
Reclassification of ASFV into 7 Biotypes Using Unsupervised Machine Learning
 , , ,
, , ,  , ,
, , 
Abstract
:1. Introduction
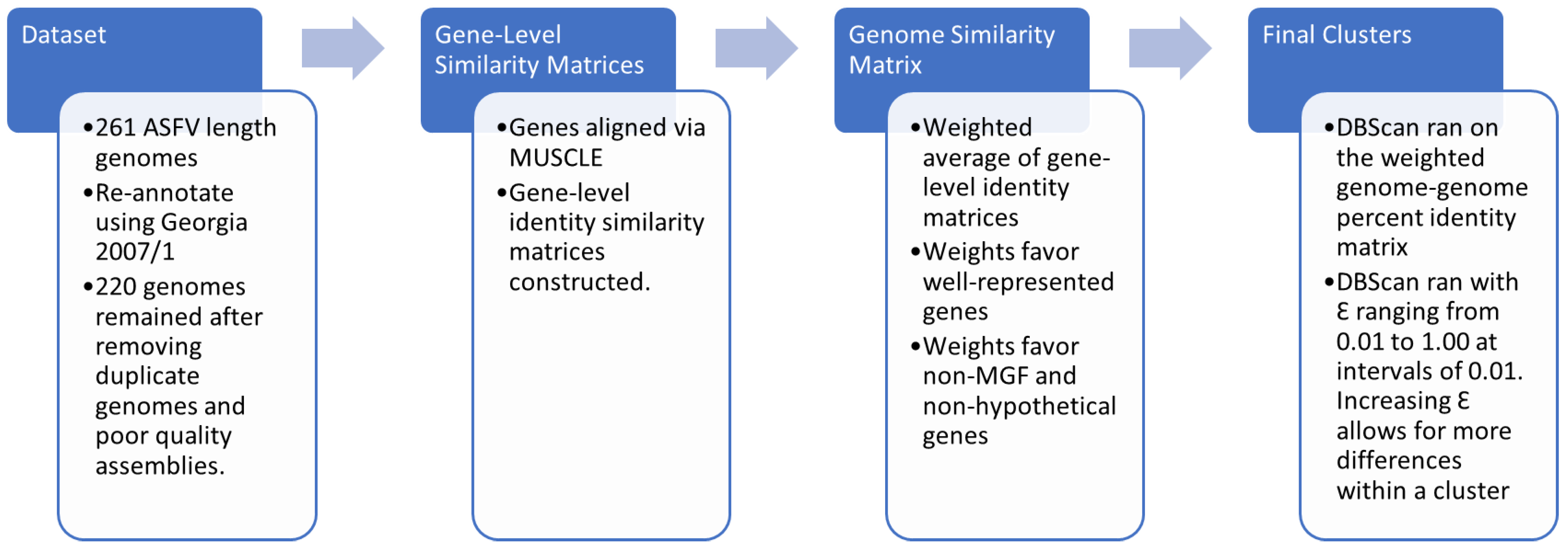
2. Materials and Methods
2.1. Description of the Dataset
2.2. Annotation of the Dataset
2.3. Curation of the Dataset
2.4. Protein Alignment
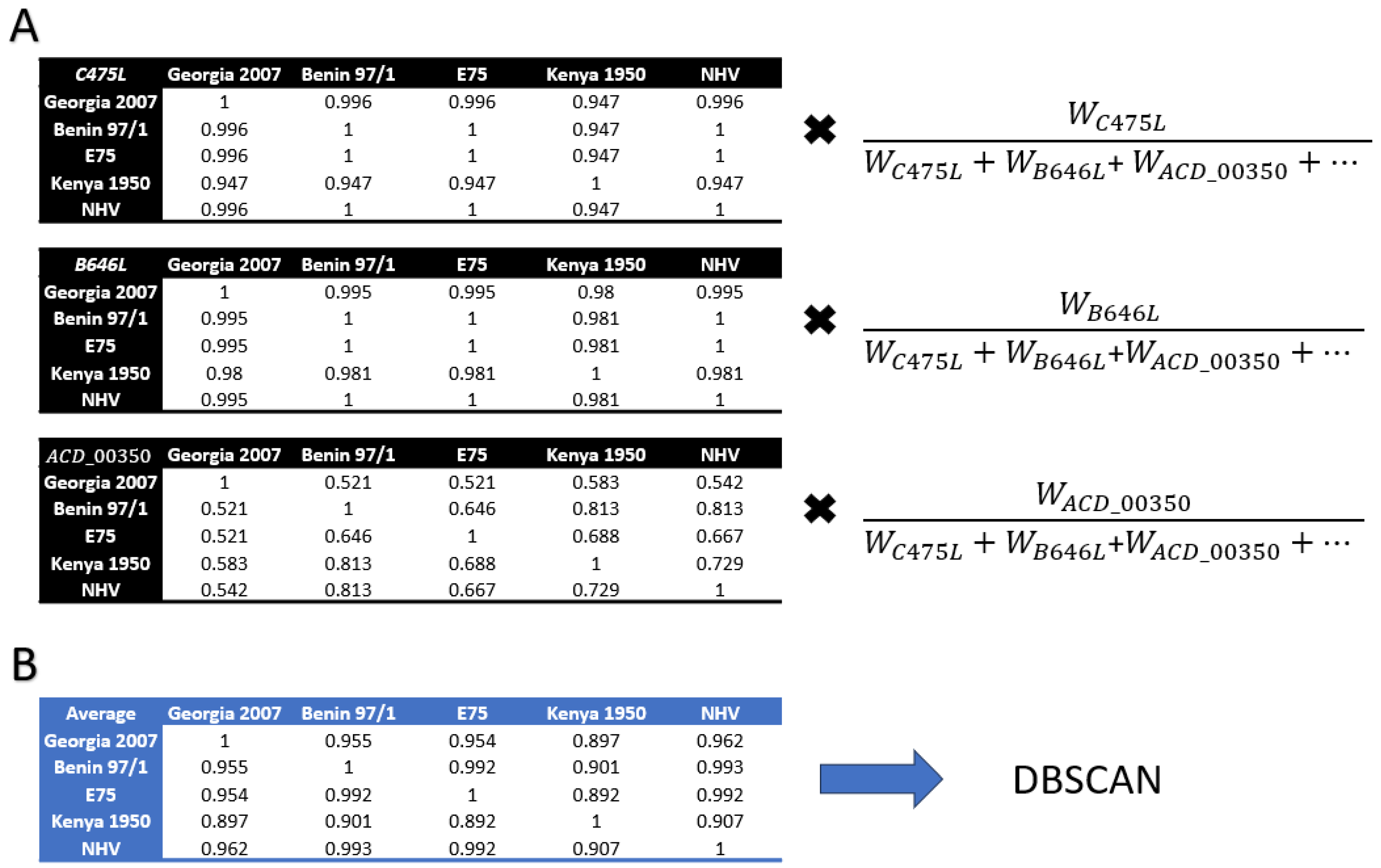
2.5. Genome Level Analysis and Clustering
3. Results
3.1. Full-Length Genomes on NCBI
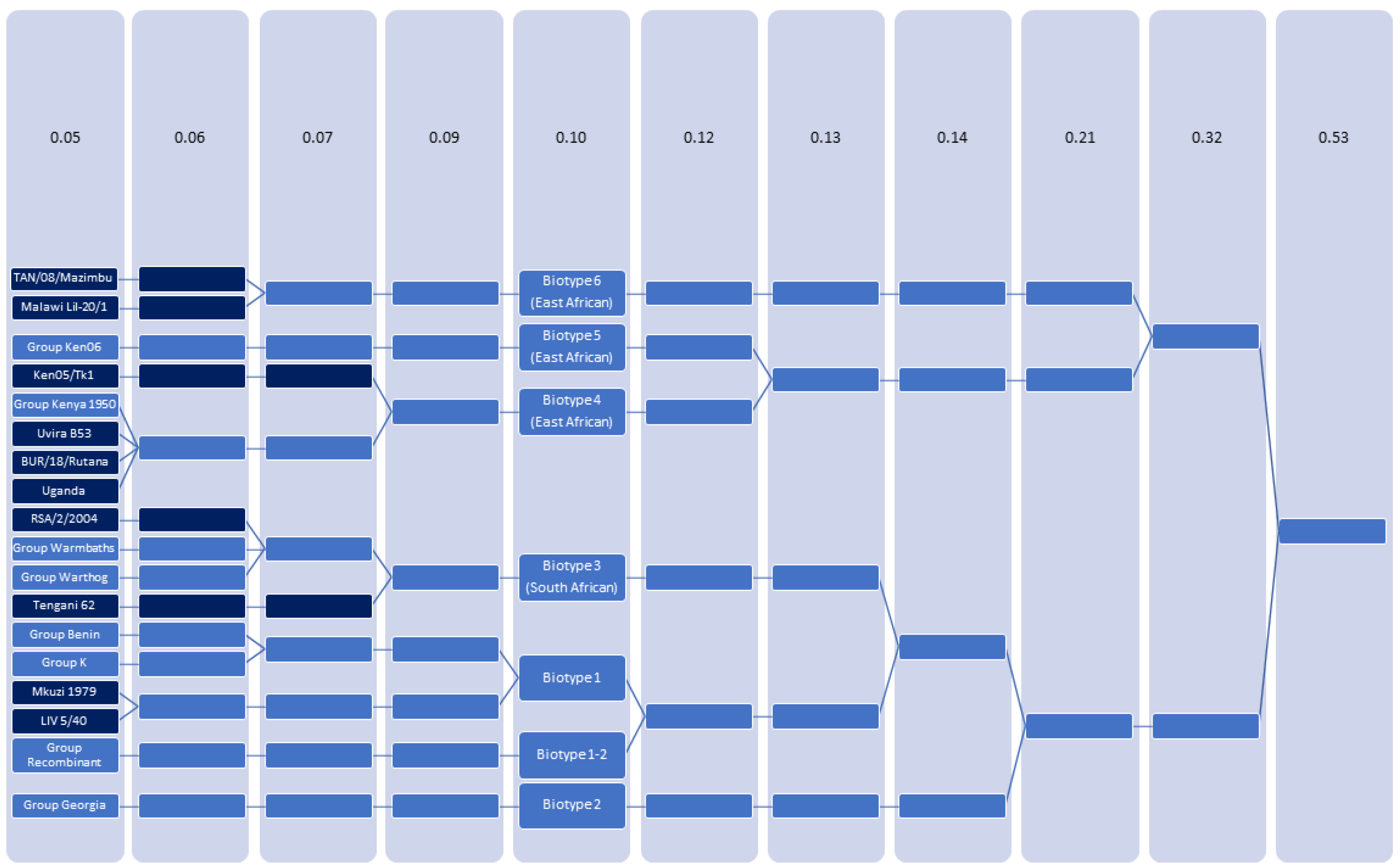
3.2. Clustering of ASFV
3.3. ASFV Can Be Classified as 7 Biotypes
3.4. Webportal for Automatic ASFV Biotyping and Genotyping
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Penrith, M.L.; Kivaria, F.M.; Masembe, C. One hundred years of African swine fever: A tribute to R. Eustace Montgomery. Transbound. Emerg. Dis. 2021, 68, 2640–2642. [Google Scholar] [CrossRef] [PubMed]
- Chapman, D.A.; Darby, A.C.; Da Silva, M.; Upton, C.; Radford, A.D.; Dixon, L.K. Genomic analysis of highly virulent Georgia 2007/1 isolate of African swine fever virus. Emerg. Infect. Dis. 2011, 17, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, W.; Moreno, C.; Duran, U.; Henao, N.; Bencosme, M.; Lora, P.; Reyes, R.; Nunez, R.; De Gracia, A.; Perez, A.M. African swine fever in the Dominican Republic. Transbound. Emerg. Dis. 2021, 68, 3018–3019. [Google Scholar] [CrossRef] [PubMed]
- Flach, B. United States Department of Agriculture (2023). First Case of African Swine Fever Found in Wild Boars in Sweden. Sweden. 2023. Available online: https://apps.fas.usda.gov/newgainapi/api/Report/DownloadReportByFileName?fileName=First%20Case%20of%20African%20Swine%20Fever%20Found%20in%20Wild%20Boars%20in%20Sweden%20_The%20Hague_Sweden_SW2023-0002.pdf (accessed on 1 December 2023).
- Zhao, D.; Sun, E.; Huang, L.; Ding, L.; Zhu, Y.; Zhang, J.; Shen, D.; Zhang, X.; Zhang, Z.; Ren, T.; et al. Highly lethal genotype I and II recombinant African swine fever viruses detected in pigs. Nat. Commun. 2023, 14, 3096. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Medina, E.; O’Donnell, V.; Silva, E.; Espinoza, N.; Velazquez-Salinas, L.; Moran, K.; Daite, D.A.; Barrette, R.; Faburay, B.; Holland, R.; et al. Experimental Infection of Domestic Pigs with an African Swine Fever Virus Field Strain Isolated in 2021 from the Dominican Republic. Viruses 2022, 14, 1090. [Google Scholar] [CrossRef] [PubMed]
- Adeola, A.C.; Luka, P.D.; Jiang, X.X.; Cai, Z.F.; Oluwole, O.O.; Shi, X.; Oladele, B.M.; Olorungbounmi, T.O.; Boladuro, B.; Omotosho, O.; et al. Target capture sequencing for the first Nigerian genotype I ASFV genome. Microb. Genom. 2023, 9, 001069. [Google Scholar] [CrossRef] [PubMed]
- Zani, L.; Forth, J.H.; Forth, L.; Nurmoja, I.; Leidenberger, S.; Henke, J.; Carlson, J.; Breidenstein, C.; Viltrop, A.; Hoper, D.; et al. Deletion at the 5’-end of Estonian ASFV strains associated with an attenuated phenotype. Sci. Rep. 2018, 8, 6510. [Google Scholar] [CrossRef]
- Spinard, E.; Rai, A.; Osei-Bonsu, J.; O’Donnell, V.; Ababio, P.T.; Tawiah-Yingar, D.; Arthur, D.; Baah, D.; Ramirez-Medina, E.; Espinoza, N.; et al. The 2022 Outbreaks of African Swine Fever Virus Demonstrate the First Report of Genotype II in Ghana. Viruses 2023, 15, 1722. [Google Scholar] [CrossRef]
- Ambagala, A.; Goonewardene, K.; Lamboo, L.; Goolia, M.; Erdelyan, C.; Fisher, M.; Handel, K.; Lung, O.; Blome, S.; King, J.; et al. Characterization of a Novel African Swine Fever Virus p72 Genotype II from Nigeria. Viruses 2023, 15, 915. [Google Scholar] [CrossRef]
- Okwasiimire, R.; Flint, J.F.; Kayaga, E.B.; Lakin, S.; Pierce, J.; Barrette, R.W.; Faburay, B.; Ndoboli, D.; Ekakoro, J.E.; Wampande, E.M.; et al. Whole Genome Sequencing Shows that African Swine Fever Virus Genotype IX Is Still Circulating in Domestic Pigs in All Regions of Uganda. Pathogens 2023, 12, 912. [Google Scholar] [CrossRef]
- Bisimwa, P.N.; Ongus, J.R.; Tiambo, C.K.; Machuka, E.M.; Bisimwa, E.B.; Steinaa, L.; Pelle, R. First detection of African swine fever (ASF) virus genotype X and serogroup 7 in symptomatic pigs in the Democratic Republic of Congo. Virol. J. 2020, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Achenbach, J.E.; Gallardo, C.; Nieto-Pelegrin, E.; Rivera-Arroyo, B.; Degefa-Negi, T.; Arias, M.; Jenberie, S.; Mulisa, D.D.; Gizaw, D.; Gelaye, E.; et al. Identification of a New Genotype of African Swine Fever Virus in Domestic Pigs from Ethiopia. Transbound. Emerg. Dis. 2017, 64, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Spinard, E.; Dinhobl, M.; Tesler, N.; Birtley, H.; Signore, A.V.; Ambagala, A.; Masembe, C.; Borca, M.V.; Gladue, D.P. A Re-Evaluation of African Swine Fever Genotypes Based on p72 Sequences Reveals the Existence of Only Six Distinct p72 Groups. Viruses 2023, 15, 2246. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Ge, S.; Zhang, Y.; Wu, X.; Wang, Z. A systematic review of genotypes and serogroups of African swine fever virus. Virus Genes 2022, 58, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Wesley, R.D.; Tuthill, A.E. Genome Relatedness among African Swine Fever Virus Field Isolates by Restriction Endonuclease Analysis. Prev. Vet. Med. 1984, 2, 53–62. [Google Scholar] [CrossRef]
- Blasco, R.; Aguero, M.; Almendral, J.M.; Vinuela, E. Variable and Constant Regions in African Swine Fever Virus-DNA. Virology 1989, 168, 330–338. [Google Scholar] [CrossRef] [PubMed]
- de Villiers, E.P.; Gallardo, C.; Arias, M.; da Silva, M.; Upton, C.; Martin, R.; Bishop, R.P. Phylogenomic analysis of 11 complete African swine fever virus genome sequences. Virology 2010, 400, 128–136. [Google Scholar] [CrossRef]
- Bao, J.; Zhang, Y.; Shi, C.; Wang, Q.; Wang, S.; Wu, X.; Cao, S.; Xu, F.; Wang, Z. Genome-Wide Diversity Analysis of African Swine Fever Virus Based on a Curated Dataset. Animals 2022, 12, 2446. [Google Scholar] [CrossRef]
- Aslanyan, L.; Avagyan, H.; Karalyan, Z. Whole-genome-based phylogeny of African swine fever virus. Vet. World 2020, 13, 2118–2125. [Google Scholar] [CrossRef]
- Shen, Z.J.; Jia, H.; Xie, C.D.; Shagainar, J.; Feng, Z.; Zhang, X.; Li, K.; Zhou, R. Bayesian Phylodynamic Analysis Reveals the Dispersal Patterns of African Swine Fever Virus. Viruses 2022, 14, 889. [Google Scholar] [CrossRef]
- Forth, J.H.; Forth, L.F.; King, J.; Groza, O.; Hubner, A.; Olesen, A.S.; Hoper, D.; Dixon, L.K.; Netherton, C.L.; Rasmussen, T.B.; et al. A Deep-Sequencing Workflow for the Fast and Efficient Generation of High-Quality African Swine Fever Virus Whole-Genome Sequences. Viruses 2019, 11, 846. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Zhang, X.; Xiong, J.; Yu, J.; Wei, H. Rapid genome-wide sequence typing of African swine fever virus based on alleles. Virus Res. 2021, 297, 198357. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Machado, M.P.; Silva, D.N.; Rossi, M.; Moran-Gilad, J.; Santos, S.; Ramirez, M.; Carrico, J.A. chewBBACA: A complete suite for gene-by-gene schema creation and strain identification. Microb. Genom. 2018, 4, e000166. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.G.; Ho, T.K. Machine Learning Made Easy: A Review of Scikit-learn Package in Python Programming Language. J. Educ. Behav. Stat. 2019, 44, 348–361. [Google Scholar] [CrossRef]
- Sander, J.; Ester, M.; Kriegel, H.P.; Xu, X.W. Density-based clustering in spatial databases: The algorithm GDBSCAN and its applications. Data Min. Knowl. Discov. 1998, 2, 169–194. [Google Scholar] [CrossRef]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nunez, D.; Castillo-Rosa, E.; Vigara-Astillero, G.; Garcia-Belmonte, R.; Gallardo, C.; Revilla, Y. Identification and Isolation of Two Different Subpopulations Within African Swine Fever Virus Arm/07 Stock. Vaccines 2020, 8, 625. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Martin Ester, H.-P.K.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clustersin Large Spatial Databases with Noise. In Proceedings of the 2nd International Conference on Knowledge Discovery and Data Mining, Portland, OR, USA, 2–4 August 1996. [Google Scholar]
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.D.; Penrith, M.L.; Cruciere, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; Thomson, G.R. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.K.; Xu, Z.Y.; Gao, H.; Xu, S.J.; Liu, J.; Xing, J.B.; Kuang, Q.Y.; Chen, Y.; Wang, H.; Zhang, G.H. Detection of a Novel African Swine Fever Virus with Three Large-Fragment Deletions in Genome, China. Microbiol. Spectr. 2022, 10, e0215522. [Google Scholar] [CrossRef]
- Forth, J.H.; Forth, L.F.; Lycett, S.; Bell-Sakyi, L.; Keil, G.M.; Blome, S.; Calvignac-Spencer, S.; Wissgott, A.; Krause, J.; Hoper, D.; et al. Identification of African swine fever virus-like elements in the soft tick genome provides insights into the virus’ evolution. BMC Biol. 2020, 18, 136. [Google Scholar] [CrossRef]
- Quembo, C.J.; Jori, F.; Vosloo, W.; Heath, L. Genetic characterization of African swine fever virus isolates from soft ticks at the wildlife/domestic interface in Mozambique and identification of a novel genotype. Transbound. Emerg. Dis. 2018, 65, 420–431. [Google Scholar] [CrossRef]
- Masembe, C.; Phan, M.V.T.; Robertson, D.L.; Cotten, M. Increased resolution of African swine fever virus genome patterns based on profile HMMs of protein domains. Virus Evol. 2020, 6, veaa044. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Ɛ | Example Isolate | Weighted Similarity | p72 Similarity |
|---|---|---|---|
| 0.05 | NHV (KM262845) | 0.99313 (412) | 1.00000 (0) |
| 0.07 | K49 (MZ202520) | 0.98256 (1046) | 1.00000 (0) |
| 0.14 | Warmbaths (AY261365) | 0.96428 (2143) | 0.99536 (3) |
| 0.21 | ASFV Georgia 2007/1 (FR682468) | 0.95485 (2709) | 0.99536 (3) |
| 0.53 | Malawi Lil-20/1 (AY261361) | 0.92482 (4511) | 0.98297 (11) |
| Group | Biotype | Updated Genotype 1 | Historic Genotype 2 | Clade 2 |
|---|---|---|---|---|
| Group Benin | 1 | 1 | I | Alpha |
| Mkuzi 1979 (AY261362) | 1 | 1 | I/VII 3 | Alpha |
| LIV 5/40 (MN318203) | 1 | 1 | I/VII | Epsilon 4 |
| Group K | 1 | 1 | I | N/A |
| Group Recombinant | 1–2 R | 2 | II | N/A |
| Group Georgia | 2 | 2 | II | Beta |
| RSA/2/2004 (MN641877) | 3 | 2 | XX | N/A |
| Group Warmbaths | 3 | 2 | III | Epsilon |
| Group Warthog | 3 | 2 | IV | Epsilon |
| Tengani 62 (NC_044951) | 3 | 2 | V | Delta |
| Ken05/Tk1 (KM111294) | 4 | 9 | X 5 | Gamma |
| Uvira B53 (MT956648) | 4 | 9 | X | N/A |
| BUR/18/Rutana (MW856067) | 4 | 9 | X | N/A |
| Uganda (UnPub002) | 4 | 9 | X | N/A |
| Group Kenya 1950 | 4 | 9 | X | Gamma |
| Group Kenya 06 | 5 | 9 | IX | Gamma |
| Malawi Lil-20/1 (1983) (NC_044954) | 6 | 8 | VIII | Gamma |
| TAN/08/Mazimbu (ON409981) | 6 | 15 | XV | N/A |
| Biotype | Isolate |
|---|---|
| 1 | 15998, 22649, 23221, 30322, 31208, 34403, 44076, 46830, 47039, 51268, 53706, 56140, 74377, 98039, 103917/18, 139/Nu/1981, 140/Or/1985, 141/Nu/1990, 142/Nu/1995, 19155_WB, 2019 WB, 22653/Ca/2014, 22943_2008, 26/Ss/2004, 26544/OG10, 28784WB, 33262WB, 33747 WB, 47/Ss/2008, 49179 WB, 4996 WB, 55234/18, 56/Ca/1978, 57/Ca/1979, 60/Nu/1997, 63525 WB, 7212WB, 72398 WB, 72407/Ss/2005, 72912 WB, 85/Ca/1985, 97/Ot/2012, BA71, BA71V, Benin 97/1, Ca1978_2, DR-1980, E75, Ghana2021-95, K49, KK262, L60, LIV/5/40, LO2018 major, LO2018 minor, Mkuzi 1979, NHV, Nu1979, Nu1986, Nu1990_1, Nu1991_2, Nu1991_3, Nu1991_7, Nu1993_2, Nu1995_3, Or_1984, Or1993_1, OURT 88/3, Pig/HeN/ZZ-P1/2021, Pig/SD/DY-I/2021 |
| 1–2 Recombinant | Pig/Henan/123014/2022, Pig/Inner Mongolia/DQDM/2022, Pig/Jiangsu/LG/2021 |
| 2 | 2020ASP01832, 2020ASP02103, 2020ASP02805, 2020ASP02894, 2021ASP00484, 2021ASP00703, 2021ASP00902, 2021ASP00921, 2021ASP01917, 2021ASP01919, 2021ASP01957, 2021ASP02148, 2021ASP02207, 2021ASP02665, 2021ASP03144, 2021ASP03251, 2021ASP03380, 2021ASP03384, 2021ASP03643, 2021ASP03658, 2021ASP03711, 2021ASP03740, 20355/RM/2022_Italy, 2802/AL/2022 Italy, A4, ABTCVSCK_ASF001, ABTCVSCK_ASF007, AQS-C-1-21, AQS-C-1-22, AQS-P-201202, AQS-P-20901-1, Arm/07/CBM/c2, ASF-MNG19, ASFV Belgium 2018/1, ASFV CzechRepublic 2017/1, ASFV Georgia 2007/1, ASFV Germany 2020/1, ASFV Korea/pig/Yeoncheon1/2019, ASFV Wuhan 2019-1, ASFV/Amur 19/WB-6905, ASFV/ARRIAH/CV-1/30, ASFV/ARRIAH/CV-1/50, ASFV/Kabardino-Balkaria 19/WB-964, ASFV/Kaliningrad_17/WB-13869, ASFV/Kaliningrad_18/WB-12516, ASFV/Kaliningrad_18/WB-12523, ASFV/Kaliningrad_18/WB-12524, ASFV/Kaliningrad_18/WB-9734, ASFV/Kaliningrad_18/WB-9735, ASFV/Kaliningrad_19/WB-10168, ASFV/Korea/pig/PaJu1/2019, ASFV/LT14/1490, ASFV/pig/China/CAS19-01/2019, ASFV/POL/2015/Podlaskie, ASFV/Primorsky 19/WB-6723, ASFV/Primorsky_19/DP-8235, ASFV/Timor-Leste/2019/1, ASFV/Ulyanovsk 19/WB-5699, ASFV/Zabaykali/WB-5314/2020, ASFV/Zabaykali_20/DP-4905, ASFV_Hanoi_2019, ASFV_NgheAn_2019, ASFV-SY18, ASFV-wbBS01, ASFV-wbShX01, Belgium/Etalle/wb/2018, CADC_HN09, China/GD/2019, China/LN/2018/1, CN/2019/InnerMongolia-AES01, Estonia 2014, Ghana2022-35, GZ201801, GZ201801_2, HB03A, HB31A, HuB20, IND/AR/SD-61/2020, IND/AS/SD-02/2020, JX21, Kashino 04/13, Korea/HC224/2020, Korea/YC1/2019, LYG18, MAL/19/Karonga, Nigeria-RV502, Odintsovo_02/14, OP823268, OP823269, Pig/Heilongjiang/HRB1/2020, Pig/HLJ/2018, Pol16_20186_o7, Pol16_20538_o9, Pol16_20540_o10, Pol16_29413_o23, Pol17_03029_C201, Pol17_04461_C210, Pol17_05838_C220, Pol17_31177_O81, Pol17_55892_C754, Pol18_28298_O111, Pol19_53050_C1959/19, SY-1, SY-2, TAN/17/Kibaha, TAN/17/Mbagala, TAN/20/Morogoro, Tanzania/Rukwa/2017/1, Vietnam/Pig/RG-1/01, Vietnam/Pig/RG-2/01, Vietnam/Pig/RG-3/01, Vietnam/Pig/RG-4/01, Vietnam/Pig/RG-5/01, Vietnam/Pig/RG-6/01, Vietnam/Pig/RG-7/01, VN/HY-ASFV1(2019), VN/QP-ASFV1(2019), VNUA-ASFV-05L1/HaNam/VN/2020, VNUA-LAVL2, wild boar/SNJ/2020, Yangzhou, YNFN202103, |
| 3 | Pretoriuskop/96/4, RSA/2/2004, RSA/2/2008, RSA/W1/1999, SPEC_57, Tengani 62, Warmbaths, Warthog, Zaire |
| 4 | ASFV Ken.rie1, BUR/18/Rutana, Ken05/Tk1, Kenya 1950, Uganda, Uvira B53, |
| 5 | Ken06.Bus, Ken1033, N10, R25, R35, R7, R8, TAN/16/Magu, |
| 6 | Malawi Lil-20/1, TAN/08/Mazimbu |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dinhobl, M.; Spinard, E.; Tesler, N.; Birtley, H.; Signore, A.; Ambagala, A.; Masembe, C.; Borca, M.V.; Gladue, D.P. Reclassification of ASFV into 7 Biotypes Using Unsupervised Machine Learning. Viruses 2024, 16, 67. https://doi.org/10.3390/v16010067
Dinhobl M, Spinard E, Tesler N, Birtley H, Signore A, Ambagala A, Masembe C, Borca MV, Gladue DP. Reclassification of ASFV into 7 Biotypes Using Unsupervised Machine Learning. Viruses. 2024; 16(1):67. https://doi.org/10.3390/v16010067
Chicago/Turabian StyleDinhobl, Mark, Edward Spinard, Nicolas Tesler, Hillary Birtley, Anthony Signore, Aruna Ambagala, Charles Masembe, Manuel V. Borca, and Douglas P. Gladue. 2024. "Reclassification of ASFV into 7 Biotypes Using Unsupervised Machine Learning" Viruses 16, no. 1: 67. https://doi.org/10.3390/v16010067