
Generation and Characterization of an Influenza D Reporter Virus

 , and
, and 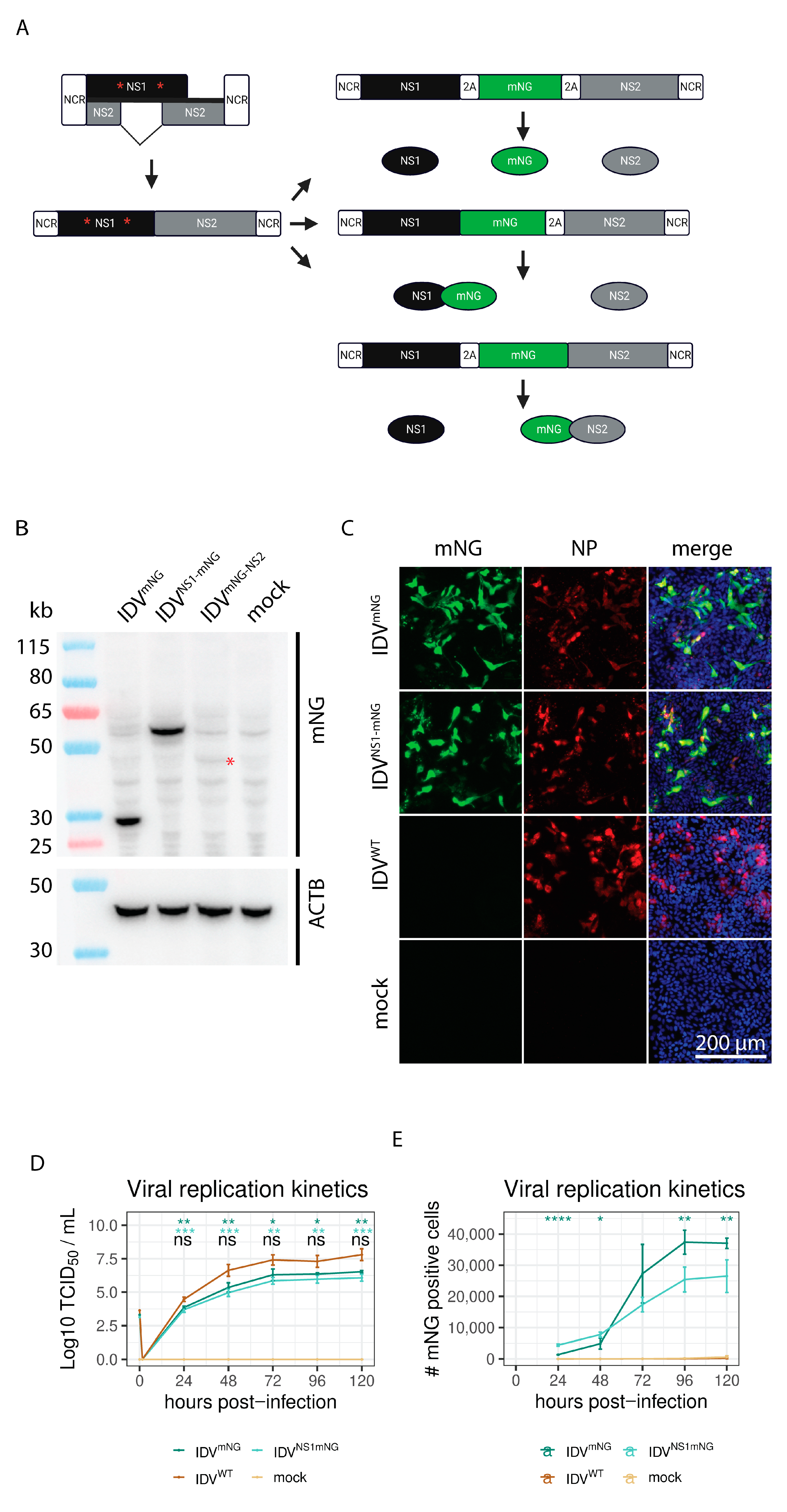{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Construction of the NS Reporter Plasmid
2.3. Western Blot
2.4. Viral Rescue
2.5. Viral Titration
2.6. Immunofluorescence
2.7. Viral Replication Kinetics
2.8. Virus Passaging
2.9. Plaque Purification
2.10. Antiviral Compound Screening
3. Results
3.1. Generation of a Reporter IDV
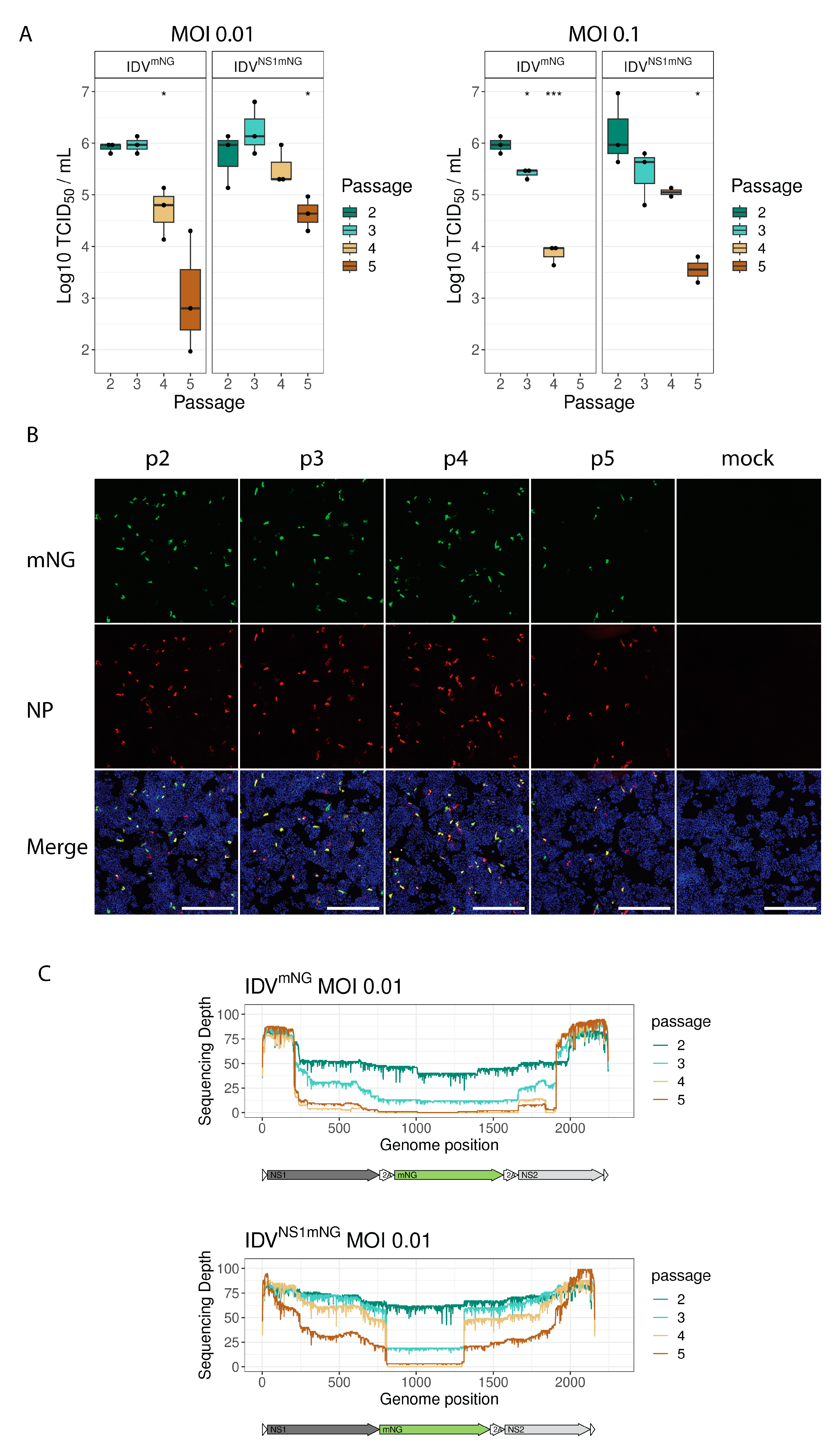
3.2. mNG Gene Expression Is Maintained for the First Three Passages
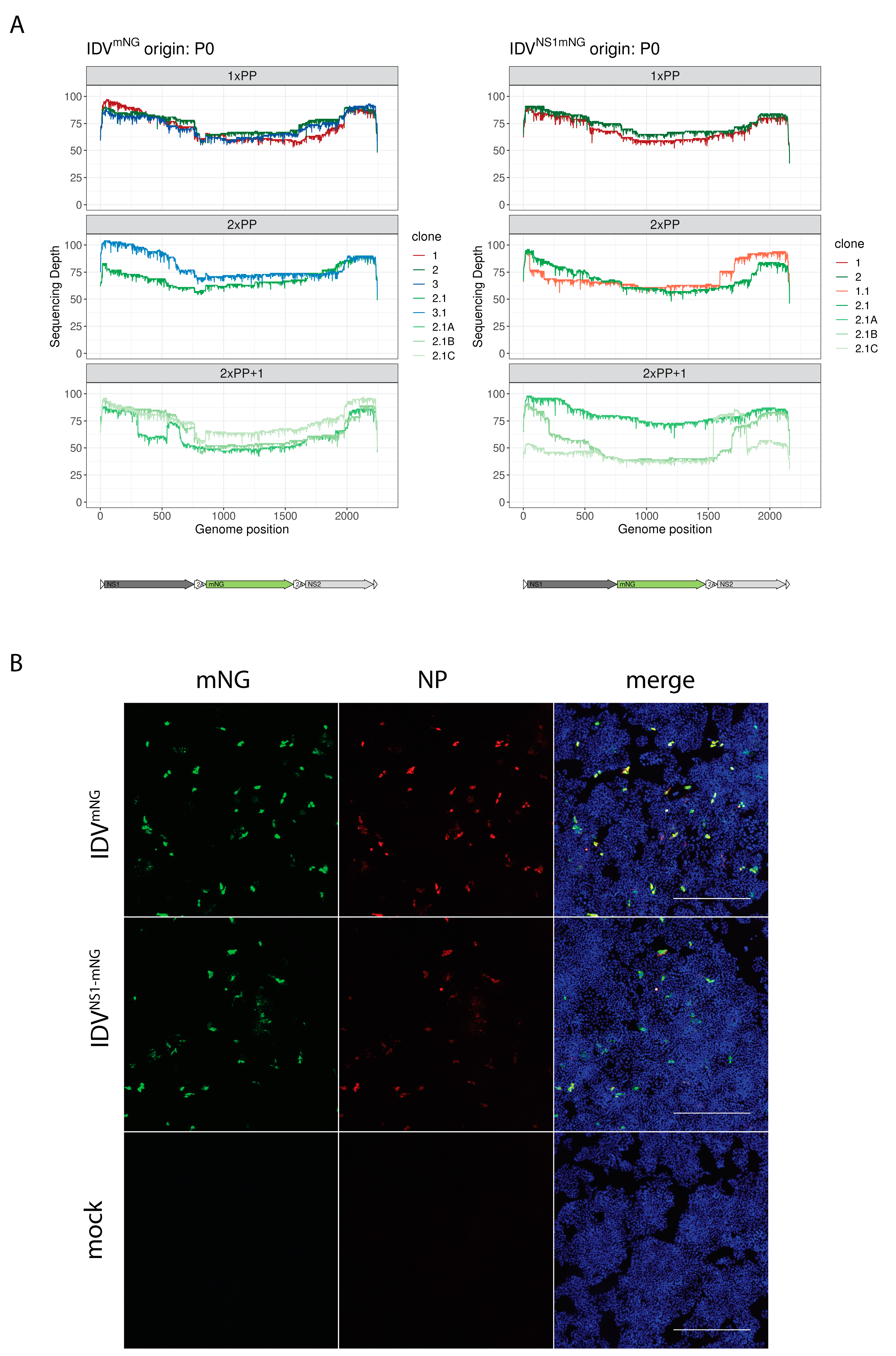
3.3. Plaque Purification Increases Retention of the Full-Length Reporter Segment
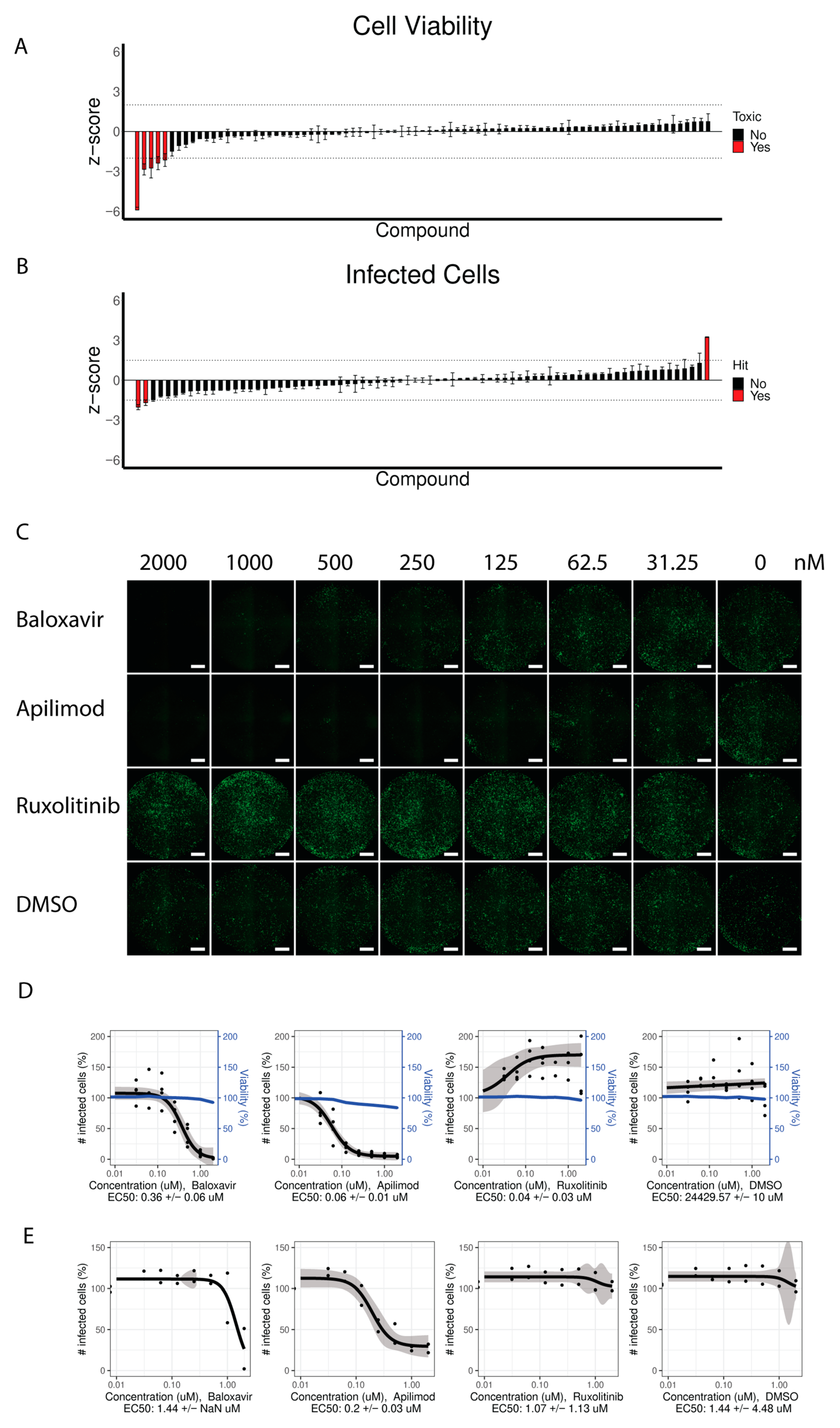
3.4. Identification of Compounds Inhibiting and Promoting Viral Replication
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hause, B.M.; Ducatez, M.; Collin, E.A.; Ran, Z.; Liu, R.; Sheng, Z.; Armien, A.; Kaplan, B.; Chakravarty, S.; Hoppe, A.D.; et al. Isolation of a Novel Swine Influenza Virus from Oklahoma in 2011 Which Is Distantly Related to Human Influenza C Viruses. PLoS Pathog. 2013, 9, e1003176. [Google Scholar] [CrossRef] [PubMed]
- Hause, B.M.; Collin, E.A.; Liu, R.; Huang, B.; Sheng, Z.; Lu, W.; Wang, D.; Nelson, E.A.; Li, F. Characterization of a Novel Influenza Virus in Cattle and Swine: Proposal for a New Genus in the Orthomyxoviridae Family. mBio 2014, 5, e00031-14. [Google Scholar] [CrossRef] [PubMed]
- Quast, M.; Sreenivasan, C.; Sexton, G.; Nedland, H.; Singrey, A.; Fawcett, L.; Miller, G.; Lauer, D.; Voss, S.; Pollock, S.; et al. Serological Evidence for the Presence of Influenza D Virus in Small Ruminants. Vet. Microbiol. 2015, 180, 281–285. [Google Scholar] [CrossRef] [PubMed]
- White, S.K.; Ma, W.; McDaniel, C.J.; Gray, G.C.; Lednicky, J.A. Serologic Evidence of Exposure to Influenza D Virus among Persons with Occupational Contact with Cattle. J. Clin. Virol. 2016, 81, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Salem, E.; Cook, E.A.J.; Lbacha, H.A.; Oliva, J.; Awoume, F.; Aplogan, G.L.; Hymann, E.C.; Muloi, D.; Deem, S.L.; Alali, S.; et al. Serologic Evidence for Influenza C and D Virus among Ruminants and Camelids, Africa, 1991–2015. Emerg. Infect. Dis. 2017, 23, 1556–1559. [Google Scholar] [CrossRef] [PubMed]
- Oliva, J.; Eichenbaum, A.; Belin, J.; Gaudino, M.; Guillotin, J.; Alzieu, J.P.; Nicollet, P.; Brugidou, R.; Gueneau, E.; Michel, E.; et al. Serological Evidence of Influenza D Virus Circulation Among Cattle and Small Ruminants in France. Viruses 2019, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.L.; Zhang, H.; Chen, S.N.; Zhou, X.; Lin, T.; Liu, R.; Lv, D.H.; Wen, X.H.; Wei, W.K.; Wang, D.; et al. Influenza D Virus in Animal Species in Guangdong Province, Southern China. Emerg. Infect. Dis. 2017, 23, 1392–1396. [Google Scholar] [CrossRef]
- Holwerda, M.; Kelly, J.; Laloli, L.; Stürmer, I.; Portmann, J.; Stalder, H.; Dijkman, R. Determining the Replication Kinetics and Cellular Tropism of Influenza D Virus on Primary Well-Differentiated Human Airway Epithelial Cells. Viruses 2019, 11, 377. [Google Scholar] [CrossRef]
- Leibler, J.H.; Abdelgadir, A.; Seidel, J.; White, R.F.; Johnson, W.E.; Reynolds, S.J.; Gray, G.C.; Schaeffer, J.W. Influenza D Virus Exposure among US Cattle Workers: A Call for Surveillance. Zoonoses Public Health 2022, 70, 166–170. [Google Scholar] [CrossRef]
- Reuther, P.; Göpfert, K.; Dudek, A.H.; Heiner, M.; Herold, S.; Schwemmle, M. Generation of a Variety of Stable Influenza A Reporter Viruses by Genetic Engineering of the NS Gene Segment. Sci. Rep. 2015, 5, 11346. [Google Scholar] [CrossRef]
- Manicassamy, B.; Manicassamy, S.; Belicha-Villanueva, A.; Pisanelli, G.; Pulendran, B.; García-Sastre, A. Analysis of in Vivo Dynamics of Influenza Virus Infection in Mice Using a GFP Reporter Virus. Proc. Natl. Acad. Sci. USA 2010, 107, 11531–11536. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Baker, S.F.; Martínez-Sobrido, L. Replication-Competent Influenza A Viruses Expressing a Red Fluorescent Protein. Virology 2015, 476, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Chen, B.; Xing, L.; Cai, X.; Liang, S.; Zhang, L.; Wang, X.; Song, W. Generation of a PdmH1N1 2018 Influenza A Reporter Virus Carrying a MCherry Fluorescent Protein in the PA Segment. Front. Cell Infect. Microbiol. 2022, 11, 827790. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Schotsaert, M.; Rathnasinghe, R.; DeDiego, M.L.; García-Sastre, A.; Martinez-Sobrido, L. Replication-Competent ΔNS1 Influenza A Viruses Expressing Reporter Genes. Viruses 2021, 13, 698. [Google Scholar] [CrossRef] [PubMed]
- De Baets, S.; Verhelst, J.; Van Den Hoecke, S.; Smet, A.; Schotsaert, M.; Job, E.R.; Roose, K.; Schepens, B.; Fiers, W.; Saelens, X. A GFP Expressing Influenza a Virus to Report in Vivo Tropism and Protection by a Matrix Protein 2 Ectodomain-Specific Monoclonal Antibody. PLoS ONE 2015, 10, e0121491. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Aydillo, T.; Ávila-Pérez, G.; Escalera, A.; Chiem, K.; Cadagan, R.; DeDiego, M.L.; Li, F.; García-Sastre, A.; Martínez-Sobrido, L. Functional Characterization and Direct Comparison of Influenza A, B, C, and D NS1 Proteins in Vitro and in Vivo. Front. Microbiol. 2019, 10, 2862. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xia, H.; Huang, J.; Zheng, Y.; Liu, C.; Su, J.; Ping, J. Features of Nuclear Export Signals of NS2 Protein of Influenza D Virus. Viruses 2020, 12, 1100. [Google Scholar] [CrossRef]
- Nogales, A.; Rodríguez-Sánchez, I.; Monte, K.; Lenschow, D.J.; Perez, D.R.; Martínez-Sobrido, L. Replication-Competent Fluorescent-Expressing Influenza B Virus. Virus Res. 2016, 213, 69–81. [Google Scholar] [CrossRef]
- Fulton, B.O.; Palese, P.; Heaton, N.S. Replication-Competent Influenza B Reporter Viruses as Tools for Screening Antivirals and Antibodies. J. Virol. 2015, 89, 12226–12231. [Google Scholar] [CrossRef]
- Holwerda, M.; Laloli, L.; Wider, M.; Schönecker, L.; Becker, J.; Meylan, M.; Dijkman, R. Establishment of a Reverse Genetic System from a Bovine Derived Influenza D Virus Isolate. Viruses 2021, 13, 502. [Google Scholar] [CrossRef]
- Ishida, H.; Murakami, S.; Kamiki, H.; Matsugo, H.; Takenaka-Uema, A.; Horimoto, T. Establishment of a Reverse Genetics System for Influenza D Virus. J. Virol. 2020, 94, e01767-19. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Murakami, S.; Kamiki, H.; Matsugo, H.; Katayama, M.; Sekine, W.; Ohira, K.; Takenaka-Uema, A.; Horimoto, T. Construction of an Influenza D Virus with an Eight-Segmented Genome. Viruses 2021, 13, 2166. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Liu, R.; Zhou, B.; Chou, T.; Ghedin, E.; Sheng, Z.; Gao, R.; Zhai, S.; Wang, D.; Li, F. Development and Characterization of a Reverse-Genetics System for Influenza D Virus. J. Virol. 2019, 93, e01186-19. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A Bright Monomeric Green Fluorescent Protein Derived from Branchiostoma Lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef] [PubMed]
- García-Nafría, J.; Watson, J.F.; Greger, I.H. IVA Cloning: A Single-Tube Universal Cloning System Exploiting Bacterial In Vivo Assembly. Sci. Rep. 2016, 6, 27459. [Google Scholar] [CrossRef] [PubMed]
- Kärber, G. Beitrag Zur Kollektiven Behandlung Pharmakologischer Reihenversuche. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Mutterer, J.; Zinck, E. Quick-and-Clean Article Figures with FigureJ. J. Microsc. 2013, 252, 89–91. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef]
- Mishin, V.P.; Patel, M.C.; Chesnokov, A.; De La Cruz, J.; Nguyen, H.T.; Lollis, L.; Hodges, E.; Jang, Y.; Barnes, J.; Uyeki, T.; et al. Susceptibility of Influenza A, B, C, and D Viruses to Baloxavir. Emerg. Infect. Dis. 2019, 25, 1969–1972. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Yan, F.; Doronina, V.A.; Escuin-Ordinas, H.; Ryan, M.D.; Brown, J.D. 2A Peptides Provide Distinct Solutions to Driving Stop-Carry on Translational Recoding. Nucleic Acids Res. 2012, 40, 3143–3151. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, M.; V’kovski, P.; Wider, M.; Thiel, V.; Dijkman, R. Identification of an Antiviral Compound from the Pandemic Response Box That Efficiently Inhibits SARS-CoV-2 Infection In Vitro. Microorganisms 2020, 8, 1872. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Xu, Y.; Cheung, A.K.; Tomlinson, R.C.; Alcázar-Román, A.; Murphy, L.; Billich, A.; Zhang, B.; Feng, Y.; Klumpp, M.; et al. PIKfyve, a Class III PI Kinase, Is the Target of the Small Molecular IL-12/IL-23 Inhibitor Apilimod and a Player in Toll-like Receptor Signaling. Chem. Biol. 2013, 20, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Vaddi, K.; Liu, P.; Manshouri, T.; Li, J.; Scherle, P.A.; Caulder, E.; Wen, X.; Li, Y.; Waeltz, P.; et al. Preclinical Characterization of the Selective JAK1/2 Inhibitor INCB018424: Therapeutic Implications for the Treatment of Myeloproliferative Neoplasms. Blood 2010, 115, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.; Poole, D.S.; Jeffery, J.J.; Sheahan, T.P.; Creech, D.; Yevtodiyenko, A.; Peat, A.J.; Francis, K.P.; You, S.; Mehle, A. Multi-Modal Imaging with a Toolbox of Influenza A Reporter Viruses. Viruses 2015, 7, 5319–5327. [Google Scholar] [CrossRef]
- Oliva, J.; Mettier, J.; Sedano, L.; Delverdier, M.; Bourgès-Abella, N.; Hause, B.; Loupias, J.; Pardo, I.; Bleuart, C.; Bordignon, P.J.; et al. Murine Model for the Study of Influenza D Virus. J. Virol. 2019, 94, e01662-19. [Google Scholar] [CrossRef]
- Alnaji, F.G.; Reiser, W.K.; Rivera-Cardona, J.; te Velthuis, A.J.W.; Brooke, C.B. Influenza A Virus Defective Viral Genomes Are Inefficiently Packaged into Virions Relative to Wild-Type Genomic RNAs. mBio 2021, 12, e0295921. [Google Scholar] [CrossRef]
- Nelson, E.A.; Dyall, J.; Hoenen, T.; Barnes, A.B.; Zhou, H.; Liang, J.Y.; Michelotti, J.; Dewey, W.H.; DeWald, L.E.; Bennett, R.S.; et al. The Phosphatidylinositol-3-Phosphate 5-Kinase Inhibitor Apilimod Blocks Filoviral Entry and Infection. PLoS Negl. Trop. Dis. 2017, 11, e0005540. [Google Scholar] [CrossRef]
- Laloli, L.; Licheri, M.F.; Probst, L.; Licheri, M.; Gultom, M.; Holwerda, M.; V’kovski, P.; Dijkman, R. Time-Resolved Characterization of the Innate Immune Response in the Respiratory Epithelium of Human, Porcine, and Bovine during Influenza Virus Infection. Front. Immunol. 2022, 13, 970325. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Probst, L.; Laloli, L.; Licheri, M.F.; Licheri, M.; Gultom, M.; Holwerda, M.; V’kovski, P.; Dijkman, R. Generation and Characterization of an Influenza D Reporter Virus. Viruses 2023, 15, 2444. https://doi.org/10.3390/v15122444
Probst L, Laloli L, Licheri MF, Licheri M, Gultom M, Holwerda M, V’kovski P, Dijkman R. Generation and Characterization of an Influenza D Reporter Virus. Viruses. 2023; 15(12):2444. https://doi.org/10.3390/v15122444
Chicago/Turabian StyleProbst, Lukas, Laura Laloli, Manon Flore Licheri, Matthias Licheri, Mitra Gultom, Melle Holwerda, Philip V’kovski, and Ronald Dijkman. 2023. "Generation and Characterization of an Influenza D Reporter Virus" Viruses 15, no. 12: 2444. https://doi.org/10.3390/v15122444