
High Prevalence, Genetic Diversity, and Recombination of Porcine Sapelovirus in Pig Farms in Fujian, Southern China
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples, Strain, and Materials
2.2. Primer Design and Synthesis
2.3. Preparation of Standard Plasmid
2.4. Establishment of qPCR Method and Standard Curve
2.5. Specificity, Sensitivity, and Repeatability Test
2.6. Full-Length Genome Amplification, Sequencing, and Annotation of PSV
2.7. PSV2020 Genome Homology, Genetic Evolution, and Recombination Analysis
3. Results
3.1. Standard Curve
3.2. Specificity, Sensitivity, and Repeatability of PCR Assay
3.3. The Prevalence of PSV in Pig Farms in Fujian Province
3.4. Whole-Genome Amplification, Sequencing, and Annotation of PSV2020
3.5. Phylogenetic Analyses of Complete ORF, VP1, and 3D Gene
3.6. Homology Analyses of Complete ORF
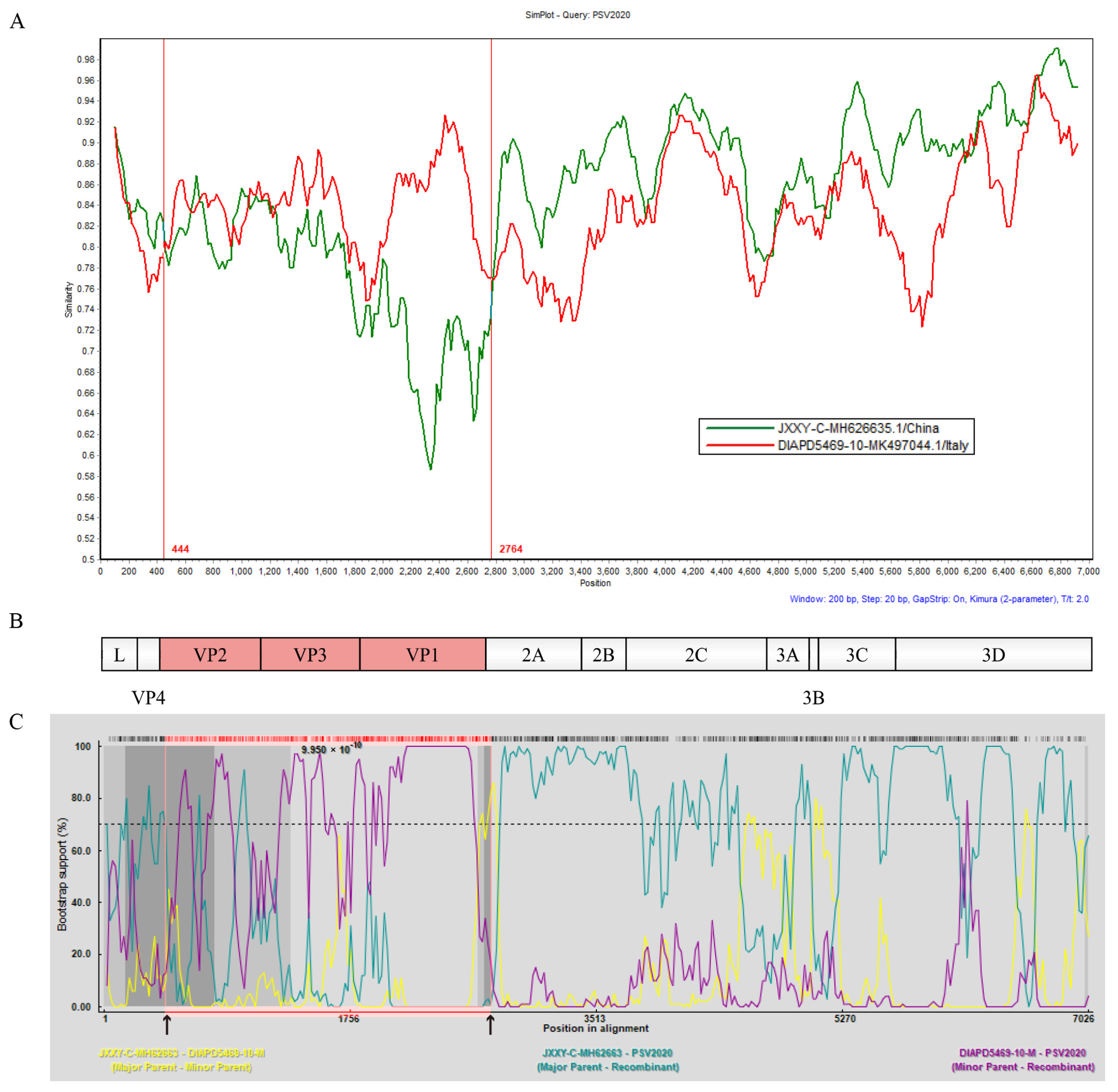
3.7. Recombination Analyses of PSV2020
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef] [PubMed]
- Lamont, P.H.; Betts, A.O. Studies on Enteroviruses of the Pig—IV: The Isolation in Tissue Culture of a Possible Enteric Cytopathogenic Swine Orphan (ECSO) Virus (V 13) from the Faeces of a Pig. Res. Vet. Sci. 1960, 1, 152–161. [Google Scholar] [CrossRef]
- Harima, H.; Kajihara, M.; Simulundu, E.; Bwalya, E.; Qiu, Y.; Isono, M.; Okuya, K.; Gonzalez, G.; Yamagishi, J.; Hang’ombe, B.M.; et al. Genetic and Biological Diversity of Porcine Sapeloviruses Prevailing in Zambia. Viruses 2020, 12, 180. [Google Scholar] [CrossRef] [PubMed]
- Sunaga, F.; Masuda, T.; Ito, M.; Akagami, M.; Naoi, Y.; Sano, K.; Katayama, Y.; Omatsu, T.; Oba, M.; Sakaguchi, S.; et al. Complete genomic analysis and molecular characterization of Japanese porcine sapeloviruses. Virus Genes 2019, 55, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Yu, X.; Yan, M.; Luo, B.; Li, R.; Qu, T.; Luo, Z.; Ge, M.; Zhao, D. Molecular characterization of Porcine sapelovirus in Hunan, China. J. Gen. Virol. 2017, 98, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Suo, X.; Cao, L.; Yuan, C.; Shi, L.; Duan, Y.; Zheng, H.; Wang, Q. Virome Analysis for Identification of a Novel Porcine Sapelovirus Isolated in Western China. Microbiol. Spectr. 2022, 10, e0180122. [Google Scholar] [CrossRef] [PubMed]
- Son, K.Y.; Kim, D.S.; Matthijnssens, J.; Kwon, H.J.; Park, J.G.; Hosmillo, M.; Alfajaro, M.M.; Ryu, E.H.; Kim, J.Y.; Kang, M.I.; et al. Molecular epidemiology of Korean porcine sapeloviruses. Arch. Virol. 2014, 159, 1175–1180. [Google Scholar] [CrossRef]
- Bak, G.Y.; Kang, M.I.; Son, K.Y.; Park, J.G.; Kim, D.S.; Seo, J.Y.; Kim, J.Y.; Alfajaro, M.M.; Soliman, M.; Baek, Y.B.; et al. Occurrence and molecular characterization of Sapelovirus A in diarrhea and non-diarrhea feces of different age group pigs in one Korean pig farm. J. Vet. Med. Sci. 2017, 78, 1911–1914. [Google Scholar] [CrossRef]
- Boros, Á.; László, Z.; Pankovics, P.; Marosi, A.; Albert, M.; Cságola, A.; Bíró, H.; Fahsbender, E.; Delwart, E.; Reuter, G. High prevalence, genetic diversity and a potentially novel genotype of Sapelovirus A (Picornaviridae) in enteric and respiratory samples in Hungarian swine farms. J. Gen. Virol. 2020, 101, 609–621. [Google Scholar] [CrossRef]
- Yang, T.; Lu, Y.; Zhang, L. Proposed genotype definition of Porcine sapelovirus. Pol. J. Vet. Sci. 2021, 24, 307–312. [Google Scholar] [CrossRef]
- Alexandersen, S.; Knowles, N.J.; Belsham, G.J.; Dekker, A.; Nfon, C.; Zhang, Z.; Koenen, F. Picornaviruses. In Diseases of Swine, 11th ed.; Zimmerman, J.J., Karriker, L.A., Ramirez, A., Schwartz, K.J., Stevenson, G.W., Zhang, J., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2019; Chapter 40; p. 675. [Google Scholar]
- Huang, J.; Gentry, R.F.; Zarkower, A. Experimental infection of pregnant sows with porcine enteroviruses. Am. J. Vet. Res. 1980, 41, 469–473. [Google Scholar] [PubMed]
- Lan, D.; Ji, W.; Yang, S.; Cui, L.; Yang, Z.; Yuan, C.; Hua, X. Isolation and characterization of the first Chinese porcine sapelovirus strain. Arch. Virol. 2011, 156, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Schock, A.; Gurrala, R.; Fuller, H.; Foyle, L.; Dauber, M.; Martelli, F.; Scholes, S.; Roberts, L.; Steinbach, F.; Dastjerdi, A. Investigation into an outbreak of encephalomyelitis caused by a neuroinvasive porcine sapelovirus in the United Kingdom. Vet. Microbiol. 2014, 172, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Kang, M.I.; Son, K.Y.; Bak, G.Y.; Park, J.G.; Hosmillo, M.; Seo, J.Y.; Kim, J.Y.; Alfajaro, M.M.; Soliman, M.; et al. Pathogenesis of Korean SapelovirusA in piglets and chicks. J. Gen. Virol. 2016, 97, 2566–2574. [Google Scholar] [CrossRef] [PubMed]
- Arruda, P.H.; Arruda, B.L.; Schwartz, K.J.; Vannucci, F.; Resende, T.; Rovira, A.; Sundberg, P.; Nietfeld, J.; Hause, B.M. Detection of a novel sapelovirus in central nervous tissue of pigs with polioencephalomyelitis in the USA. Transbound. Emerg. Dis. 2017, 64, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Liu, J.; Fang, L.; Kataoka, M.; Takeda, N.; Wakita, T.; Li, T.C. Characterization of porcine sapelovirus isolated from Japanese swine with PLC/PRF/5 cells. Transbound. Emerg. Dis. 2018, 65, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Sozzi, E.; Barbieri, I.; Lavazza, A.; Lelli, D.; Moreno, A.; Canelli, E.; Bugnetti, M.; Cordioli, P. Molecular characterization and phylogenetic analysis of VP1 of porcine enteric picornaviruses isolates in Italy. Transbound. Emerg. Dis. 2010, 57, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Du, L.; Jin, T.; Cheng, Y.; Zhang, X.; Jiao, S.; Huang, T.; Zhang, Y.; Yan, Y.; Gu, J.; et al. Characterization and epidemiological survey of porcine sapelovirus in China. Vet. Microbiol. 2019, 232, 13–21. [Google Scholar] [CrossRef]
- Zhang, B.; Tang, C.; Yue, H.; Ren, Y.; Song, Z. Viral metagenomics analysis demonstrates the diversity of viral flora in piglet diarrhoeic faeces in China. J. Gen. Virol. 2014, 95, 1603–1611. [Google Scholar] [CrossRef]
- Lee, C.; Kim, J.; Shin, S.G.; Hwang, S. Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J. Biotechnol. 2006, 123, 273–280. [Google Scholar] [CrossRef]
- Piorkowski, G.; Capai, L.; Falchi, A.; Casabianca, F.; Maestrini, O.; Gallian, P.; Barthélémy, K.; Py, O.; Charrel, R.; de Lamballerie, X. First Identification and Genomic Characterization of a Porcine Sapelovirus from Corsica, France, 2017. Microbiol. Resour. Announc. 2018, 7, e01049-18. [Google Scholar] [CrossRef] [PubMed]
- Van Dung, N.; Anh, P.H.; Van Cuong, N.; Hoa, N.T.; Carrique-Mas, J.; Hien, V.B.; Campbell, J.; Baker, S.; Farrar, J.; Woolhouse, M.E.; et al. Prevalence, genetic diversity and recombination of species G enteroviruses infecting pigs in Vietnam. J. Gen. Virol. 2014, 95, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Donin, D.G.; Leme, R.d.A.; Alfieri, A.F.; Alberton, G.C.; Alfieri, A.A. Molecular survey of porcine teschovirus, porcine sapelovirus, and enterovirus G in captive wild boars (Sus scrofa scrofa) of Paraná state, Brazil. Pesqui. Veterinária Bras. 2015, 35, 403–408. [Google Scholar] [CrossRef]
- Prodělalová, J. The survey of porcine teschoviruses, sapeloviruses and enteroviruses B infecting domestic pigs and wild boars in the Czech Republic between 2005 and 2011. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2012, 12, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, F.; Zhou, Q.; Li, W.; Chen, Y.; Song, Y.; Zhang, X.; Xue, C.; Bi, Y.; Cao, Y. Development of a minor groove binder assay for real-time PCR detection of porcine Sapelovirus. J. Virol. Methods 2014, 198, 69–74. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, L.; Lu, Y.; Guo, M.; Zhang, Z.; Lin, A. Characterization of porcine sapelovirus prevalent in western Jiangxi, China. BMC Vet. Res. 2021, 17, 273. [Google Scholar] [CrossRef]
- Foo, D.G.; Alonso, S.; Phoon, M.C.; Ramachandran, N.P.; Chow, V.T.; Poh, C.L. Identification of neutralizing linear epitopes from the VP1 capsid protein of Enterovirus 71 using synthetic peptides. Virus Res. 2007, 125, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Horsington, J.J.; Gilkerson, J.R.; Hartley, C.A. Mapping B-cell epitopes in equine rhinitis B viruses and identification of a neutralising site in the VP1 C-terminus. Vet. Microbiol. 2012, 155, 128–136. [Google Scholar] [CrossRef]
- Mateu, M.G. Antibody recognition of picornaviruses and escape from neutralization: A structural view. Virus Res. 1995, 38, 1–24. [Google Scholar] [CrossRef]
- Zell, R.; Dauber, M.; Krumbholz, A.; Henke, A.; Birch-Hirschfeld, E.; Stelzner, A.; Prager, D.; Wurm, R. Porcine teschoviruses comprise at least eleven distinct serotypes: Molecular and evolutionary aspects. J. Virol. 2001, 75, 1620–1631. [Google Scholar] [CrossRef]
- Subramaniam, S.; Mohapatra, J.K.; Das, B.; Sharma, G.K.; Biswal, J.K.; Mahajan, S.; Misri, J.; Dash, B.B.; Pattnaik, B. Capsid coding region diversity of re-emerging lineage C foot-and-mouth disease virus serotype Asia1 from India. Arch. Virol. 2015, 160, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Krumbholz, A.; Dauber, M.; Henke, A.; Birch-Hirschfeld, E.; Knowles, N.J.; Stelzner, A.; Zell, R. Sequencing of porcine enterovirus groups II and III reveals unique features of both virus groups. J. Virol. 2002, 76, 5813–5821. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N. Recombination among picornaviruses. Rev. Med. Virol. 2010, 20, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Holmblat, B.; Jégouic, S.; Muslin, C.; Blondel, B.; Joffret, M.L.; Delpeyroux, F. Nonhomologous recombination between defective poliovirus and coxsackievirus genomes suggests a new model of genetic plasticity for picornaviruses. mBio 2014, 5, e01119-14. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Primer | Sequences (5′-3′) | Product Size (bp) |
|---|---|---|
| PSV-F | GATACACTTAAATGGCAGTAGCGT | 129 |
| PSV-R | CTCACTGTCTACTCTCCTGTAACCA | |
| PSV-taq | FAM-CAATTGTCGATAGCCAT-MGB |
| Plasmid | Concentration (Copies/µL) | Intra-Assay | Inter-Assay | ||
|---|---|---|---|---|---|
| CV (%) | CV (%) | ||||
| PSV-P | 4.67 × 102 | 31.53 ± 0.15 | 0.47 | 31.26 ± 0.26 | 0.83 |
| 4.67 ×104 | 25.23 ± 0.03 | 0.14 | 25.10 ± 0.19 | 0.76 | |
| 4.67 × 106 | 19.41 ± 0.07 | 0.36 | 19.47 ± 0.15 | 0.78 | |
| Age Group | No. of Farms | Farm Prevalence | No. of Samples | Sample Prevalence | , log10 Copies/mL) |
|---|---|---|---|---|---|
| Suckling pigs (0–28 days) | 25 | 60.0% (15/25) | 102 | 41.18% (42/102) | 6.99 ± 0.80 |
| Post-weaning pigs (29–60 days) | 28 | 35.71% (10/28) | 56 | 26.8% (15/56) | 7.05 ± 1.90 |
| Fattening pigs (61 days–25 weeks) | 18 | 22.22% (4/18) | 30 | 16.6% (5/30) | 6.03 ± 0.39 |
| Sow | 30 | 56.0% (14/30) | 72 | 25.0% (18/72) | 6.75 ± 0.85 |
| Total | 30 | 76.67% (23/30) | 260 | 30.8% (80/260) |
| Number | Gene | Location | Length |
|---|---|---|---|
| 1 | 5′UTR | 1–446 | 446 |
| 2 | L | 447–698 | 252 |
| 3 | VP4 | 699–857 | 159 |
| 4 | VP2 | 858–1571 | 714 |
| 5 | VP3 | 1572–2273 | 702 |
| 6 | VP1 | 2274–3164 | 891 |
| 7 | 2A | 3165–3842 | 678 |
| 8 | 2B | 3843–4157 | 315 |
| 9 | 2C | 4158–5153 | 996 |
| 10 | 3A | 5154–5453 | 300 |
| 11 | 3B | 5454–5519 | 66 |
| 12 | 3C | 5520–6065 | 546 |
| 13 | 3D | 6066–7454 | 1389 |
| 14 | 3′UTR | 7455–7550 | 96 |
| PSV2020 VS | PSV-1 | PSV-2 | Out Group | |||
|---|---|---|---|---|---|---|
| G1-Asia | G1-America | G2-Europe | G2-India | |||
| ORF nucleotides | 84.9–88.7% | 84.6–86.0% | 84.1–85.9% | 84.2–85.7% | 77.3% | 59.5% |
| ORF amino acids | 93.7–97.0% | 94.2–95.4% | 93.2–96.2% | 93.8–95.6% | 84.3% | 51.7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.-Y.; Sun, Z.-H.; Che, Y.-L.; Chen, R.-J.; Wu, X.-M.; Wu, R.-J.; Wang, L.-B.; Zhou, L.-J. High Prevalence, Genetic Diversity, and Recombination of Porcine Sapelovirus in Pig Farms in Fujian, Southern China. Viruses 2023, 15, 1751. https://doi.org/10.3390/v15081751
Chen Q-Y, Sun Z-H, Che Y-L, Chen R-J, Wu X-M, Wu R-J, Wang L-B, Zhou L-J. High Prevalence, Genetic Diversity, and Recombination of Porcine Sapelovirus in Pig Farms in Fujian, Southern China. Viruses. 2023; 15(8):1751. https://doi.org/10.3390/v15081751
Chicago/Turabian StyleChen, Qiu-Yong, Zhi-Hua Sun, Yong-Liang Che, Ru-Jing Chen, Xue-Min Wu, Ren-Jie Wu, Long-Bai Wang, and Lun-Jiang Zhou. 2023. "High Prevalence, Genetic Diversity, and Recombination of Porcine Sapelovirus in Pig Farms in Fujian, Southern China" Viruses 15, no. 8: 1751. https://doi.org/10.3390/v15081751