

Ecological, Genetic, and Phylogenetic Aspects of YFV 2017–2019 Spread in Rio de Janeiro State
 , , , , , and
, , , , , and 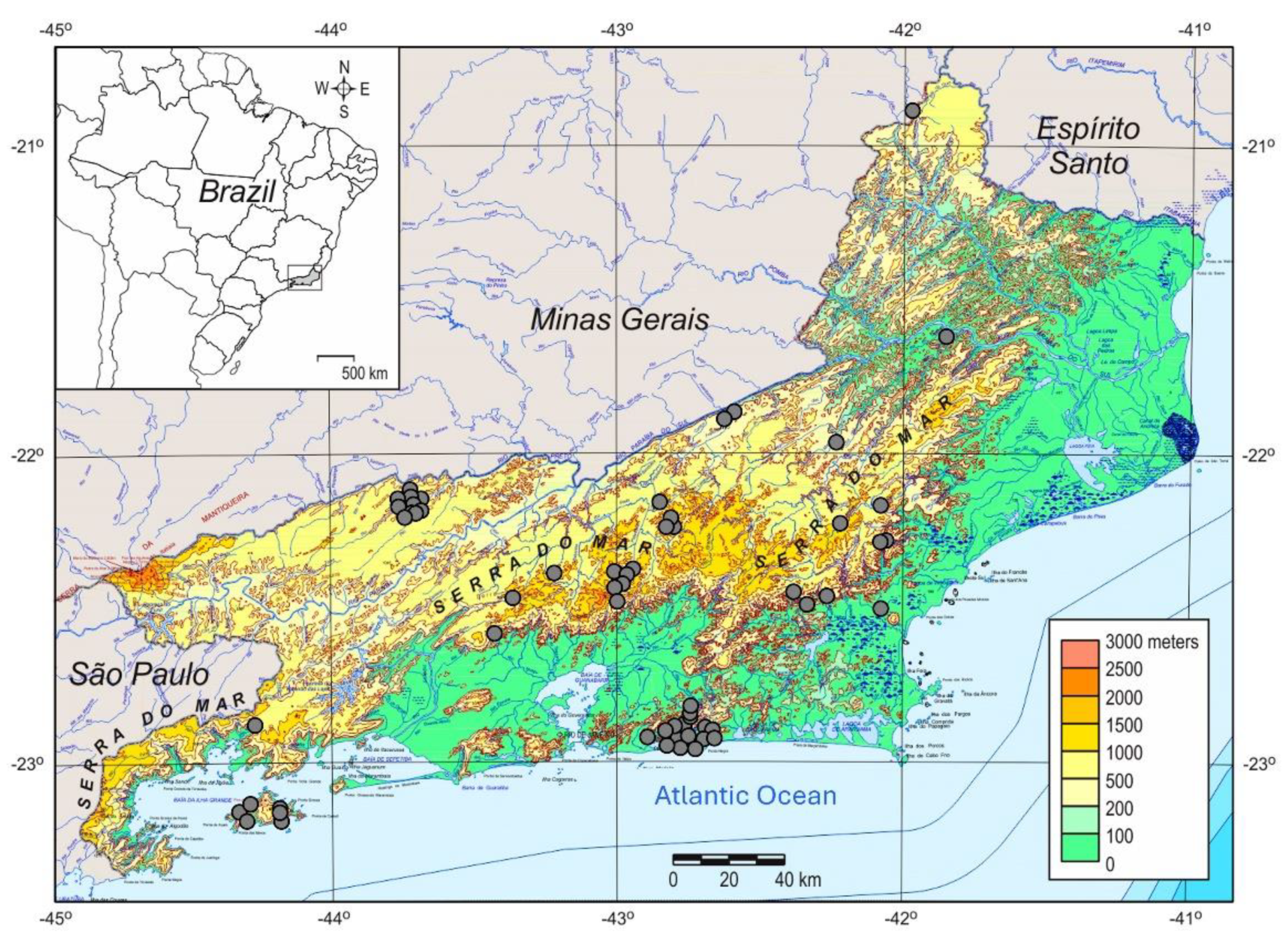{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethic Issues
2.2. Sampling Efforts, YFV Detection, and Sequencing Procedures
2.3. Evolutionary and Phylogeographic Analyses
3. Results
3.1. Spatial Patterns and YFV Isolates from the 2016–2019 Outbreak in RJ State
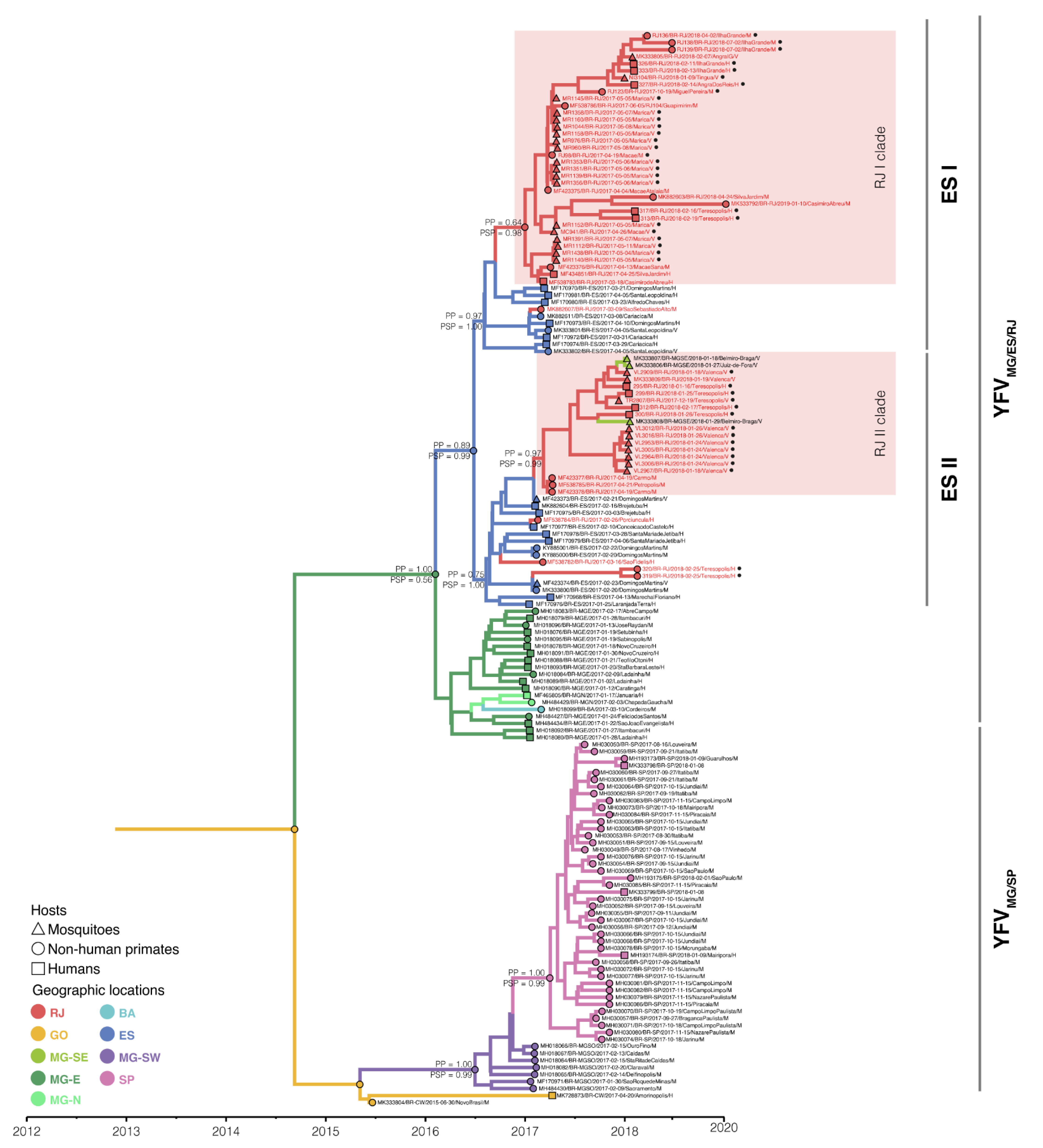
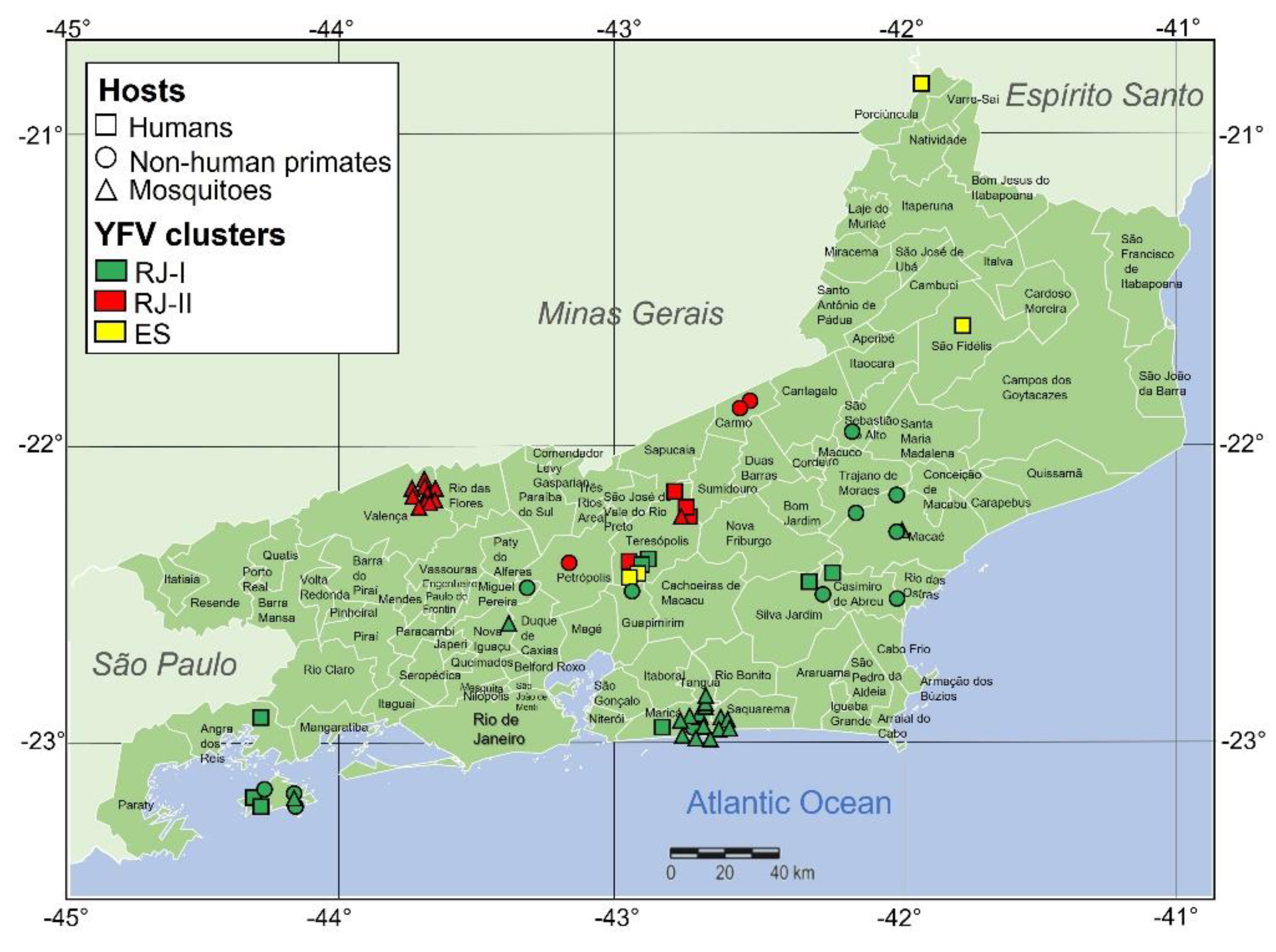
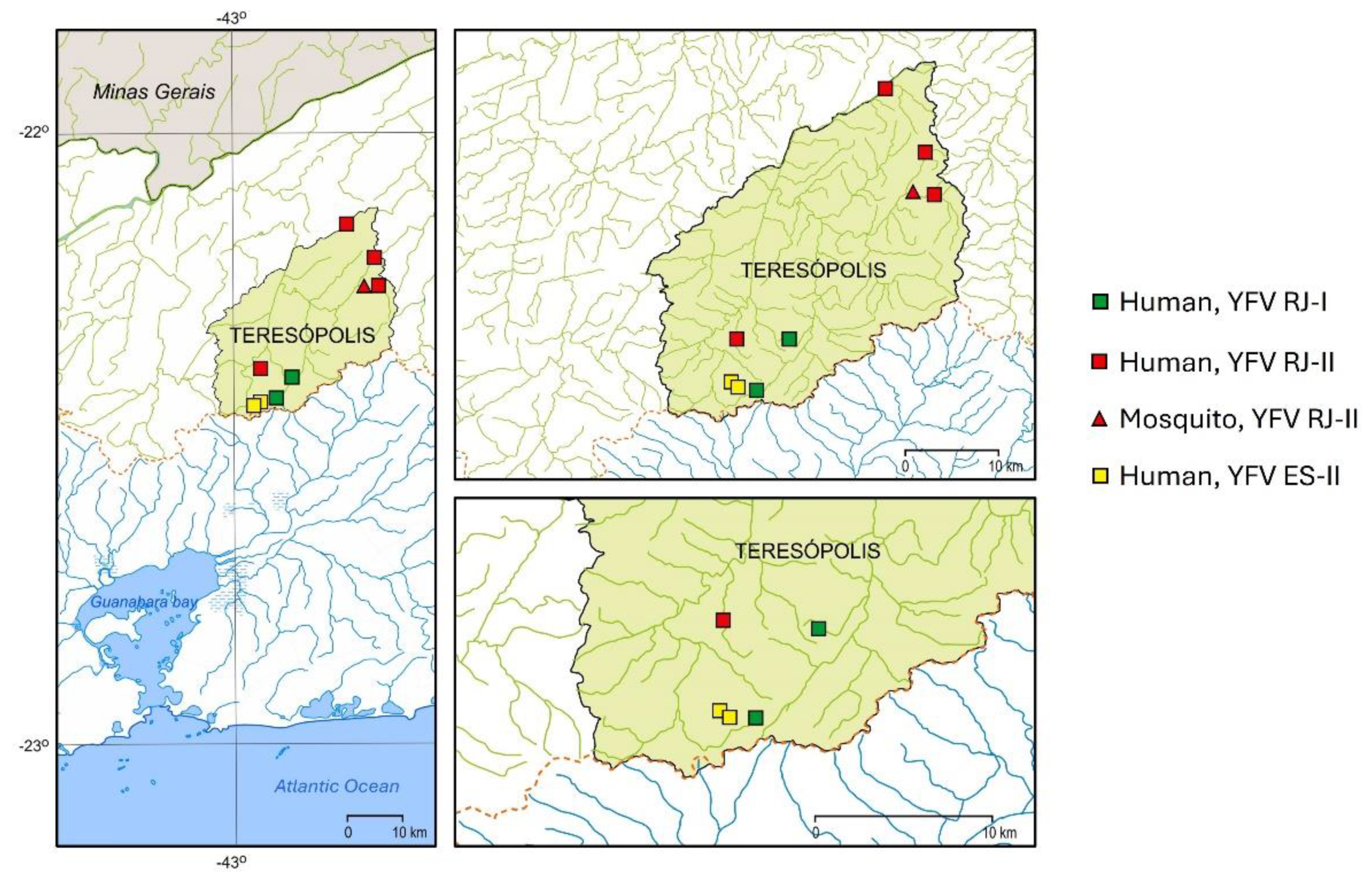
3.2. The Spatial Dissemination of the Sylvatic Transmission Chains YFV RJ-I and YFV RJ-II Clades
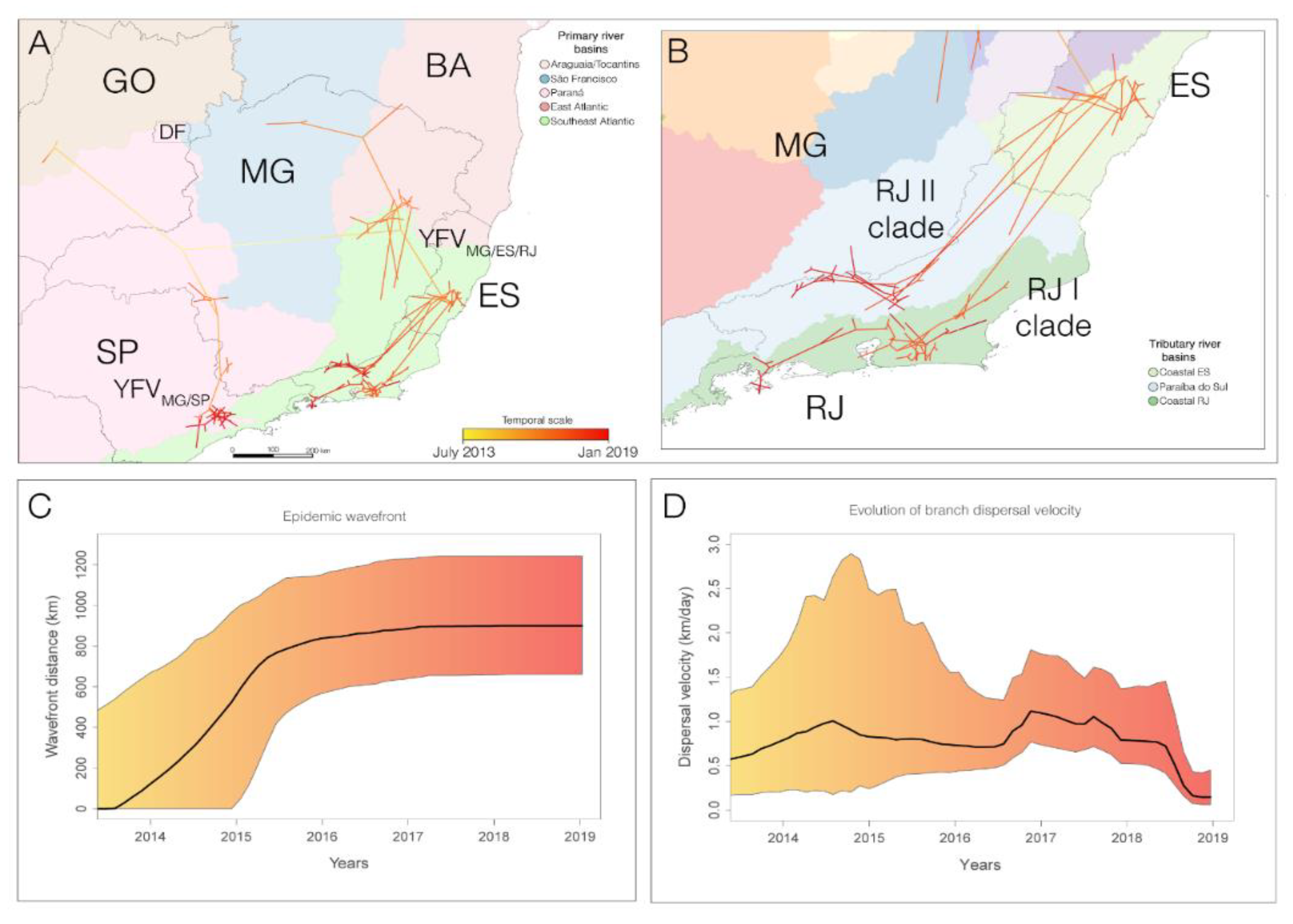
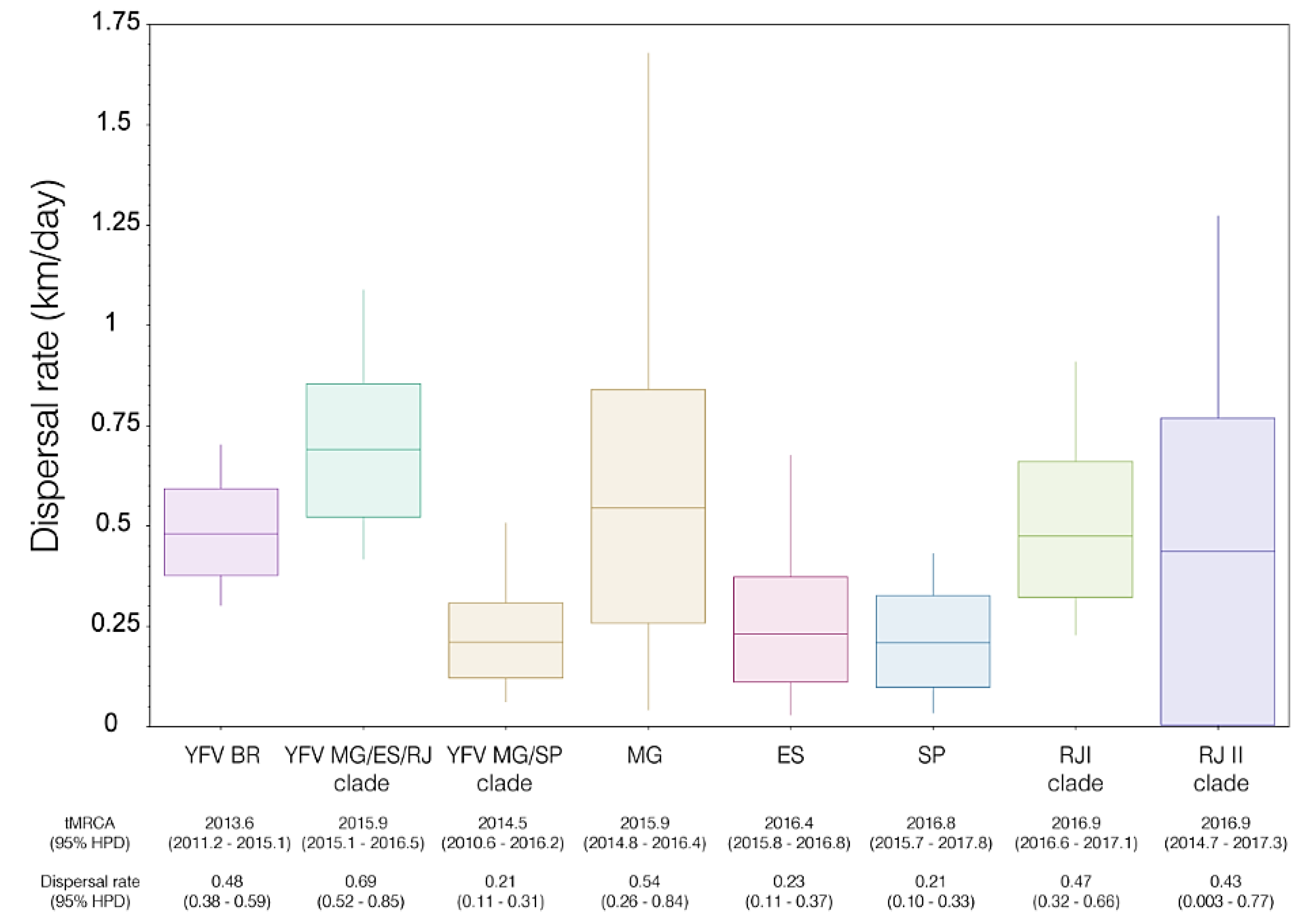
3.3. The YFV 2015–2019 Spread with Different Rates through the Southeast Region of Brazil but Not within the Rio de Janeiro State
3.4. Mapping the Genetic Diversity Raised in YFV 2016–2019 Outbreak in RJ State
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brazilian Ministry of Health. Reemergência e Manutenção Extra-Amazônica da Febre Amarela no Brasil, 2014 a 2019: Principais Desafios Para a Vigilância, a Prevenção e o Controle. Available online: https://www.saude.gov.br/images/pdf/2019/dezembro/05/Saude-Brasil-2019-imunizacao.pdf (accessed on 17 July 2022).
- Possas, C.; Lourenco-de-Oliveira, R.; Tauil, P.L.; Pinheiro, F.P.; Pissinatti, A.; Cunha, R.V.D.; Freire, M.; Martins, R.M.; Homma, A. Yellow fever outbreak in Brazil: The puzzle of rapid viral spread and challenges for immunisation. Mem. Inst. Oswaldo Cruz. 2018, 113, e180278. [Google Scholar] [CrossRef]
- Vasconcelos, P.F.; Costa, Z.G.; Travassos Da Rosa, E.S.; Luna, E.; Rodrigues, S.G.; Barros, V.L.; Dias, J.P.; Monteiro, H.A.; Oliva, O.F.; Vasconcelos, H.B.; et al. Epidemic of jungle yellow fever in Brazil, 2000: Implications of climatic alterations in disease spread. J. Med. Virol. 2001, 65, 598–604. [Google Scholar] [CrossRef]
- Romano, A.P.; Costa, Z.G.; Ramos, D.G.; Andrade, M.A.; Jayme Vde, S.; Almeida, M.A.; Vettorello, K.C.; Mascheretti, M.; Flannery, B. Yellow Fever outbreaks in unvaccinated populations, Brazil, 2008–2009. PLoS Negl. Trop. Dis. 2014, 8, e2740. [Google Scholar] [CrossRef] [PubMed]
- Cunha, M.D.P.; Duarte-Neto, A.N.; Pour, S.Z.; Ortiz-Baez, A.S.; Cerny, J.; Pereira, B.B.S.; Braconi, C.T.; Ho, Y.L.; Perondi, B.; Sztajnbok, J.; et al. Origin of the Sao Paulo Yellow Fever epidemic of 2017-2018 revealed through molecular epidemiological analysis of fatal cases. Sci. Rep. 2019, 9, 20418. [Google Scholar] [CrossRef]
- Cunha, M.D.P.; Duarte-Neto, A.N.; Pour, S.Z.; Pereira, B.B.S.; Ho, Y.L.; Perondi, B.; Sztajnbok, J.; Alves, V.A.F.; da Silva, L.F.F.; Dolhnikoff, M.; et al. Phylogeographic patterns of the yellow fever virus around the metropolitan region of Sao Paulo, Brazil, 2016–2019. PLoS Negl. Trop. Dis. 2022, 16, e0010705. [Google Scholar] [CrossRef]
- de Azevedo Fernandes, N.C.C.; Guerra, J.M.; Diaz-Delgado, J.; Cunha, M.S.; Saad, L.D.; Iglezias, S.D.; Ressio, R.A.; Dos Santos Cirqueira, C.; Kanamura, C.T.; Jesus, I.P.; et al. Differential Yellow Fever Susceptibility in New World Nonhuman Primates, Comparison with Humans, and Implications for Surveillance. Emerg. Infect. Dis. 2021, 27, 47–56. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Figueiredo, P.; Stoffella-Dutra, A.G.; Barbosa Costa, G.; Silva de Oliveira, J.; Dourado Amaral, C.; Duarte Santos, J.; Soares Rocha, K.L.; Araujo Junior, J.P.; Lacerda Nogueira, M.; Zaza Borges, M.A.; et al. Re-Emergence of Yellow Fever in Brazil during 2016-2019: Challenges, Lessons Learned, and Perspectives. Viruses 2020, 12, 1233. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.C.; de Souza, R.; Theze, J.; Claro, I.; Aguiar, R.S.; Abade, L.; Santos, F.C.P.; Cunha, M.S.; Nogueira, J.S.; Salles, F.C.S.; et al. Genomic Surveillance of Yellow Fever Virus Epizootic in Sao Paulo, Brazil, 2016–2018. PLoS Pathog 2020, 16, e1008699. [Google Scholar] [CrossRef]
- Mares-Guia, M.; Horta, M.A.; Romano, A.; Rodrigues, C.D.S.; Mendonca, M.C.L.; Dos Santos, C.C.; Torres, M.C.; Araujo, E.S.M.; Fabri, A.; de Souza, E.R.; et al. Yellow fever epizootics in non-human primates, Southeast and Northeast Brazil (2017 and 2018). Parasit. Vectors 2020, 13, 90. [Google Scholar] [CrossRef] [PubMed]
- Cupertino, M.C.; Garcia, R.; Gomes, A.P.; de Paula, S.O.; Mayers, N.; Siqueira-Batista, R. Epidemiological, prevention and control updates of yellow fever outbreak in Brazil. Asian Pac. J. Trop. Med. 2019, 12, 11. [Google Scholar] [CrossRef]
- Health, B.M.o. Health Brazil 2019: A Health Situation Analysis Focusing on Immunopreventable Diseases and Immunization. Available online: http://bvsms.saude.gov.br/bvs/publicacoes/saude_brasil_2020-2021_analise_pandemia_covid-19.pdf (accessed on 17 July 2022).
- Health, B.M.o. Boletim Epidemiológico 46. Available online: https://www.gov.br/saude/pt-br/media/pdf/2020/dezembro/09/boletim_epidemiologico_svs_46.pdf (accessed on 17 July 2022).
- Health, B.M.o. Boletim Epidemiológico 31. Available online: https://www.gov.br/saude/ptbr/centrais-de-conteudo/publicacoes/boletins/boletins-epidemiologicos/2021/boletim_epidemiologico_svs_31.pdf (accessed on 17 July 2022).
- Abreu, F.V.S.; Ribeiro, I.P.; Ferreira-de-Brito, A.; Santos, A.; Miranda, R.M.; Bonelly, I.S.; Neves, M.; Bersot, M.I.; Santos, T.P.D.; Gomes, M.Q.; et al. Haemagogus leucocelaenus and Haemagogus janthinomys are the primary vectors in the major yellow fever outbreak in Brazil, 2016–2018. Emerg. Microbes. Infect. 2019, 8, 218–231. [Google Scholar] [CrossRef]
- Stanzani, L.M.A.; Motta, M.A.; Erbisti, R.S.; Abreu, F.V.S.; Nascimento-Pereira, A.C.; Ferreira-de-Brito, A.; Neves, M.; Pereira, G.R.; Pereira, G.R.; Santos, C.B.D.; et al. Back to Where It Was First Described: Vectors of Sylvatic Yellow Fever Transmission in the 2017 Outbreak in Espirito Santo, Brazil. Viruses 2022, 14, 2805. [Google Scholar] [CrossRef]
- Barbosa, C.M.; Di Paola, N.; Cunha, M.P.; Rodrigues-Jesus, M.J.; Araujo, D.B.; Silveira, V.B.; Leal, F.B.; Mesquita, F.S.; Botosso, V.F.; Zanotto, P.M.A.; et al. Yellow Fever Virus RNA in Urine and Semen of Convalescent Patient, Brazil. Emerg. Infect. Dis. 2018, 24, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, M.C.; Gomez, M.M.; Dos Santos, A.A.; Abreu, F.V.S.; Ferreira-de-Brito, A.; Miranda, R.M.; Castro, M.G.; Lourenco-de-Oliveira, R. Genome analysis of yellow fever virus of the ongoing outbreak in Brazil reveals polymorphisms. Mem. Inst. Oswaldo Cruz. 2017, 112, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Delatorre, E.; de Abreu, F.V.S.; Ribeiro, I.P.; Gomez, M.M.; Dos Santos, A.A.C.; Ferreira-de-Brito, A.; Neves, M.; Bonelly, I.; de Miranda, R.M.; Furtado, N.D.; et al. Distinct YFV Lineages Co-circulated in the Central-Western and Southeastern Brazilian Regions From 2015 to 2018. Front. Microbiol. 2019, 10, 1079. [Google Scholar] [CrossRef]
- Gomez, M.M.; Abreu, F.V.S.; Santos, A.; Mello, I.S.; Santos, M.P.; Ribeiro, I.P.; Ferreira-de-Brito, A.; Miranda, R.M.; Castro, M.G.; Ribeiro, M.S.; et al. Genomic and structural features of the yellow fever virus from the 2016–2017 Brazilian outbreak. J. Gen. Virol. 2018, 99, 536–548. [Google Scholar] [CrossRef]
- Barrett, A.D.; Gould, E.A. Comparison of neurovirulence of different strains of yellow fever virus in mice. J. Gen. Virol. 1986, 67 Pt 4, 631–637. [Google Scholar] [CrossRef]
- Furtado, N.D.; Gomez, M.M.; de Mello, I.S.; Fernandes, D.R.; Bonaldo, M.C. Phenotypic and Genetic Studies of the Viral Lineage Associated with the Recent Yellow Fever Outbreak in Brazil. Viruses 2022, 14, 1818. [Google Scholar] [CrossRef]
- Furtado, N.D.; Raphael, L.M.; Ribeiro, I.P.; de Mello, I.S.; Fernandes, D.R.; Gomez, M.M.; Dos Santos, A.A.C.; Nogueira, M.D.S.; de Castro, M.G.; de Abreu, F.V.S.; et al. Biological Characterization of Yellow Fever Viruses Isolated From Non-human Primates in Brazil With Distinct Genomic Landscapes. Front. Microbiol. 2022, 13, 757084. [Google Scholar] [CrossRef] [PubMed]
- Noske, G.D.; Gawriljuk, V.O.; Fernandes, R.S.; Furtado, N.D.; Bonaldo, M.C.; Oliva, G.; Godoy, A.S. Structural characterization and polymorphism analysis of the NS2B-NS3 protease from the 2017 Brazilian circulating strain of Yellow Fever virus. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129521. [Google Scholar] [CrossRef]
- Franco, O. Campanhas de Oswaldo Cruz. In História da Febre Amarela no Brasil; Ministério da Saúde-Departamento Nacional de endemias rurais: Rio de Janeiro, Brazil, 1969; pp. 75–94. [Google Scholar]
- IBGE Cidades e Estados: Rio de Janeiro. Available online: https://www.ibge.gov.br/cidades-e-estados/rj.html (accessed on 14 May 2022).
- Abreu, F.V.S.; Delatorre, E.; Dos Santos, A.A.C.; Ferreira-de-Brito, A.; de Castro, M.G.; Ribeiro, I.P.; Furtado, N.D.; Vargas, W.P.; Ribeiro, M.S.; Meneguete, P.; et al. Combination of surveillance tools reveals that Yellow Fever virus can remain in the same Atlantic Forest area at least for three transmission seasons. Mem. Inst. Oswaldo Cruz. 2019, 114, e190076. [Google Scholar] [CrossRef] [PubMed]
- Abreu, F.V.S.; de Andreazzi, C.S.; Neves, M.; Meneguete, P.S.; Ribeiro, M.S.; Dias, C.M.G.; de Albuquerque Motta, M.; Barcellos, C.; Romao, A.R.; Magalhaes, M.; et al. Ecological and environmental factors affecting transmission of sylvatic yellow fever in the 2017-2019 outbreak in the Atlantic Forest, Brazil. Parasit. Vectors 2022, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Brito, T.T.; Oliveira-Júnior, J.F.; Lyra, G.B.; Gois, G.; Zeri, M. Multivariate analysis applied to monthly rainfall over Rio de Janeiro state, Brazil. Meteorol. Atmos. Phys. 2017, 129, 469–478. [Google Scholar] [CrossRef]
- Cysneiros, V.C.; Mendonça-Junior, J.O.; Gaui, T.D.; Braz, D.M. Diversity, community structure and conservation status of an Atlantic Forest fragment in Rio de Janeiro State, Brazil. Biota Neotrop. 2015, 15, 15. [Google Scholar] [CrossRef]
- Andrade, M.S.; Campos, F.S.; Campos, A.A.S.; Abreu, F.V.S.; Melo, F.L.; Seva, A.D.P.; Cardoso, J.D.C.; Dos Santos, E.; Born, L.C.; Silva, C.; et al. Real-Time Genomic Surveillance during the 2021 Re-Emergence of the Yellow Fever Virus in Rio Grande do Sul State, Brazil. Viruses 2021, 13, 1976. [Google Scholar] [CrossRef]
- Lacerda, A.B.; Del Castillo Saad, L.; Ikefuti, P.V.; Pinter, A.; Chiaravalloti-Neto, F. Diffusion of sylvatic yellow fever in the state of Sao Paulo, Brazil. Sci. Rep. 2021, 11, 16277. [Google Scholar] [CrossRef]
- Pereira Dos Santos, T.; Roiz, D.; Santos de Abreu, F.V.; Luz, S.L.B.; Santalucia, M.; Jiolle, D.; Santos Neves, M.S.A.; Simard, F.; Lourenco-de-Oliveira, R.; Paupy, C. Potential of Aedes albopictus as a bridge vector for enzootic pathogens at the urban-forest interface in Brazil. Emerg. Microbes. Infect. 2018, 7, 191. [Google Scholar] [CrossRef]
- de Abreu, F.V.S.; Dos Santos, E.; Gomes, M.Q.; Vargas, W.P.; de Oliveira Passos, P.H.; Nunes, E.S.C.; Araujo, P.C.; Pires, J.R.; Romano, A.P.M.; Teixeira, D.S.; et al. Capture of Alouatta guariba clamitans for the surveillance of sylvatic yellow fever and zoonotic malaria: Which is the best strategy in the tropical Atlantic Forest? Am. J. Primatol. 2019, 81, e23000. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Ayres, D.L.; Darling, A.; Zwickl, D.J.; Beerli, P.; Holder, M.T.; Lewis, P.O.; Huelsenbeck, J.P.; Ronquist, F.; Swofford, D.L.; Cummings, M.P.; et al. BEAGLE: An application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 2012, 61, 170–173. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Rambaut, A.; Suchard, M.A.; Lemey, P. SPREAD: Spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 2011, 27, 2910–2912. [Google Scholar] [CrossRef]
- IBGE. Portal de Mapas. Available online: https://mapas.ibge.gov.br/images/pdf/mapas/mappag99.pdf (accessed on 11 February 2021).
- ANA. Divisões Hidrográficas do Brasil. Available online: http://www3.ana.gov.br/portal/ANA/aguas-no-brasil/panorama-das-aguas/copy_of_divisoes-hidrograficas (accessed on 11 February 2021).
- Dellicour, S.; Rose, R.; Faria, N.R.; Lemey, P.; Pybus, O.G. SERAPHIM: Studying environmental rasters and phylogenetically informed movements. Bioinformatics 2016, 32, 3204–3206. [Google Scholar] [CrossRef]
- Giovanetti, M.; de Mendonca, M.C.L.; Fonseca, V.; Mares-Guia, M.A.; Fabri, A.; Xavier, J.; de Jesus, J.G.; Graf, T.; Dos Santos Rodrigues, C.D.; Dos Santos, C.C.; et al. Yellow Fever Virus Reemergence and Spread in Southeast Brazil, 2016–2019. J. Virol. 2019, 94, e01623-19. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, M.A.B.; Dos Santos, E.; Cardoso, J.D.C.; da Silva, L.G.; Rabelo, R.M.; Bicca-Marques, J.C. Predicting Yellow Fever Through Species Distribution Modeling of Virus, Vector, and Monkeys. Ecohealth 2019, 16, 95–108. [Google Scholar] [CrossRef]
- Prist, P.R.; Tambosi, L.R.; Mucci, L.F.; Pinter, A.; de Souza, R.P.; Muylaert, R.L.; Rhodes, J.R.; Comin, C.H.; Costa, L.F.; D’Agostini, T.L.; et al. Roads and forest edges facilitate yellow fever virus dispersion. J. Appl. Ecol. 2022, 59, 4–17. [Google Scholar] [CrossRef]
- Hill, S.C.; Dellicour, S.; Claro, I.M.; Sequeira, P.C.; Adelino, T.; Thézé, J.; Wu, C.H.; Moreira, F.R.R.; Giovanetti, M.; Li, S.L.; et al. Climate and land-use shape the spread of zoonotic yellow fever virus. MedRxiv 2022. [Google Scholar] [CrossRef]
- Ilacqua, R.C.; Medeiros-Sousa, A.R.; Ramos, D.G.; Obara, M.T.; Ceretti-Junior, W.; Mucci, L.F.; Marrelli, M.T.; Laporta, G.Z. Reemergence of Yellow Fever in Brazil: The Role of Distinct Landscape Fragmentation Thresholds. J. Environ. Public Health 2021, 2021, 8230789. [Google Scholar] [CrossRef]
- Wilk-da-Silva, R.; Medeiros-Sousa, A.R.; Laporta, G.Z.; Mucci, L.F.; Prist, P.R.; Marrelli, M.T. The influence of landscape structure on the dispersal pattern of yellow fever virus in the state of Sao Paulo. Acta Trop. 2022, 228, 106333. [Google Scholar] [CrossRef]
- Vasconcelos, P.F. Yellow fever in Brazil: Thoughts and hypotheses on the emergence in previously free areas. Rev. Saude Publica 2010, 44, 1144–1149. [Google Scholar] [CrossRef] [PubMed]
- Causey, O.R.; Kumm, H.W.; Laemmert, H.W., Jr. Dispersion of forest mosquitoes in Brazil; further studies. Am. J. Trop. Med. Hyg. 1950, 30, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Haddow, A.J.; Williams, M.C.; Woodall, J.P.; Simpson, D.I.; Goma, L.K. Twelve Isolations of Zika Virus from Aedes (Stegomyia) Africanus (Theobald) Taken in and above a Uganda Forest. Bull. World Health Organ. 1964, 31, 57–69. [Google Scholar] [PubMed]
- Carpio, K.L.; Barrett, A.D.T. Flavivirus NS1 and Its Potential in Vaccine Development. Vaccines 2021, 9, 622. [Google Scholar] [CrossRef]
- Muller, D.A.; Young, P.R. The flavivirus NS1 protein: Molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antivir. Res. 2013, 98, 192–208. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, I.P.; Delatorre, E.; de Abreu, F.V.S.; dos Santos, A.A.C.; Furtado, N.D.; Ferreira-de-Brito, A.; de Pina-Costa, A.; Neves, M.S.A.S.; de Castro, M.G.; Motta, M.d.A.; et al. Ecological, Genetic, and Phylogenetic Aspects of YFV 2017–2019 Spread in Rio de Janeiro State. Viruses 2023, 15, 437. https://doi.org/10.3390/v15020437
Ribeiro IP, Delatorre E, de Abreu FVS, dos Santos AAC, Furtado ND, Ferreira-de-Brito A, de Pina-Costa A, Neves MSAS, de Castro MG, Motta MdA, et al. Ecological, Genetic, and Phylogenetic Aspects of YFV 2017–2019 Spread in Rio de Janeiro State. Viruses. 2023; 15(2):437. https://doi.org/10.3390/v15020437
Chicago/Turabian StyleRibeiro, Ieda Pereira, Edson Delatorre, Filipe Vieira Santos de Abreu, Alexandre Araújo Cunha dos Santos, Nathália Dias Furtado, Anielly Ferreira-de-Brito, Anielle de Pina-Costa, Maycon Sebastião Alberto Santos Neves, Márcia Gonçalves de Castro, Monique de Albuquerque Motta, and et al. 2023. "Ecological, Genetic, and Phylogenetic Aspects of YFV 2017–2019 Spread in Rio de Janeiro State" Viruses 15, no. 2: 437. https://doi.org/10.3390/v15020437