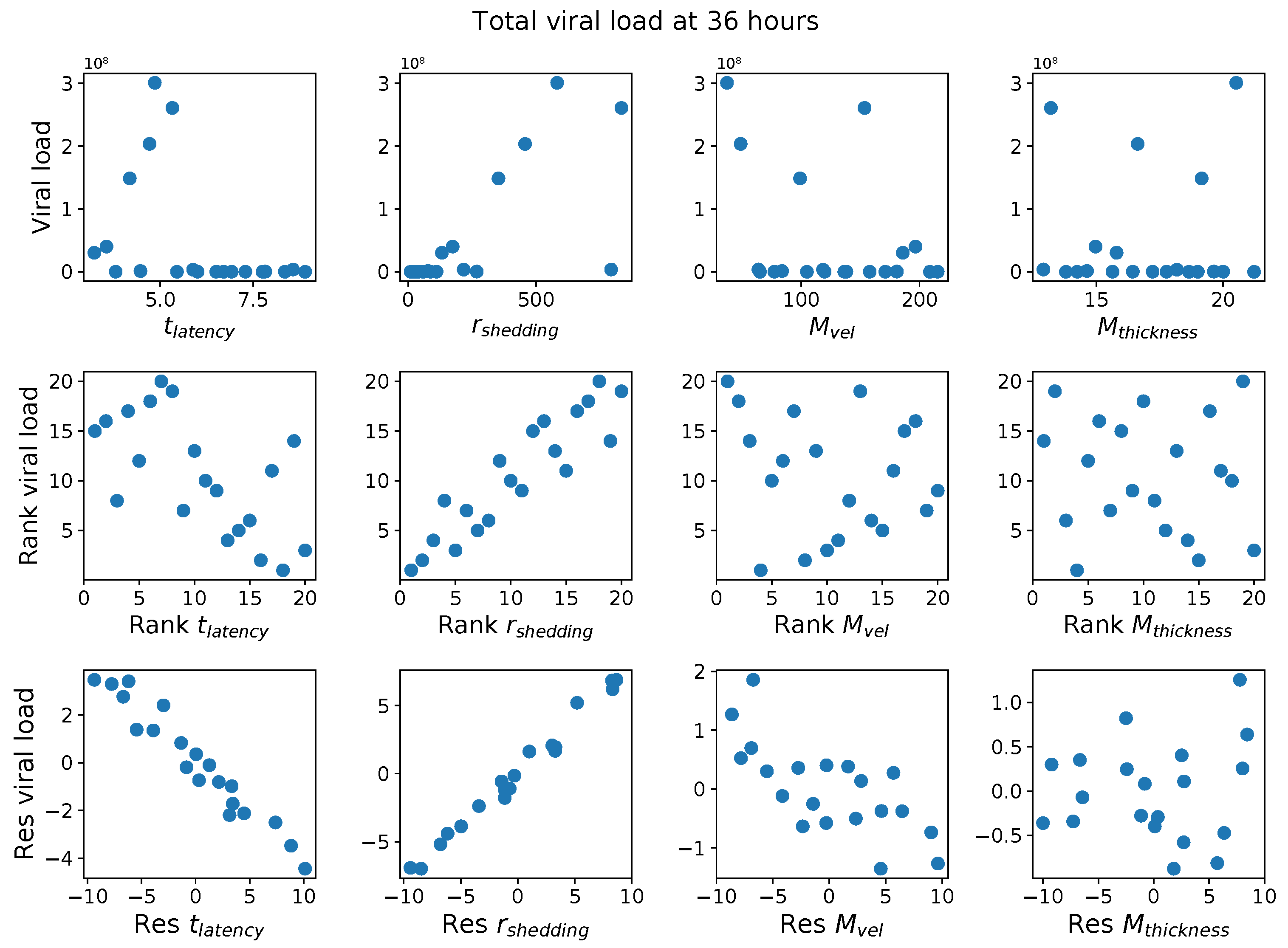
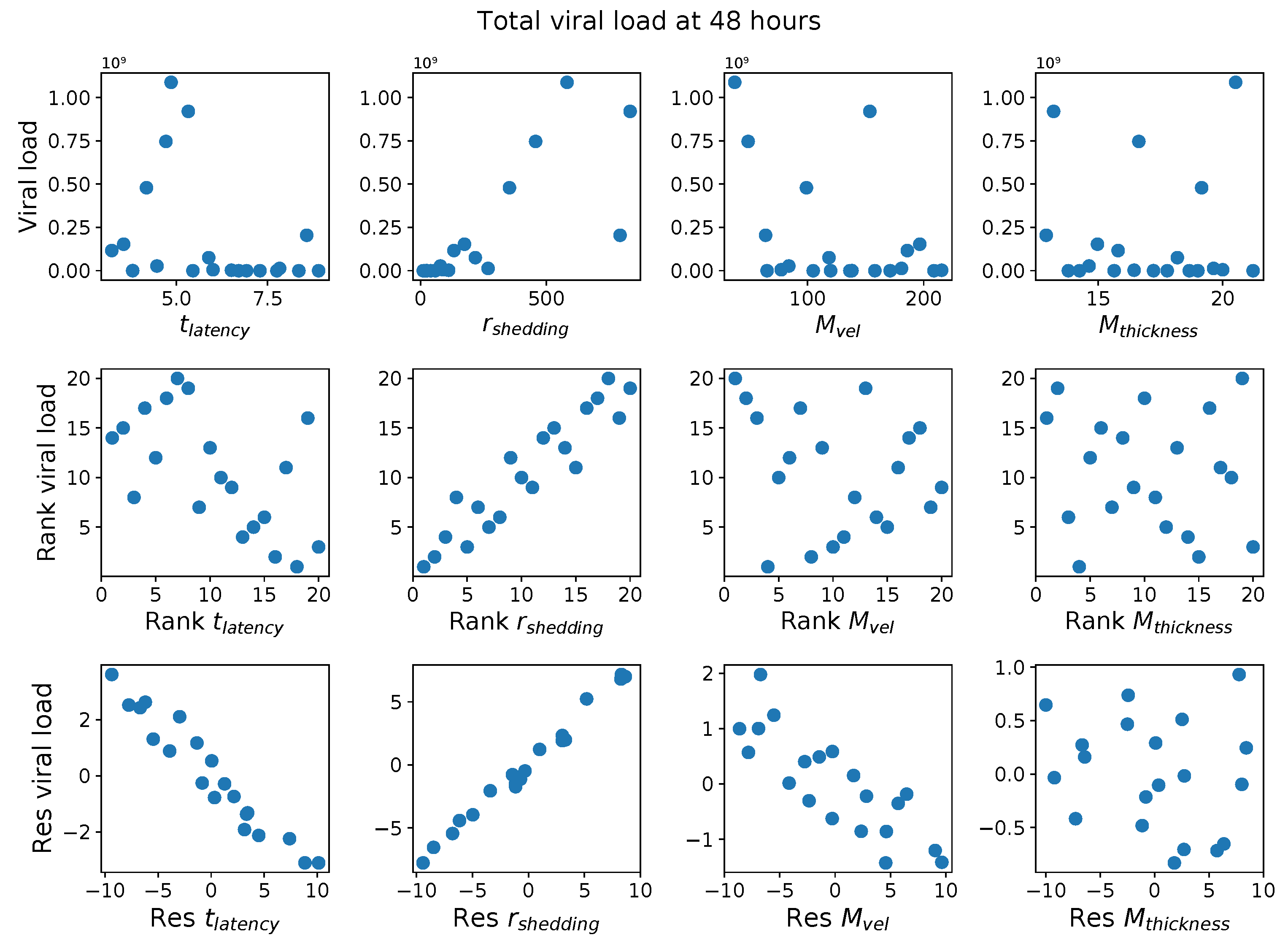
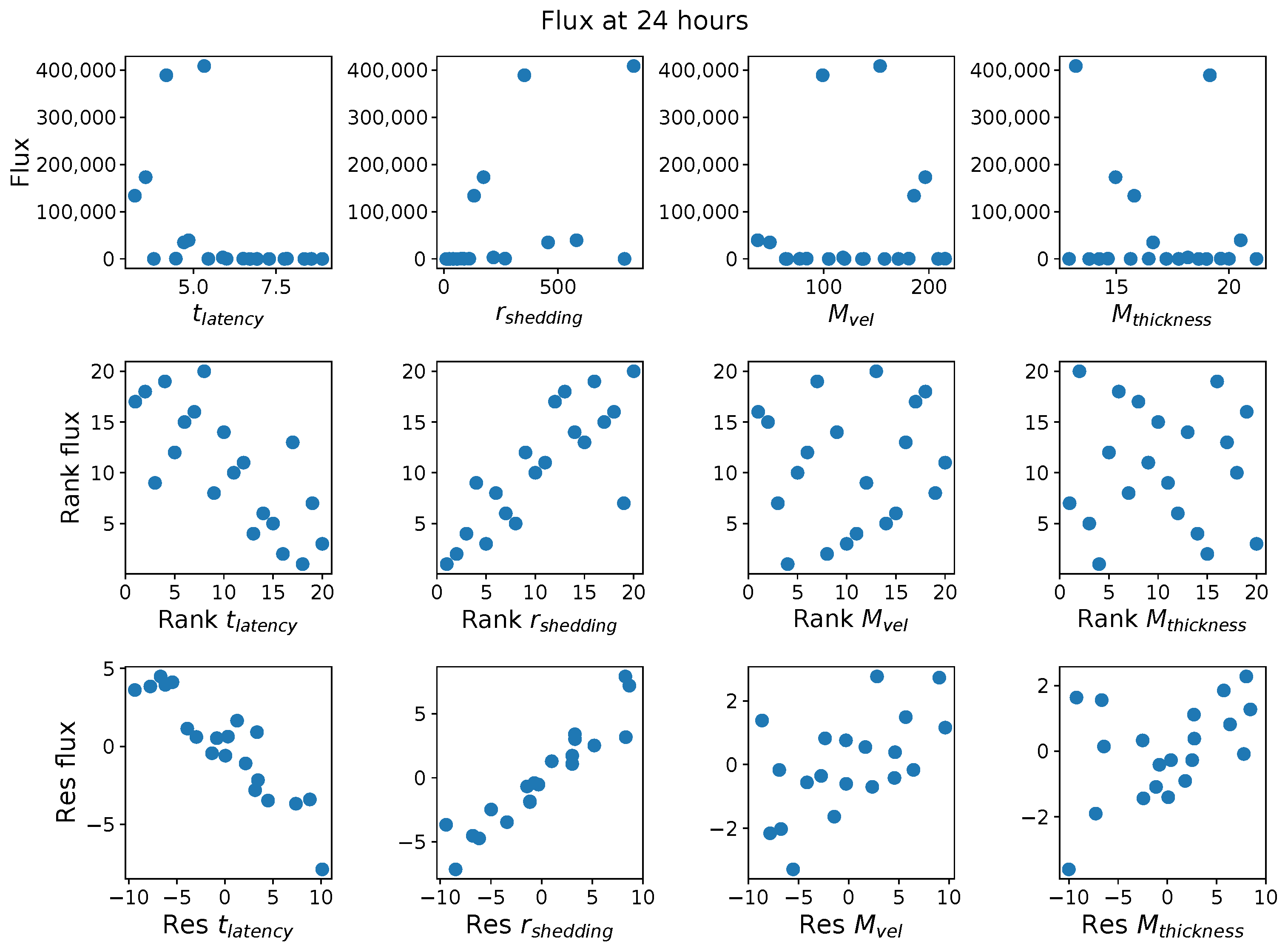
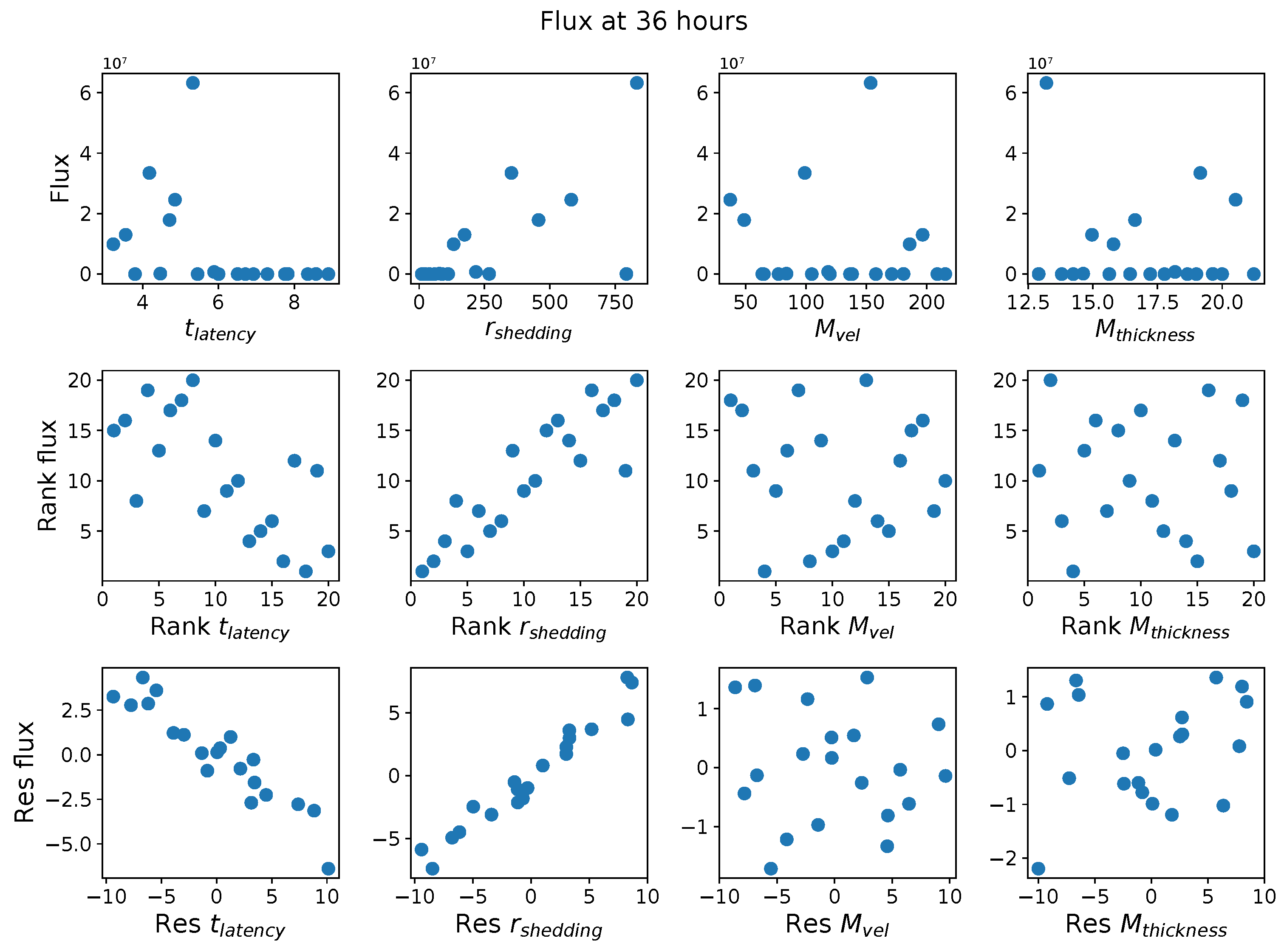
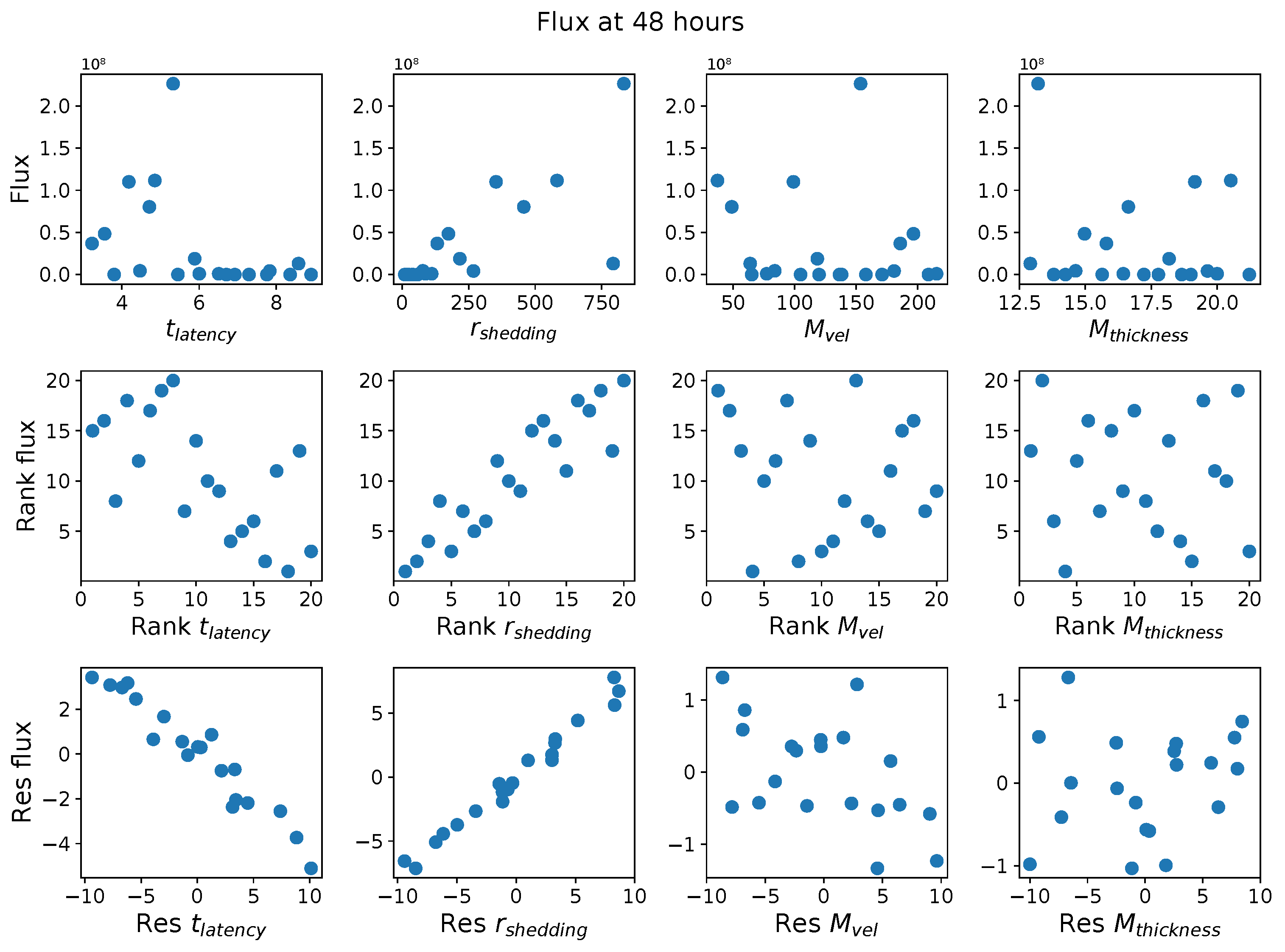
First, it was discovered that model outcomes of viral load and infected cell count are extremely robust/insensitive to variations in
, and in fact negatively correlated with
. (This result suggests that spike mutations leading to stronger binding to cell receptors may very well increase the likelihood of infection from exposure but is
not responsible for increased viral titers or infected cell counts.) As a consequence, to limit the dimension of parameter space we need to explore in this study, we fix
. Second, model outcomes are sensitive and positively correlated with
. Therefore, since the experimental and clinical data on the replication rate of
infectious RNA copies (virions) remain poorly understood, we allow for two decades of
, 10–1000 infectious virions per day by infected cells in the shedding phase. Finally, model outcomes are found to be
exponentially sensitive to linear variations in
. Therefore, based on prior [
23,
24] and continued [
25] single-cell experimental resolution data, we explore
spanning 3–9 h. (N.B. Since we fix
in this analysis, results from [
22] are presented in the Supplementary Materials to illustrate the remarkable robustness of outcomes to an order of magnitude variability in
.) Upper and lower bounds on all parameters, both physiological and in virus–cell infection kinetics, continue to be updated during the pandemic. Remarkably, none of the three cellular kinetic parameters in our model have been experimentally quantified. Therefore, we retain bounds on the sensitive parameters
and
that are consistent with the literature noted above and fix the robust kinetic parameter
. Additionally, there is strong clinical and experimental evidence [
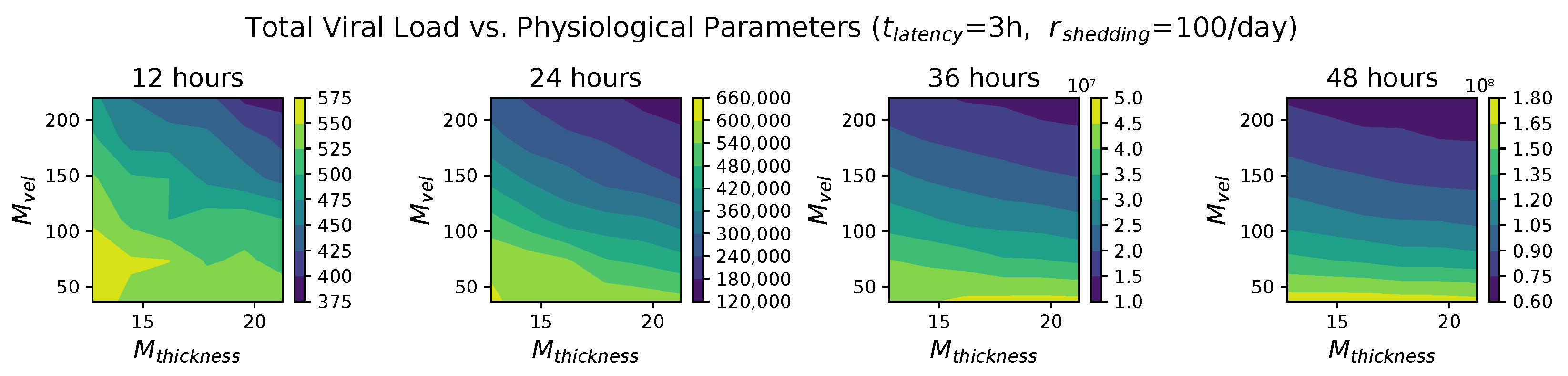
26] that two physiological parameters vary significantly with SARS-CoV-2 infection: the thickness
and the mucociliary advection velocity
of the mucus layer in the nasal passage. To our knowledge, the impact of host heterogeneity in these fundamental physiological features of nasal infection has never been explored, not just for SARS-CoV-2, but for any virus.
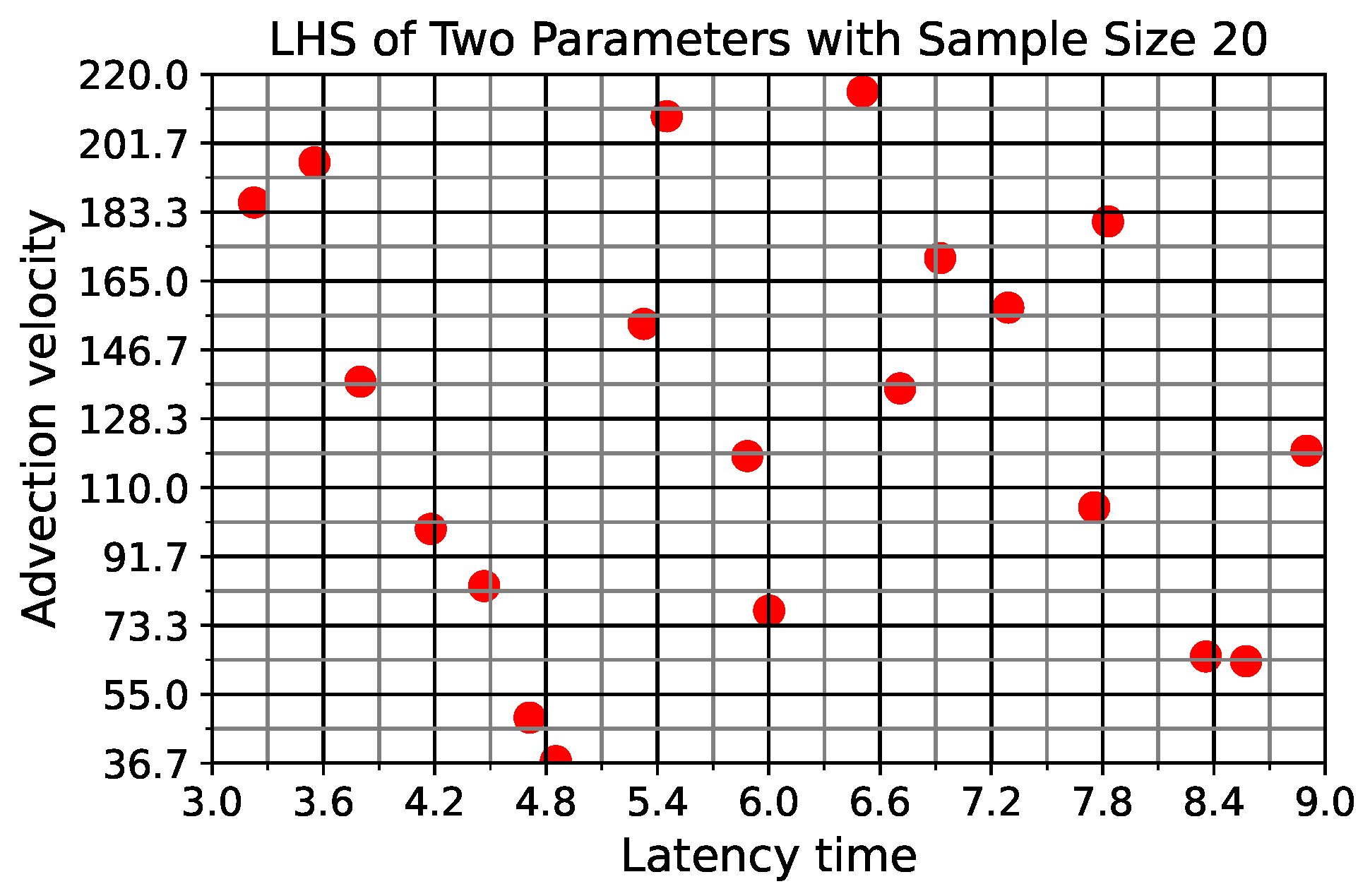
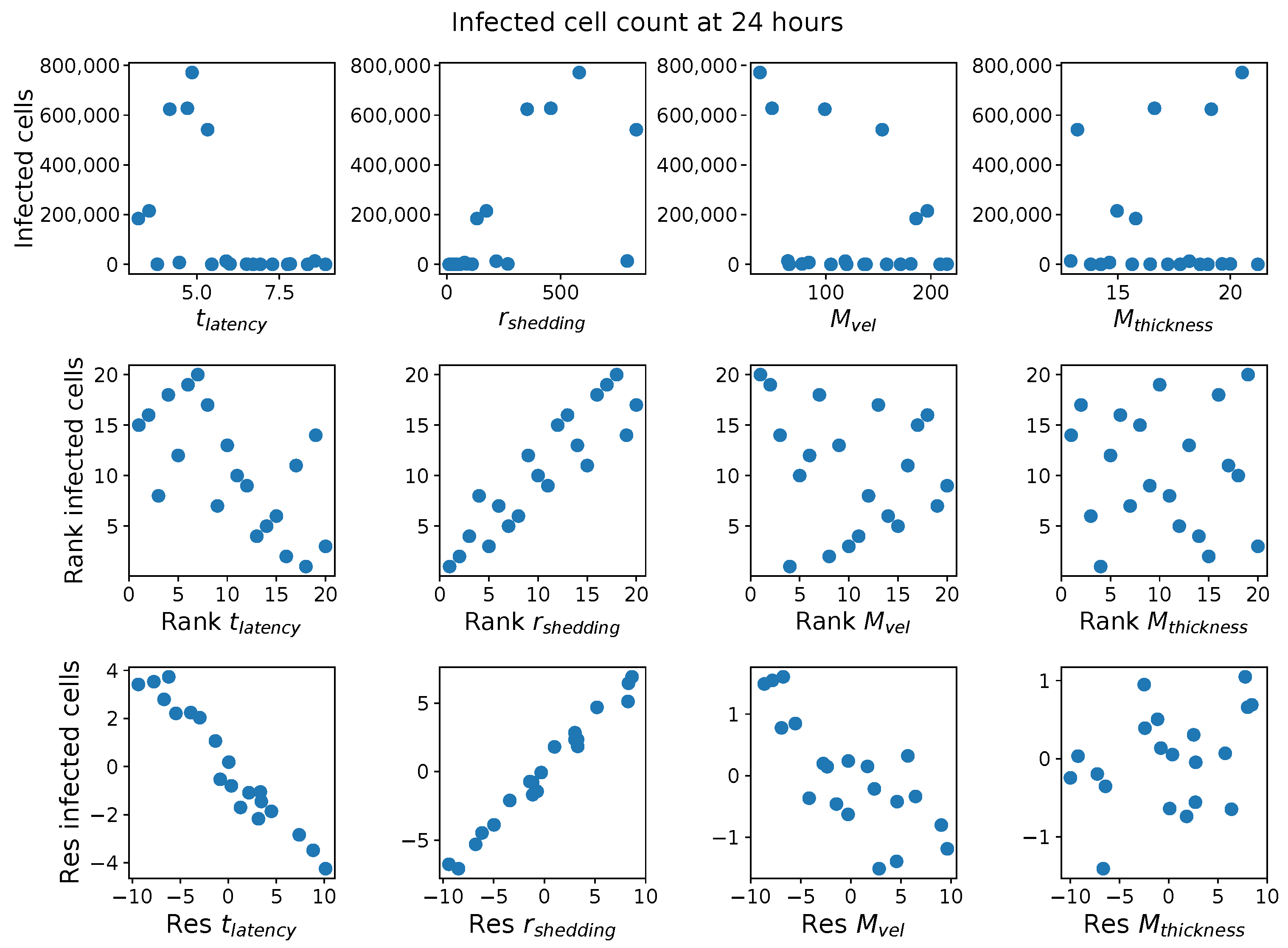
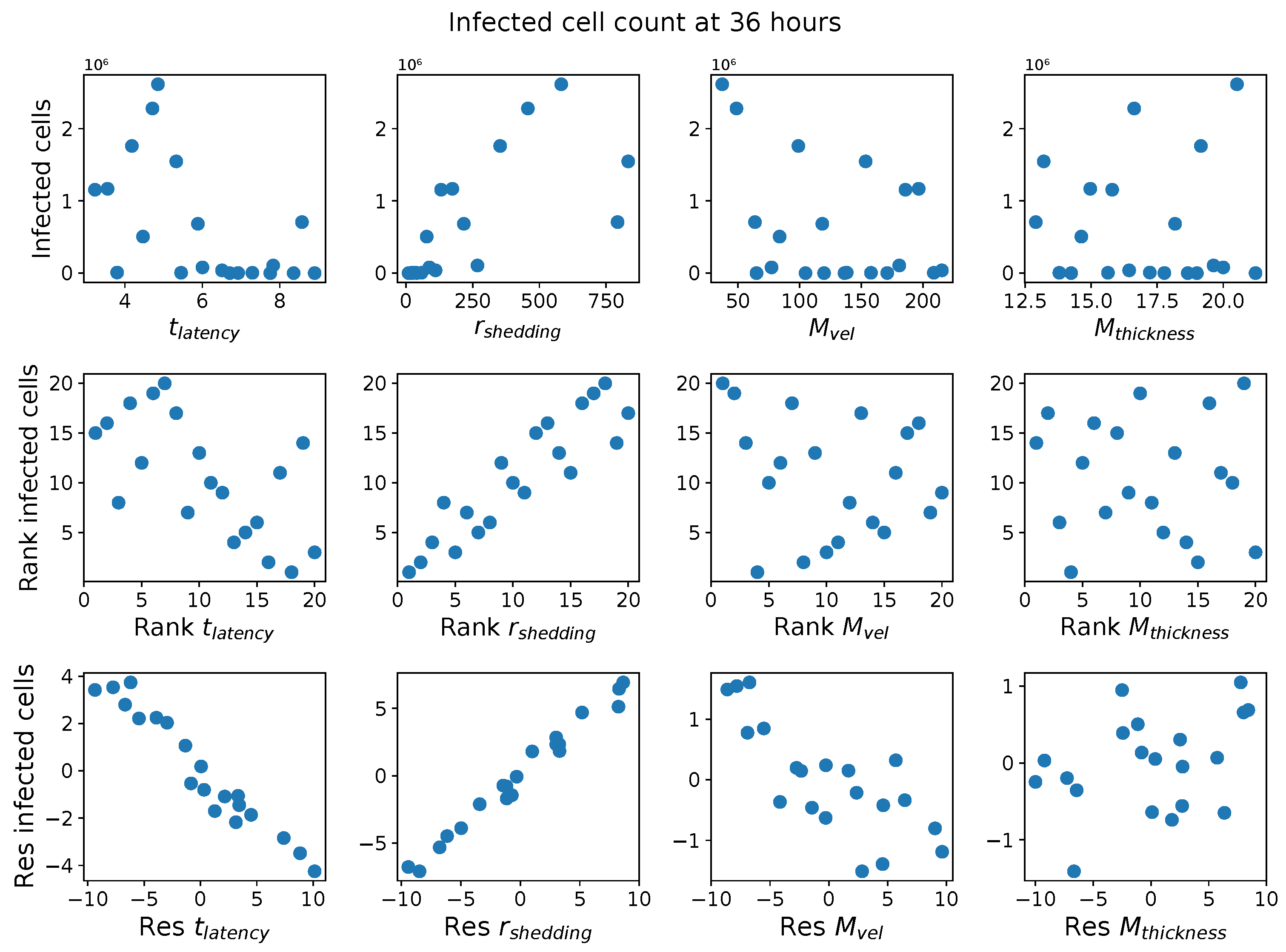
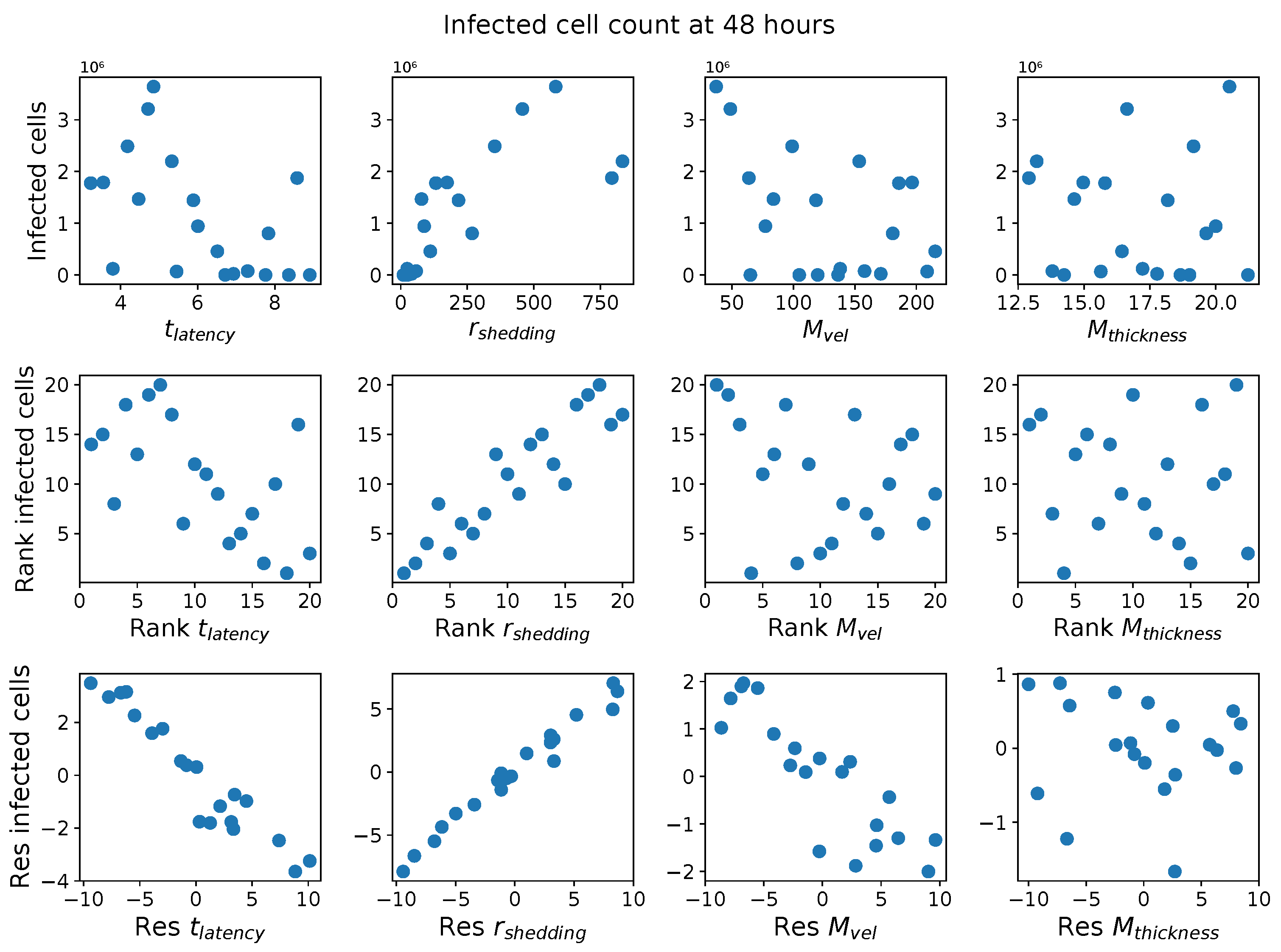
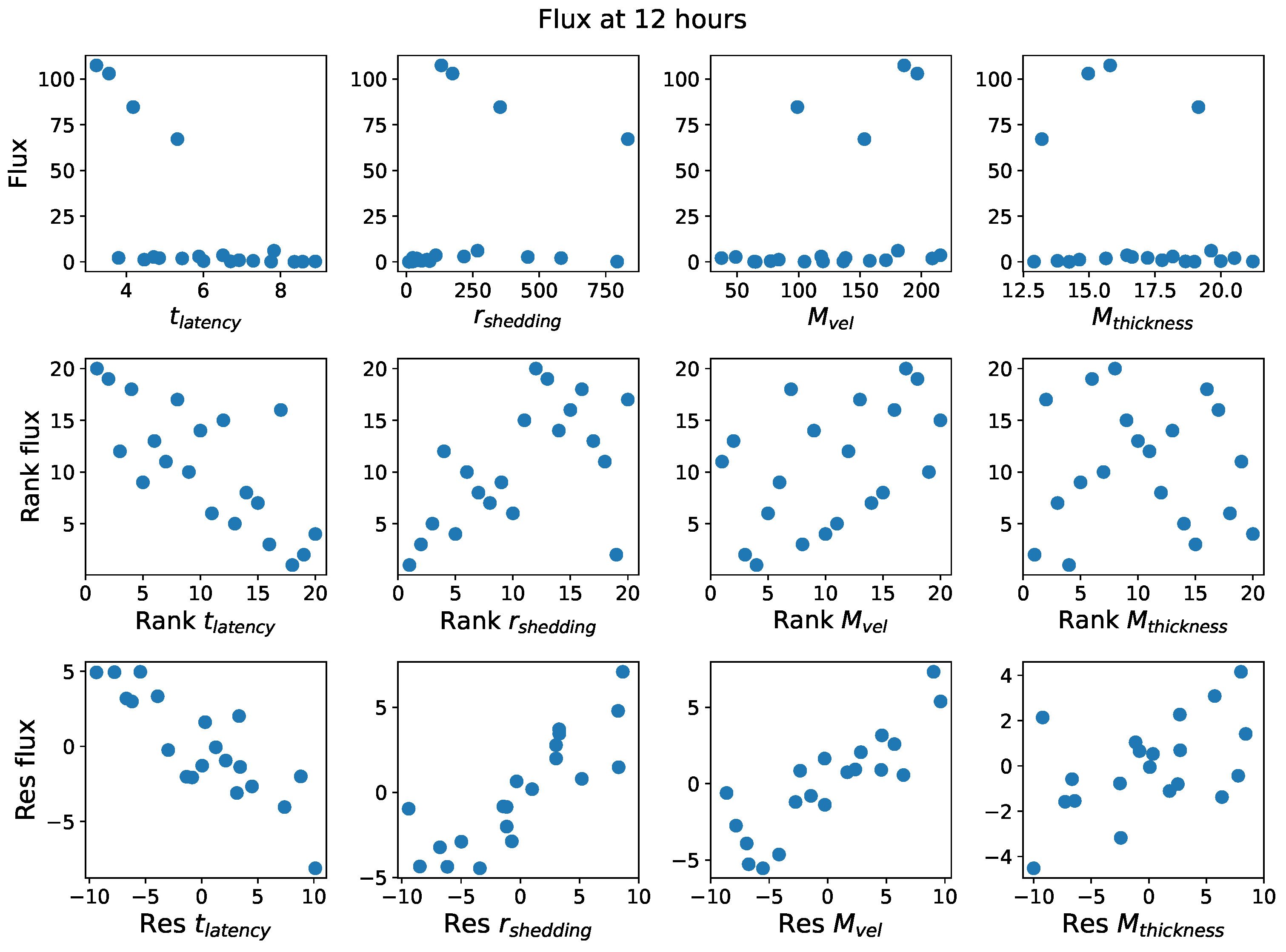
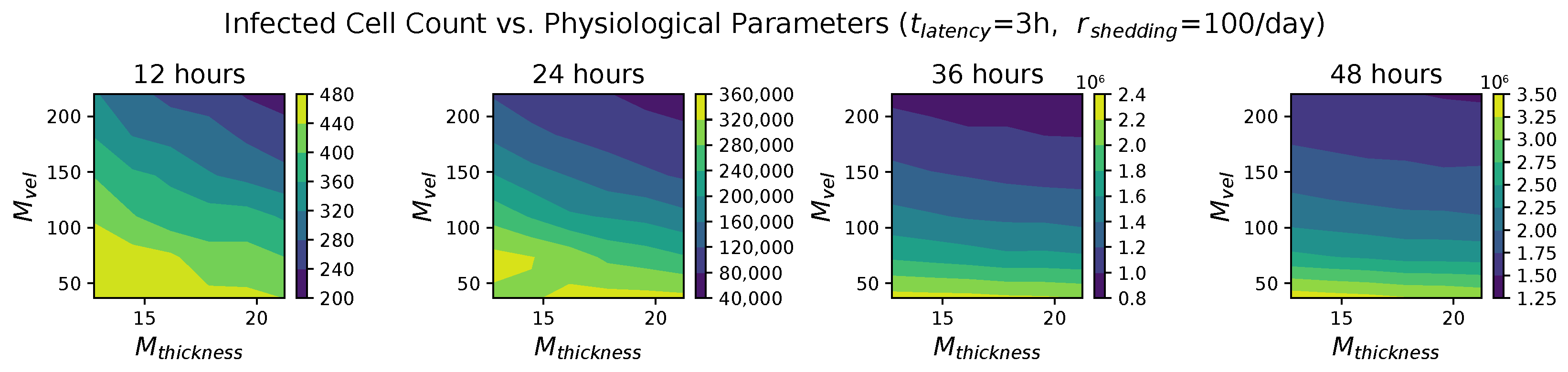
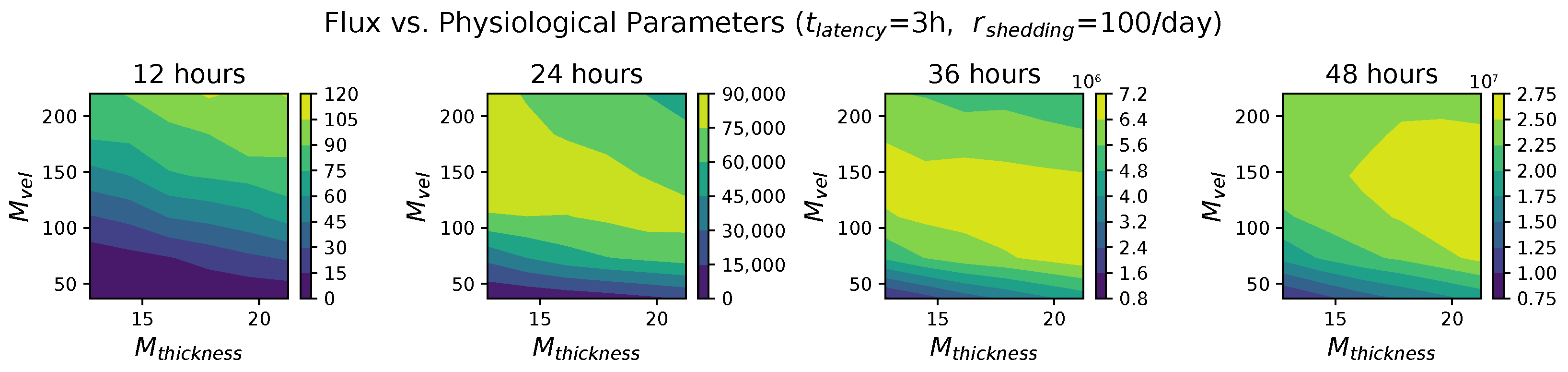
In light of the above data and results, for the present study, we explore the dynamic outcomes over 48 h in infectious viral load, total number of infected cells, and flux of infectious viral RNA copies out of the nasal passage. In this paper, we apply global sensitivity analysis techniques to our physiologically faithful, spatial respiratory infection model, focusing on the nasal passage as the source of initial infection from inhaled viruses and clinical test data from nasal swabs. As rationalized above, the global sensitivity analyses are applied across the four-parameter space of [shedding rate of infectious RNA copies, infected cell latency time, thickness, and clearance velocity of the nasal mucus layer] = [, , , ]. For this study to be self-contained, we summarize the model and the methods before presenting the results.
The Model
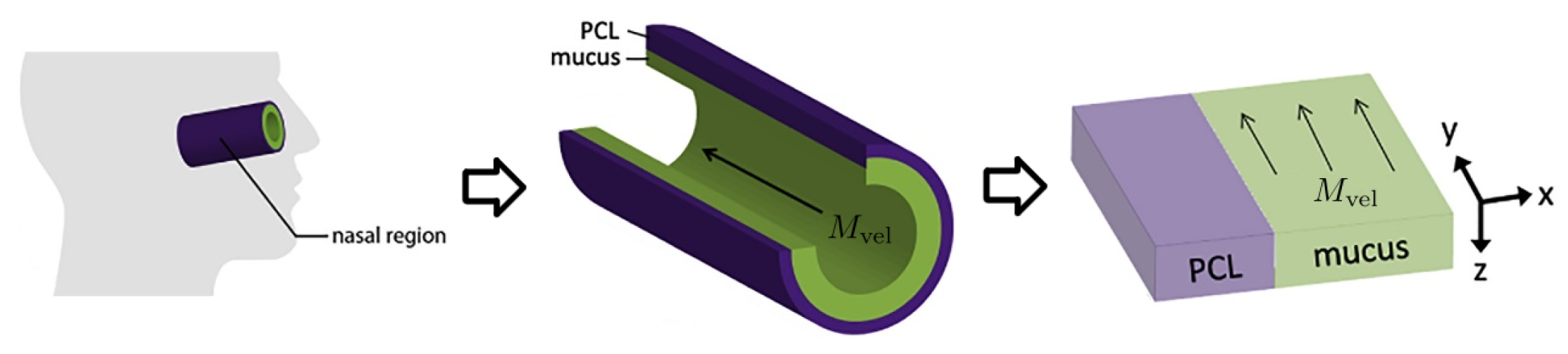
We summarize key model features from [
10] so that the present paper is self-contained. As shown in
Figure 1 from [
10], and articulated in detail in [
27], the nasal passage and all generations of the lower RT except the alveolar space are approximately cylindrical. In each generation, the epithelial cell surface is coated by a 7
m thick layer of periciliary liquid (PCL) in which cilia beat. At full extension in the power stroke, cilia penetrate the PCL-mucus interface and extend into the mucus layer up to 1
m, and the coordinated metachronal waves of cilia propel the mucus layer, “down” in the nasal passage and “up” in the lower RT, towards the esophagus to be swallowed.
We unfold this cylindrical geometry into a rectangular domain in which the
y-
z-plane falls on the epithelial cell surface.
x denotes the “radial” distance into the PCL and mucus layers, with
being the epithelium–PCL interface.
y denotes the distance along the centerline axis, which is the primary direction of mucus advection by the coordinated beating of cilia, with
representing the entry into the nasal passage.
z is the azimuthal axis of the cylinder. Infectious virions undergo diffusion in PCL and mucus and additional advection with velocity
while in the mucus layer, governed by:
where
Ciliated cells are the predominant infectable cells in the RT above the alveolar space, covering about 50% of the epithelial surface. Every epithelial cell has a degree of infectability, either non-infectable or with a prescribed probability of infection per encounter second.
In our model, a freely diffusing virion in the PCL encounters a cell when its distance from the epithelial cell surface vanishes, i.e., when . For each second during an encounter with a ciliated cell, there is a probability of an infection. If an encounter results in infection, the cell switches from uninfected to infected, and the virion is removed from the free virion population. When the stochastic virus–cell encounter does not result in infection, for infectable or non-infectable cells, the virion is reflected back into the PCL.
Each virion is tracked until it either infects a cell or exits the generation, always toward the trachea due to strong mucus advection. Once a cell switches to an infected state, it persists in an infected, non-shedding latency state for a duration , which represents cellular uptake of the virus and hijacking of the cellular machinery to replicate viral RNA copies. After has lapsed, the cell switches to a shedding state, replicating infectious virions at rate . Since infected cells typically die after 3 days post-infection, no cells switch to a death state in this 48 h study.
We assume that the kinetics of SARS-CoV-2 interactions with ciliated cells are robust within each host yet potentially highly variable between hosts, and therefore, we explore literature-supported ranges for the kinetic parameters that our previous study [
22] revealed to be sensitive. All simulations to generate data for this study start at the moment of infection of one cell at the entry of the nasal passage (axial coordinate
).
Table 1 summarizes the model parameters, fixed and variable, and the simulation details.
Table 2 summarizes the three model outcomes and associated data.
4. Concluding Remarks
The goal of this study is to use computational modeling to gain insights into the potential drivers of extreme population heterogeneity in SARS-CoV-2 viral titers from positive nasal swab tests throughout the pandemic. In the above sections, we summarized our physiologically faithful, spatially resolved computational model of viral infection in the human nasal passage [
10]. We then described the global parameter sensitivity analyses required to evaluate the absolute and relative impact of each of four hypothesized mechanistic drivers of extreme host-to-host heterogeneity in nasal titers: nasal mucus layer thickness and clearance velocity, infected cell latency time (from the moment of infection to the onset of shedding infectious viral RNA copies) and shedding rate of infectious RNA copies. We then applied the global sensitivity methods to the model-generated, virtual population database of the dynamic progression over 48 h after initial infection of viral load, infected cells, and flux of viruses out of the nasal passage. In this virtual population, each fixed, distinct set of four parameters defines a class of similar hosts. These global sensitivity methods isolate the impact unique to each parameter, de-correlated from the other parameters, and accomplish this via quasi-random sampling over the entire four-dimensional virtual population database.
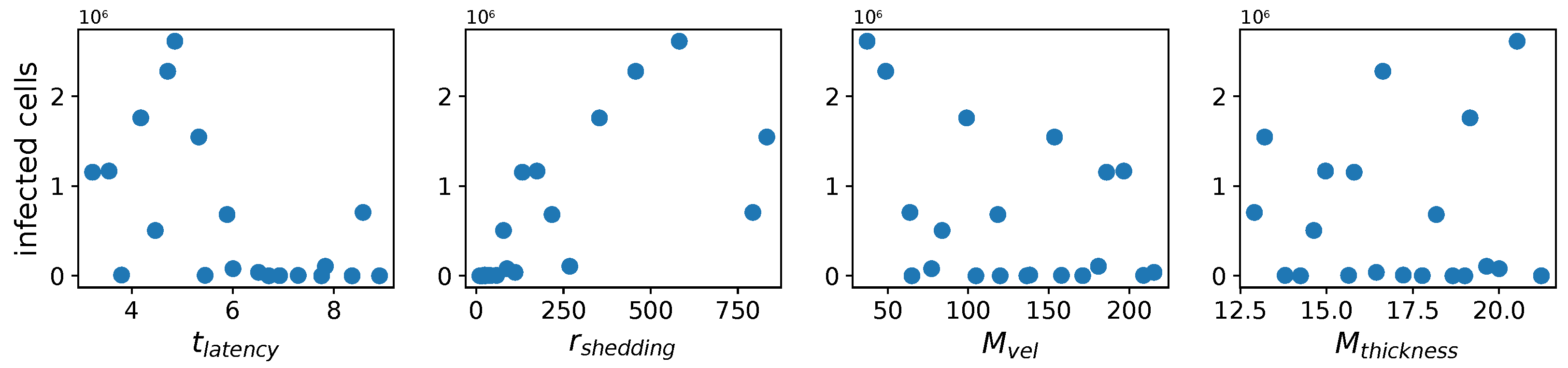
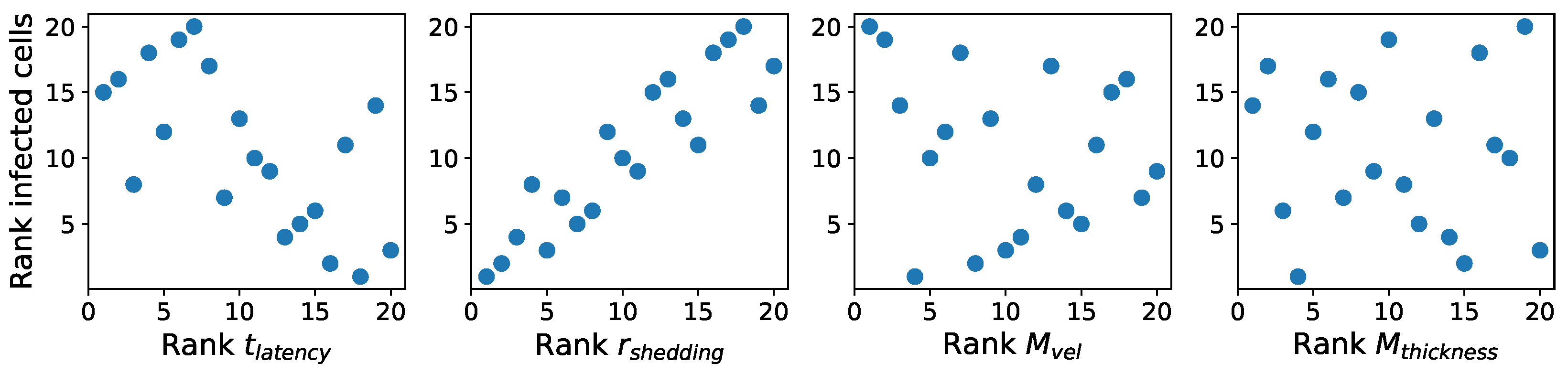
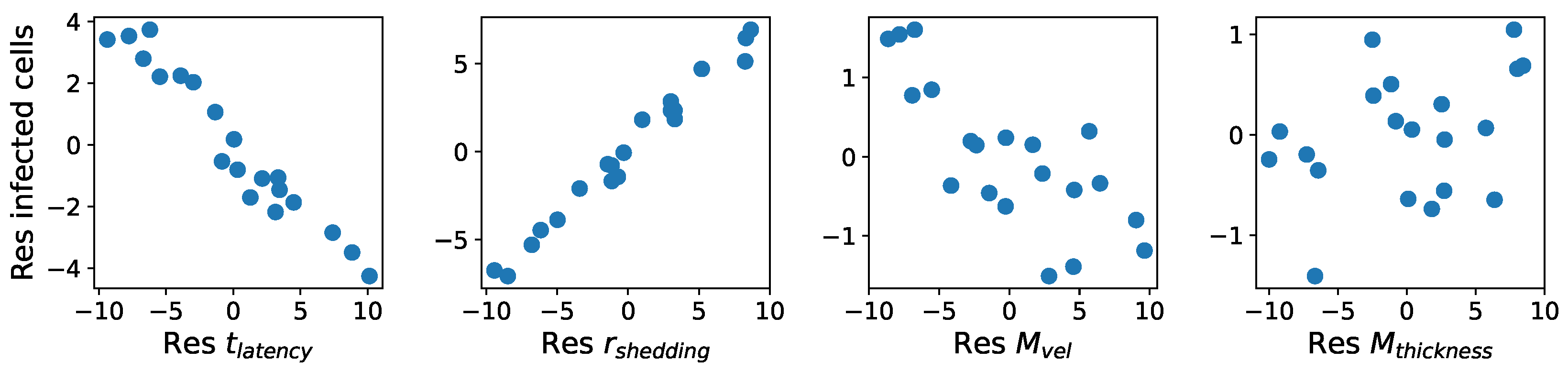
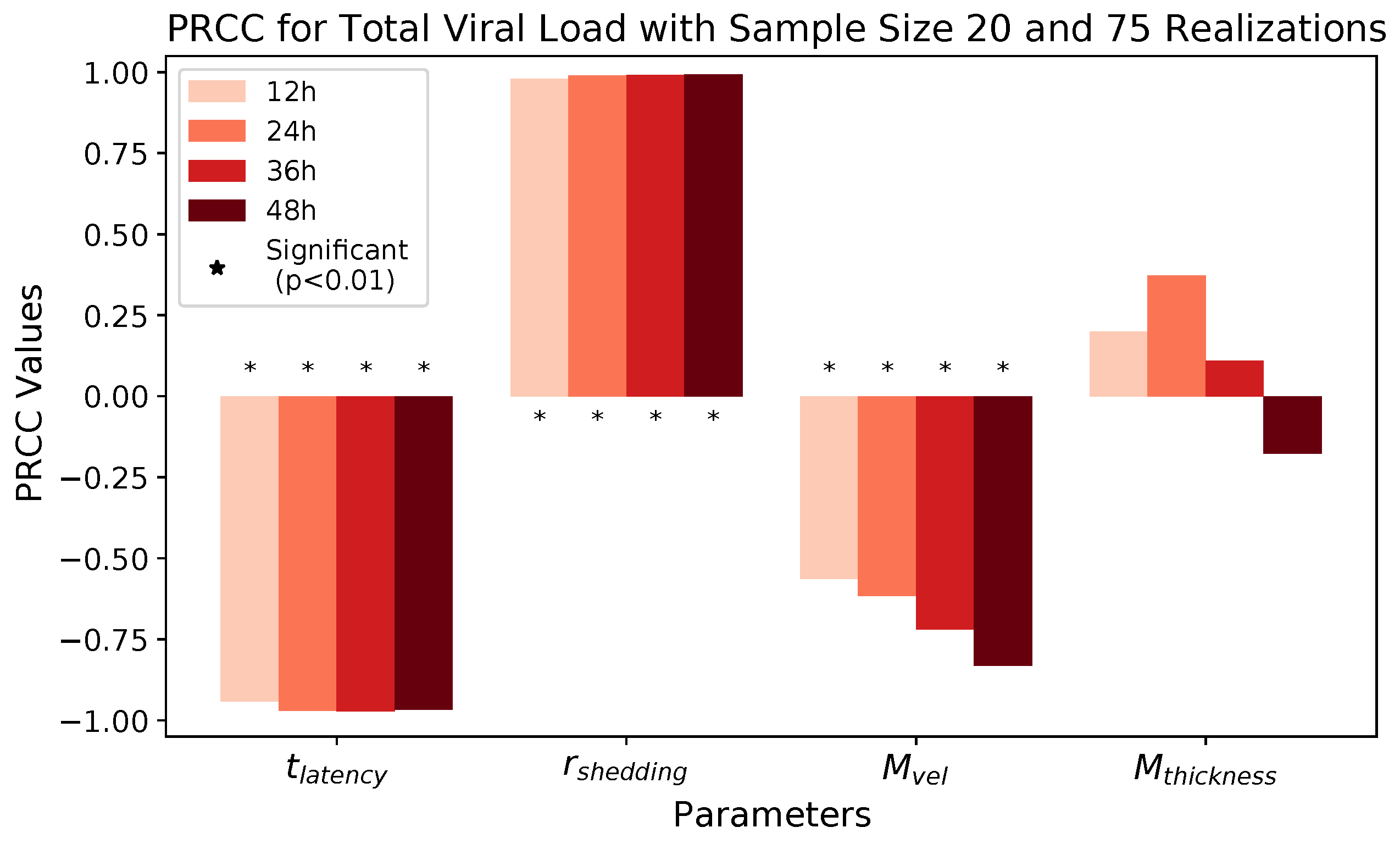
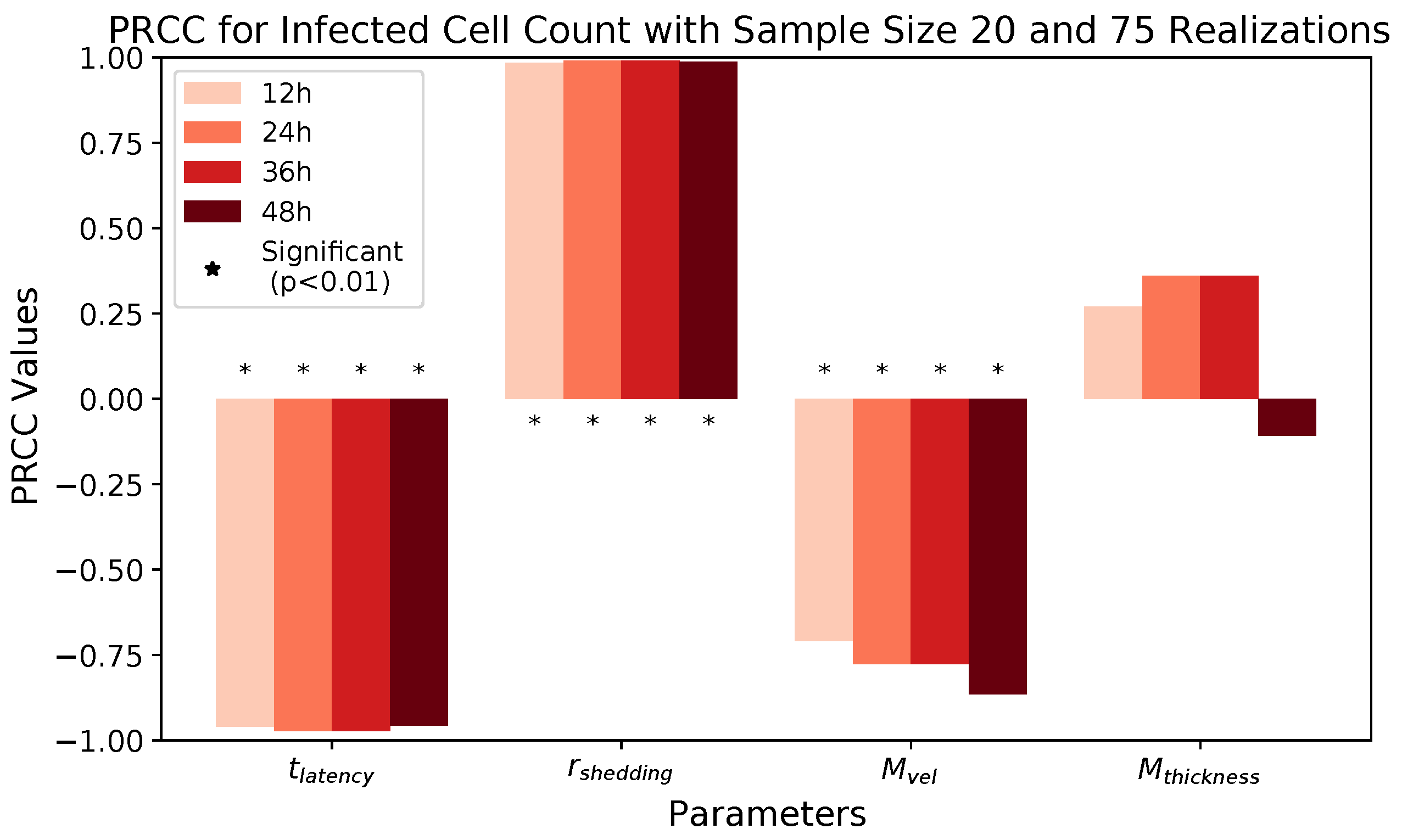
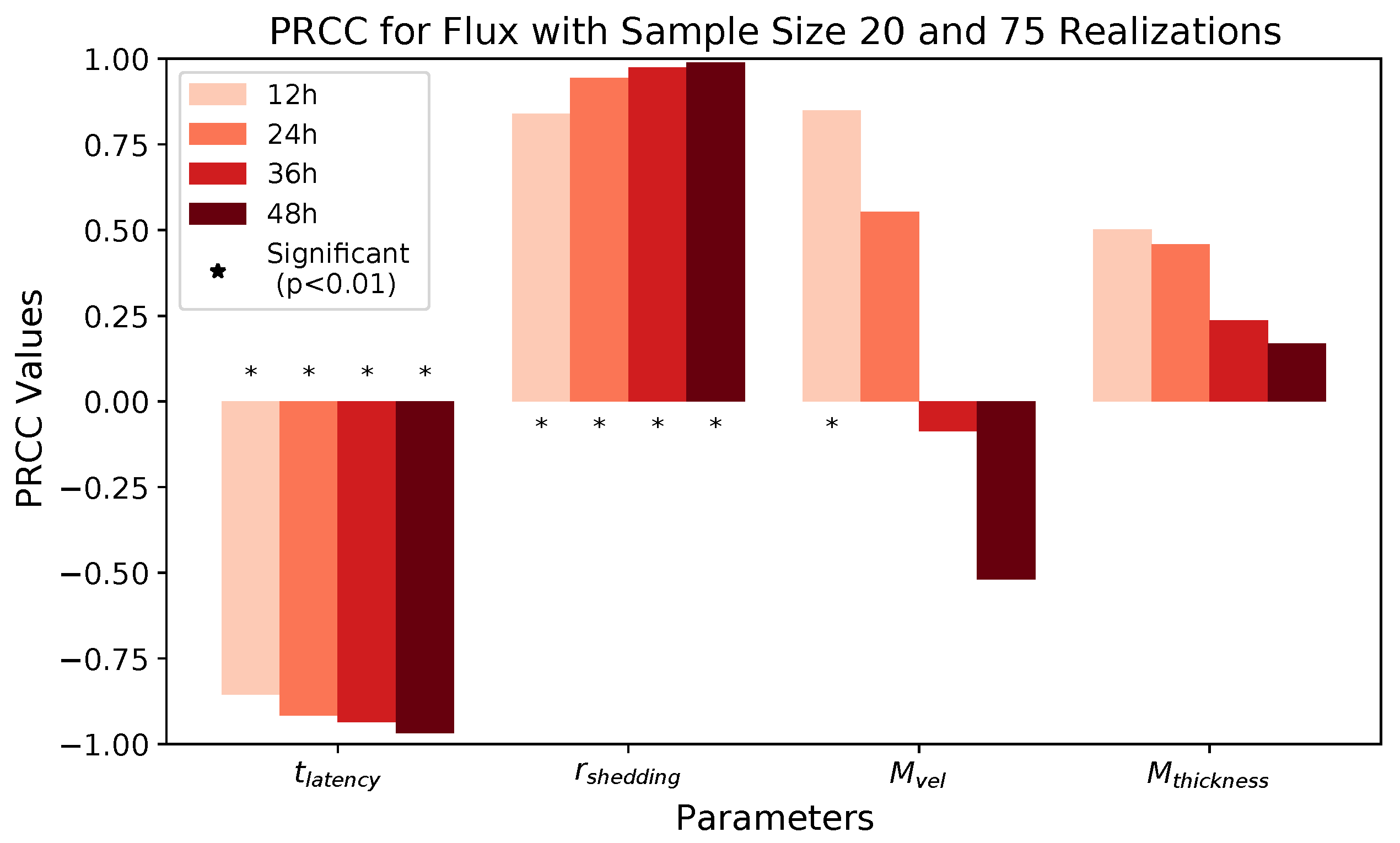
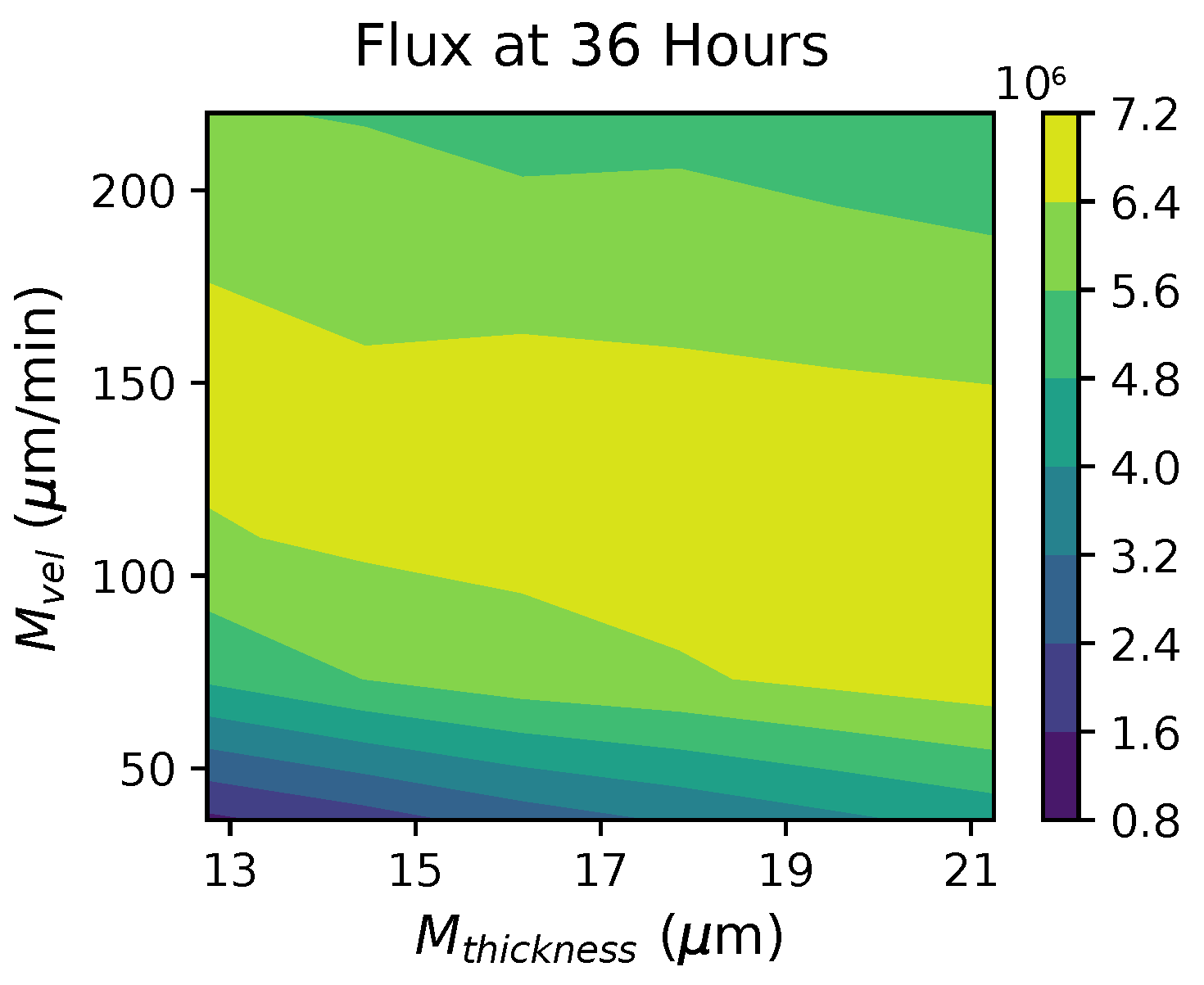
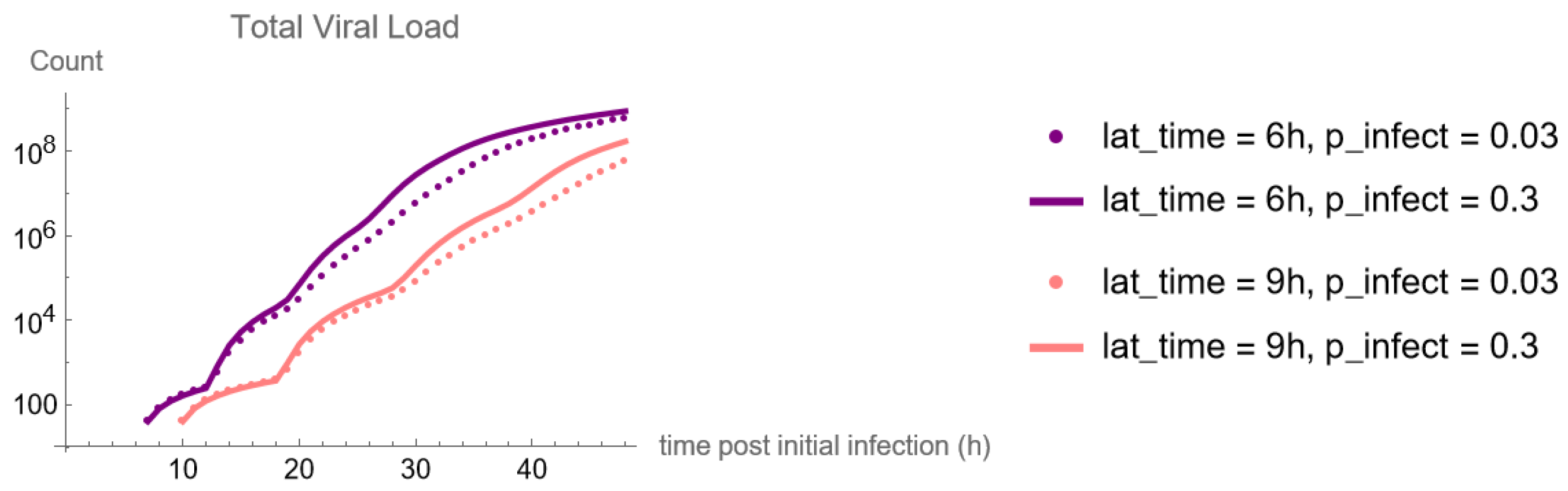
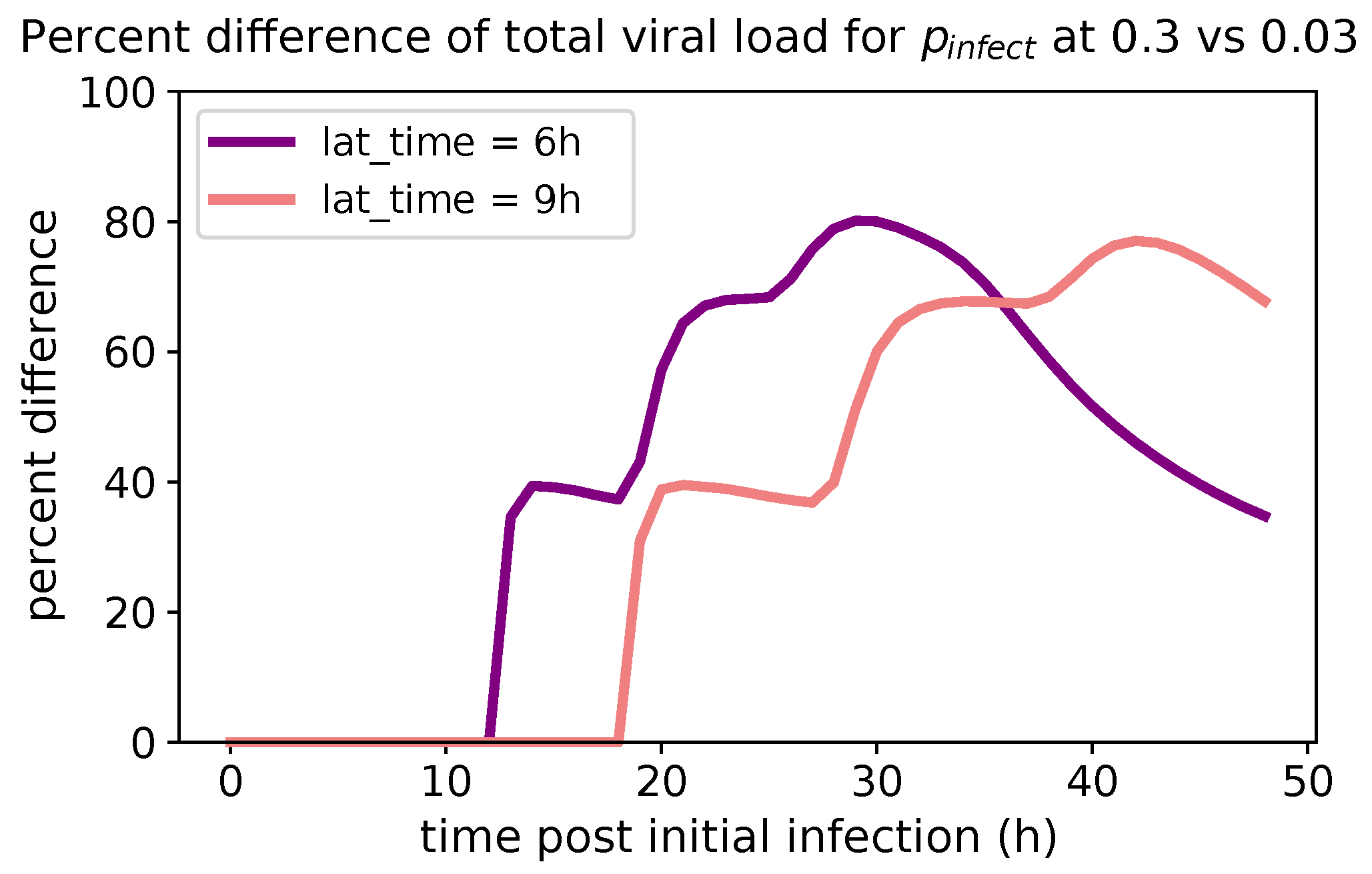
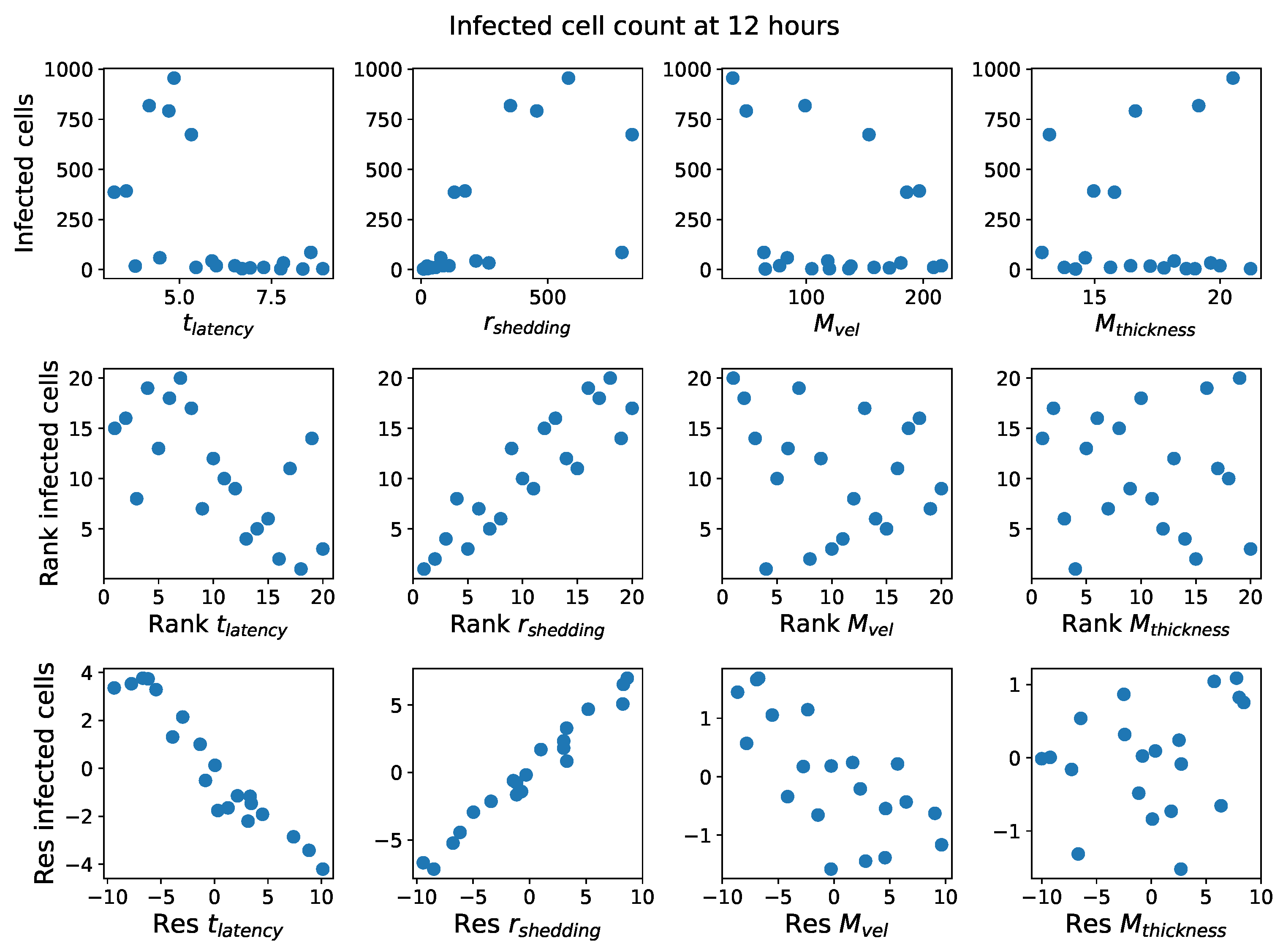
These methods produce several insightful predictions. 1. The latency time () of newly infected cells has the strongest, indeed exponential, negative correlation on total nasal viral load; i.e., linear reductions in infected cell latency time (within 9 to 3 h) produce exponential variations in total shed viral load at each 12 h timestamp, corresponding to several-orders-of-magnitude heterogeneity in viral load due solely to reduced latency time. Reduced latency time has a similar exponential impact on total infected nasal passage cell counts. 2. The viral RNA shedding rate () of infected cells in the shedding phase has a strong, proportional but not exponential, positive correlation on total viral load at each 12 h timestamp. Orders-of-magnitude increase in shedding rate produce orders-of-magnitude increase in total nasal viral load and infected cell count. 3. Nasal mucus clearance velocity () is negatively correlated with total viral load and infected cell count, with very weak impact in the immediate hours post-infection that increases through 48 h yet mildly relative to latency time and shedding rate. 4. Nasal mucus thickness () has little impact on infection outcomes.
The salient insight gained from this study is that the observed population heterogeneity in the first two days post nasal infection from inhaled exposure to SARS-CoV-2 can be reproduced by the mechanisms and physiological features within our computational model. This rules out other additional drivers of heterogeneity that are not captured within our model. However, this modeling and global sensitivity analysis clearly points to the latency time of infected cells—spanning cellular uptake of the virus and the hijacking of cellular machinery to produce viral RNA copies until the initial onset of extracellular shedding of viral RNA—as the primary driver of exponential population heterogeneity. Variations in the latency time of infected host cells potentially arise from some combination of viral RNA and cell DNA compatibility; e.g., there could be nuanced population DNA interactions to a specific SARS-CoV-2 variant or within variants. With respect to other respiratory viruses, the model and sensitivity results presented apply to any virus. However, to do so, one needs to have measurements of the virus–host kinetic interactions: the probability of infection per virus–cell encounter, latency time of infected cells prior to shedding of viral RNA copies, and shedding rate of viral RNA copies. These kinetic parameters are almost surely specific to virus and host, requiring cultures from the individual and exposure to the virus. This experimental data, coupled with the physiology of the individual, are predictive of pre-immune response in the immediate 48 h post initial nasal cell infection. Should features not incorporated into our modeling platform be shown to have a leading order effect, we are poised to incorporate those features, similar to how we have extended our pre-immune modeling platform to both innate [
11] and adaptive [
12] immunity.
These results and insights strongly suggest the need for experimental data to be collected spanning different variants of SARS-CoV-2, spanning nasal cultures grown from a diverse collection of individuals, and then careful measurements of the mechanistic parameters in our model. We note that high-resolution cell culture experiments need to focus on measurements of infection probability per virus–cell encounter, latency time, and extracellular shedding rate once an infectious virus–cell encounter takes place. The outcome metrics of total shed viral load and number of infected cells in a cell culture will not be representative of in vivo nasal infection since there is no mucus clearance in cell cultures that we know accelerates viral load. In order for these insights to be “actionable” for medical treatment, a nasal culture can determine the virus–cell infection kinetics of an individual, and single-cell measurements of latency time and replication rate could potentially guide the decision for rapid drug or antiviral therapies applied directly to the nasal passage. Lastly, the flexibility and robustness of our model and simulation platform are adaptable for future investigations into other respiratory viruses.

 ,
, 


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}