

Role of PARP-1 in Human Cytomegalovirus Infection and Functional Partners Encoded by This Virus

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Reagents
2.3. Oligonucleotides and Plasmid Construction
2.4. NAD+ Measurements
2.5. Western Blot
2.6. Cell Fraction
2.7. Immunoprecipitation
2.8. Laser Micro-Irradiation and Live Cell Image
2.9. Immunofluorescence
2.10. RNA Isolation, cDNA Synthesis and Semi-Quantitative PCR
2.11. Protein Expression and Purification
2.12. PAR Synthesis and Purification
2.13. ADP-Ribosylation Assay In Vitro
2.14. PAR Overlay Assay
2.15. Statistical Analyses
3. Results
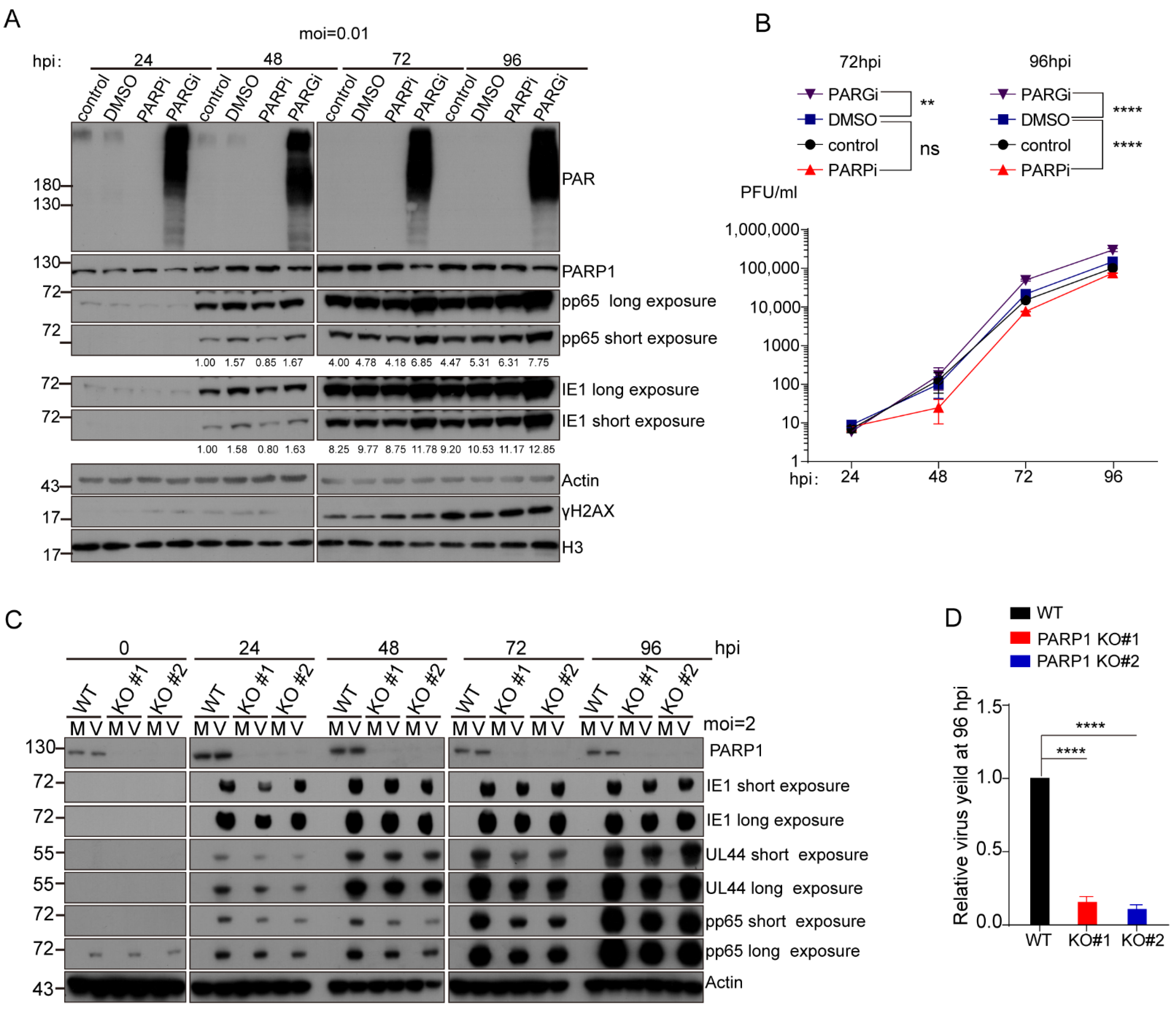
3.1. PARP-1 and Its Enzymatic Activity Were Required for Efficient HCMV Replication in Fibroblast
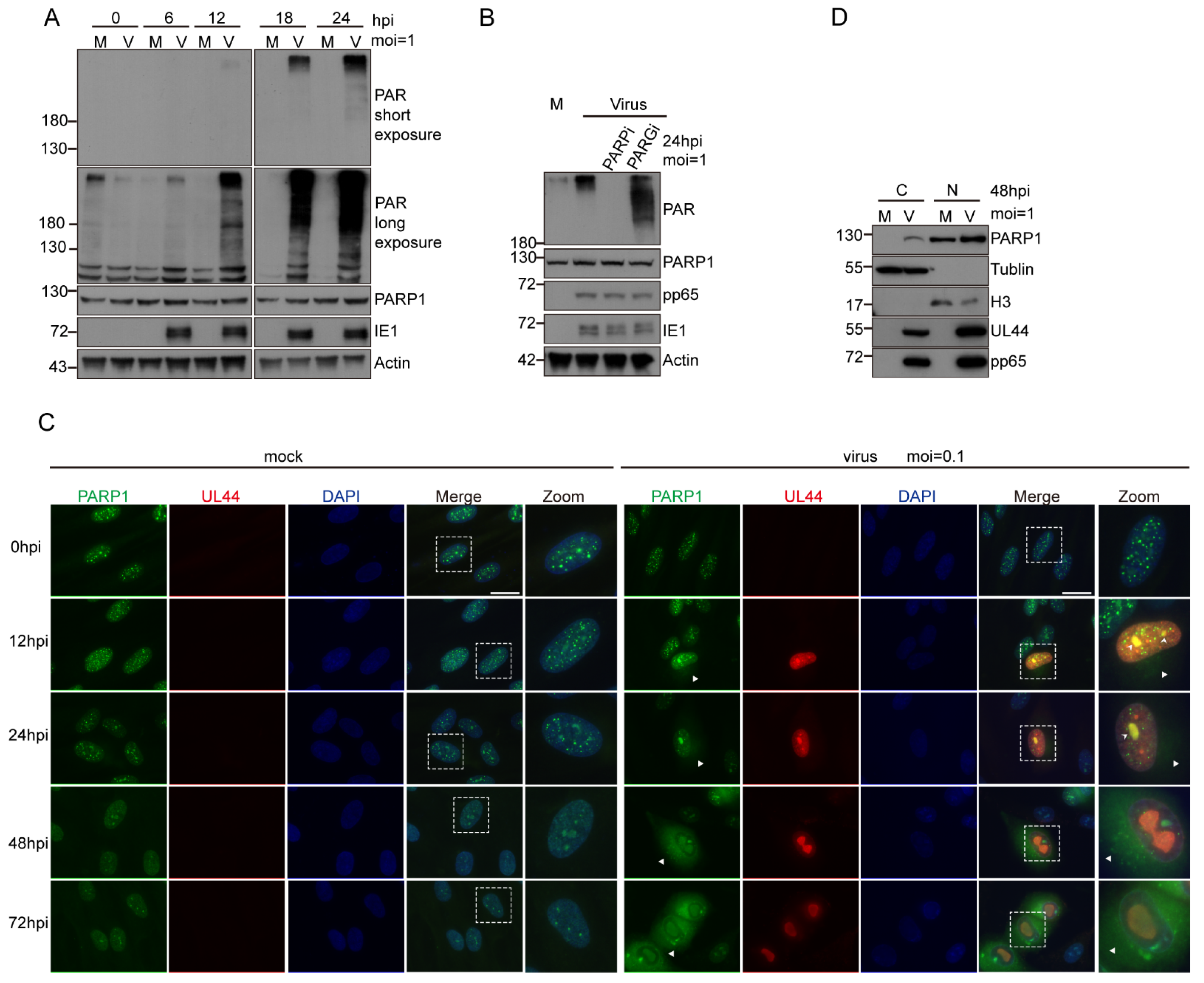
3.2. HCMV Infection Activated PARP-1 and Induced Its Translocation from Nucleus to Cytoplasm
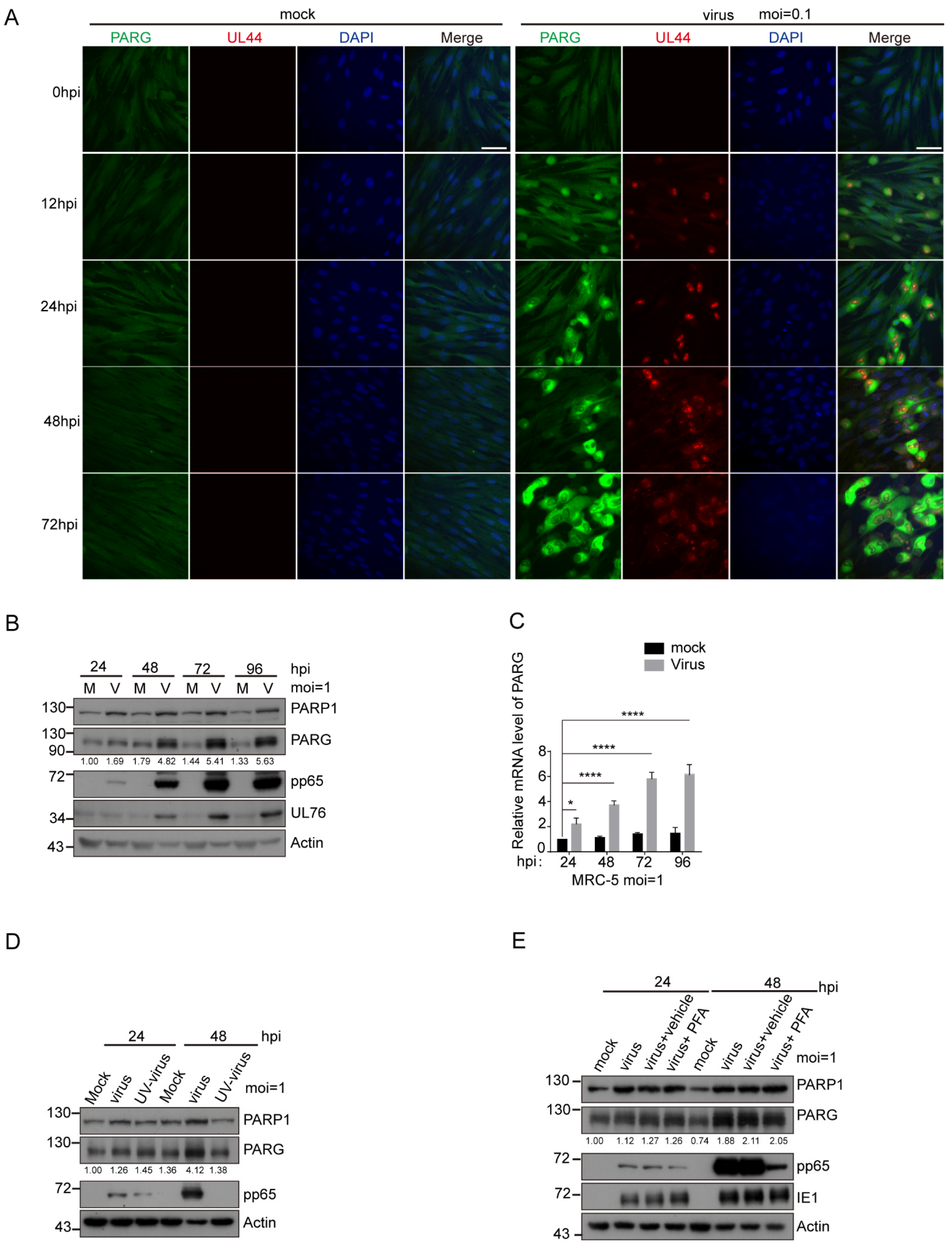
3.3. PARG Was Upregulated during HCMV Infection, and This Upregulation Was Independent of Viral DNA Replication
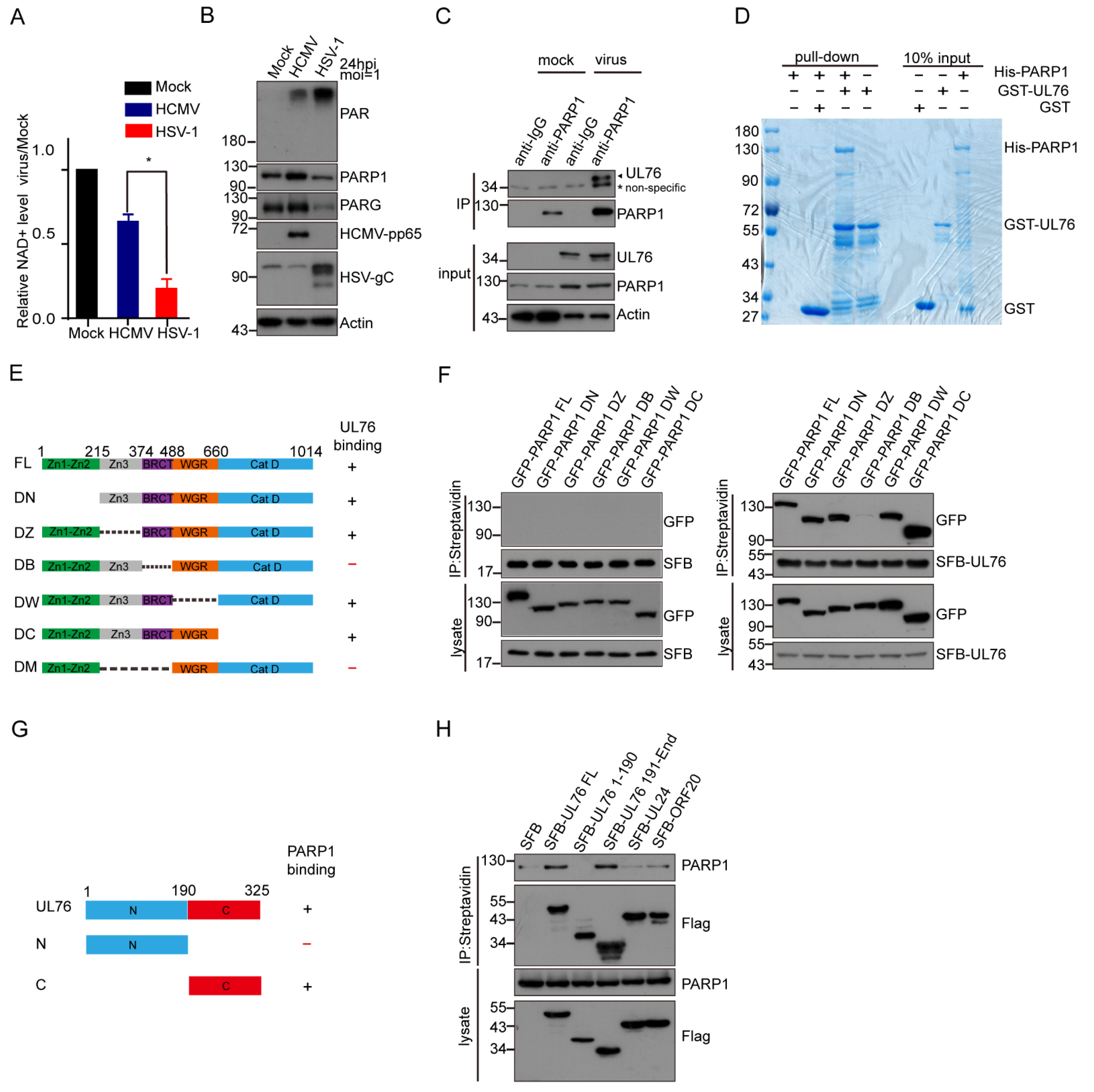
3.4. HCMV UL76 Bound to the BRCT Domain of PARP-1
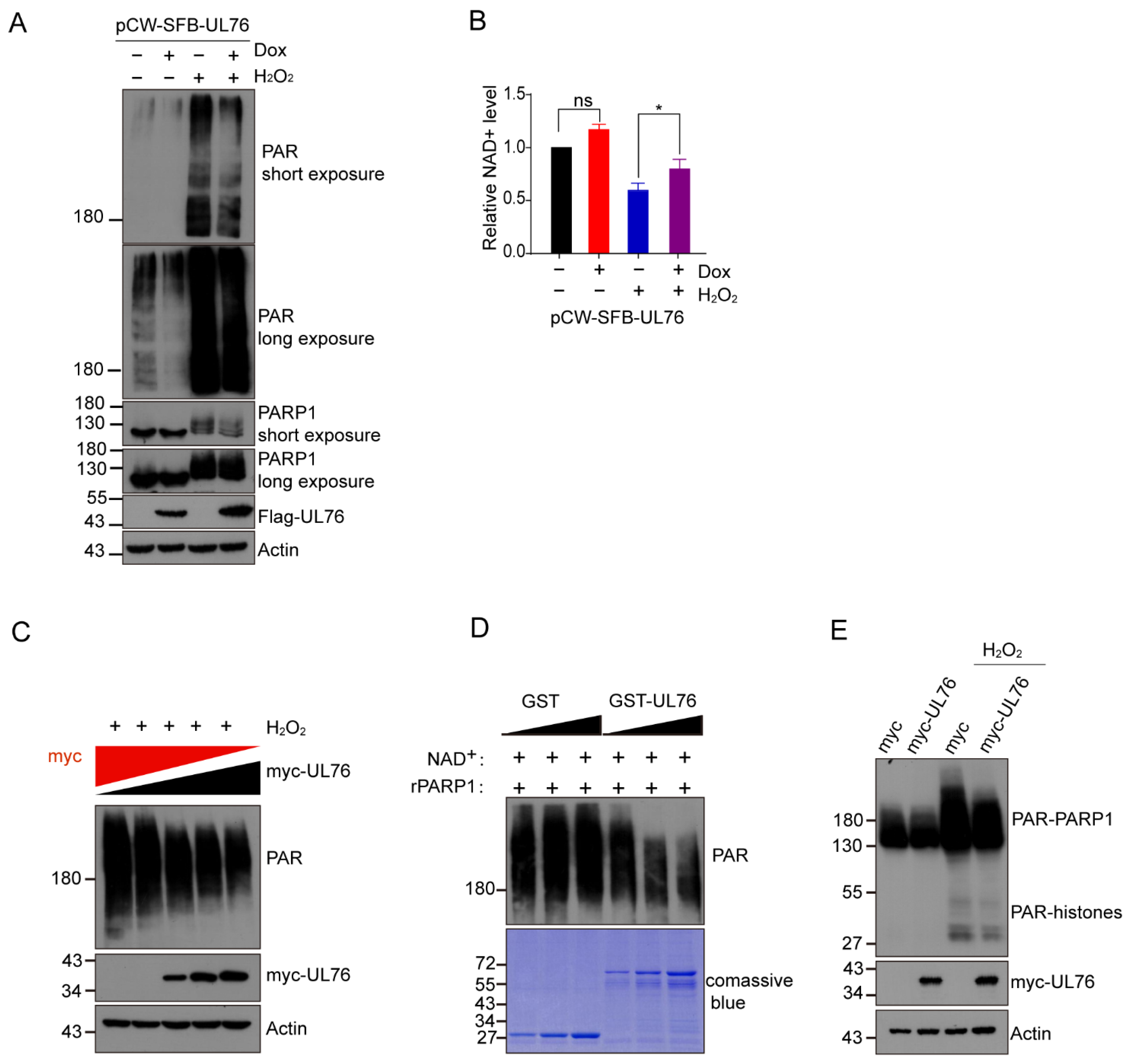
3.5. HCMV UL76 Inhibited PARP-1 Activity Both In Vivo and In Vitro
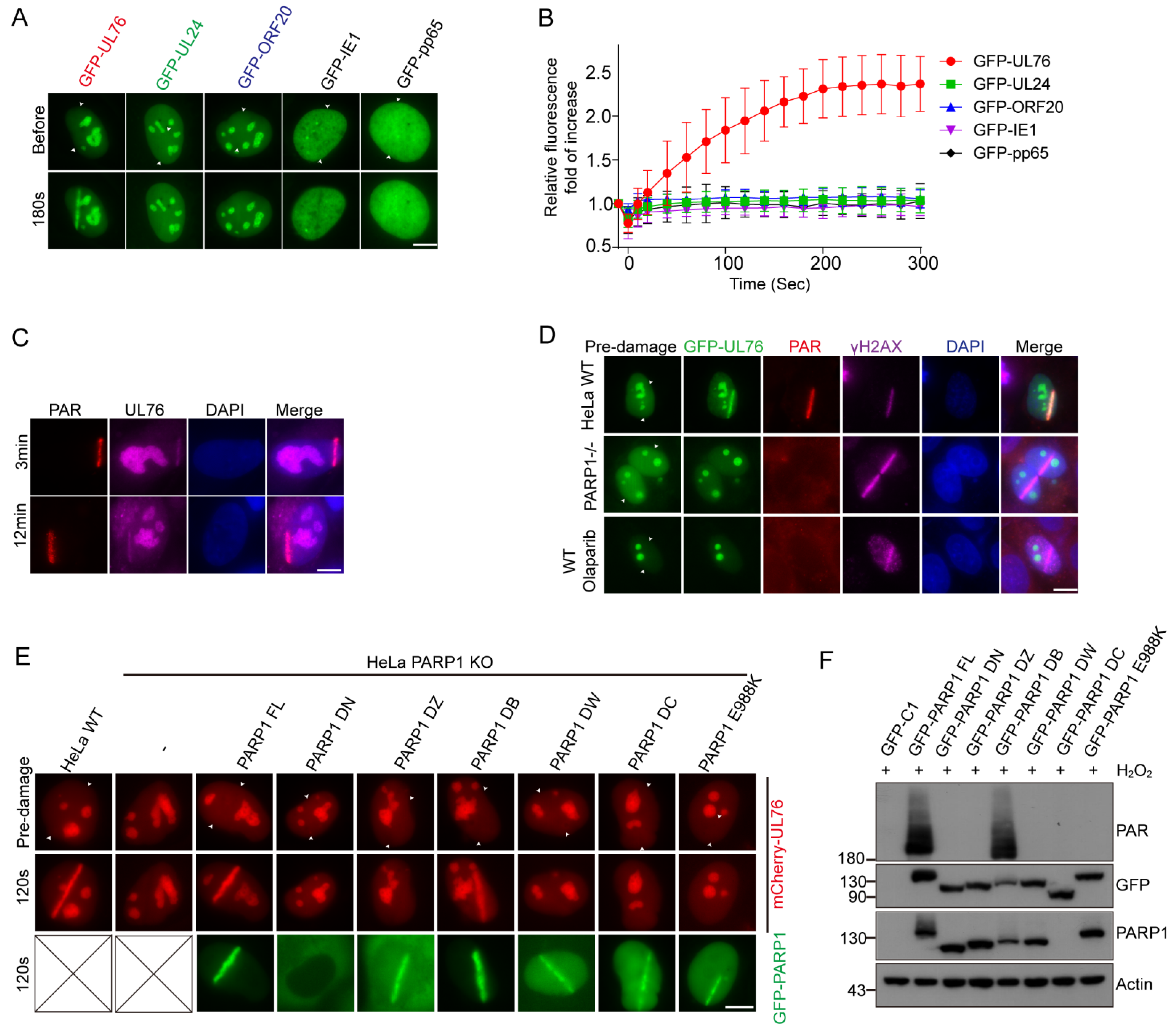
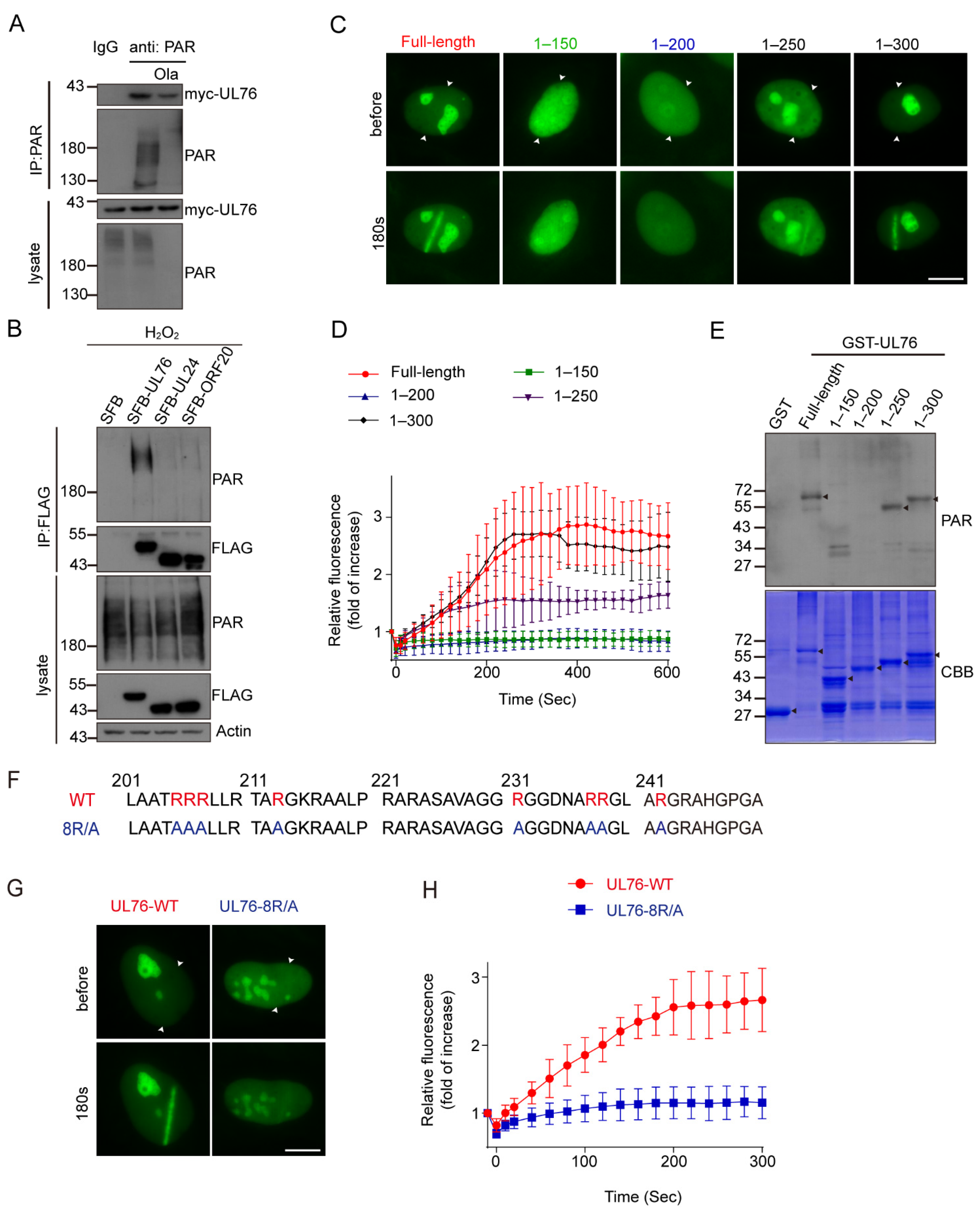
3.6. PAR Dependent Recruitment of HCMV UL76 to DNA Damage Sites
3.7. HCMV UL76 Bound to PAR via Its RG/RGG Motif in the C Terminal
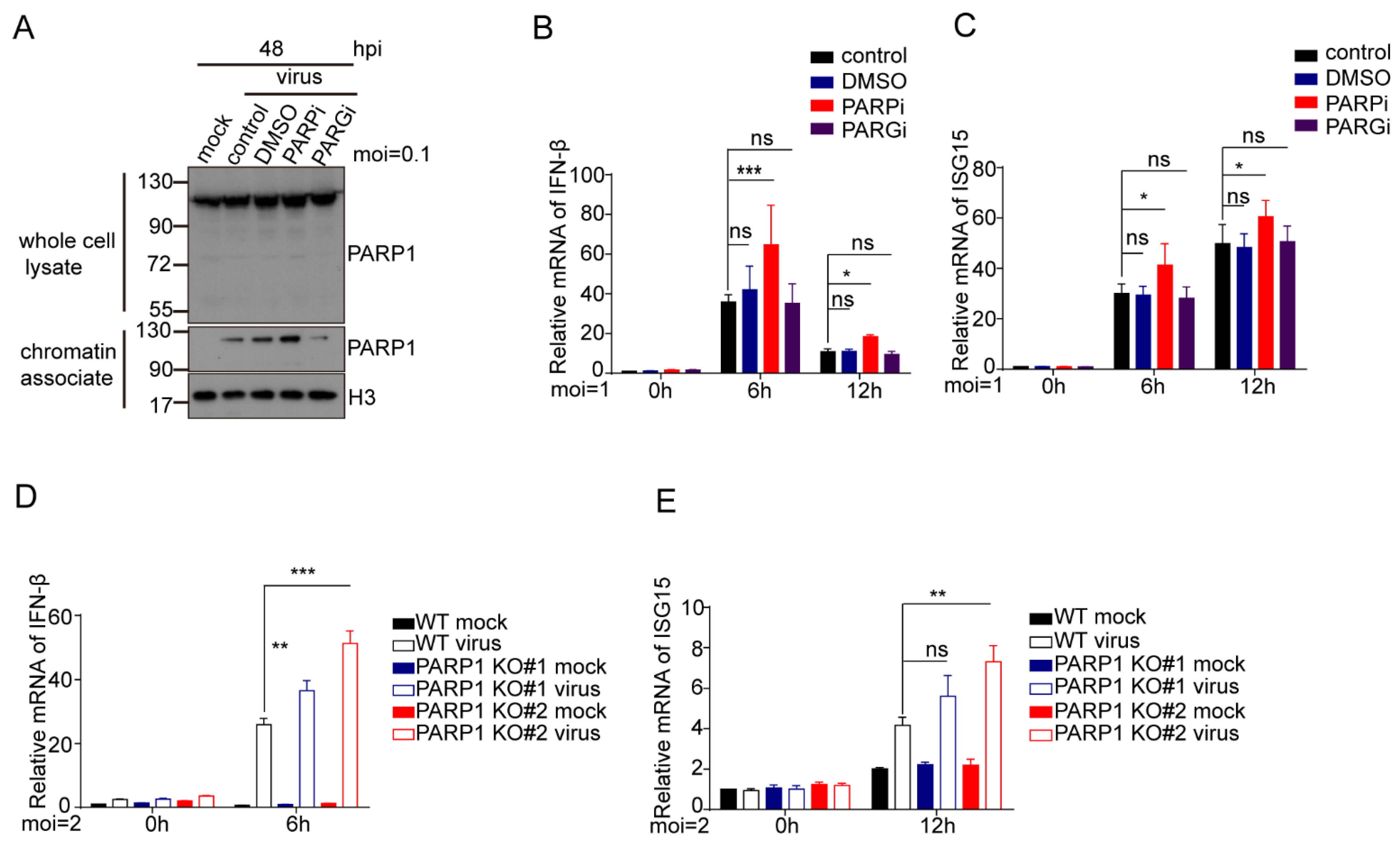
3.8. Inhibition or Depletion of PARP-1 Increases Type 1 IFN Response in HCMV Infected Cells
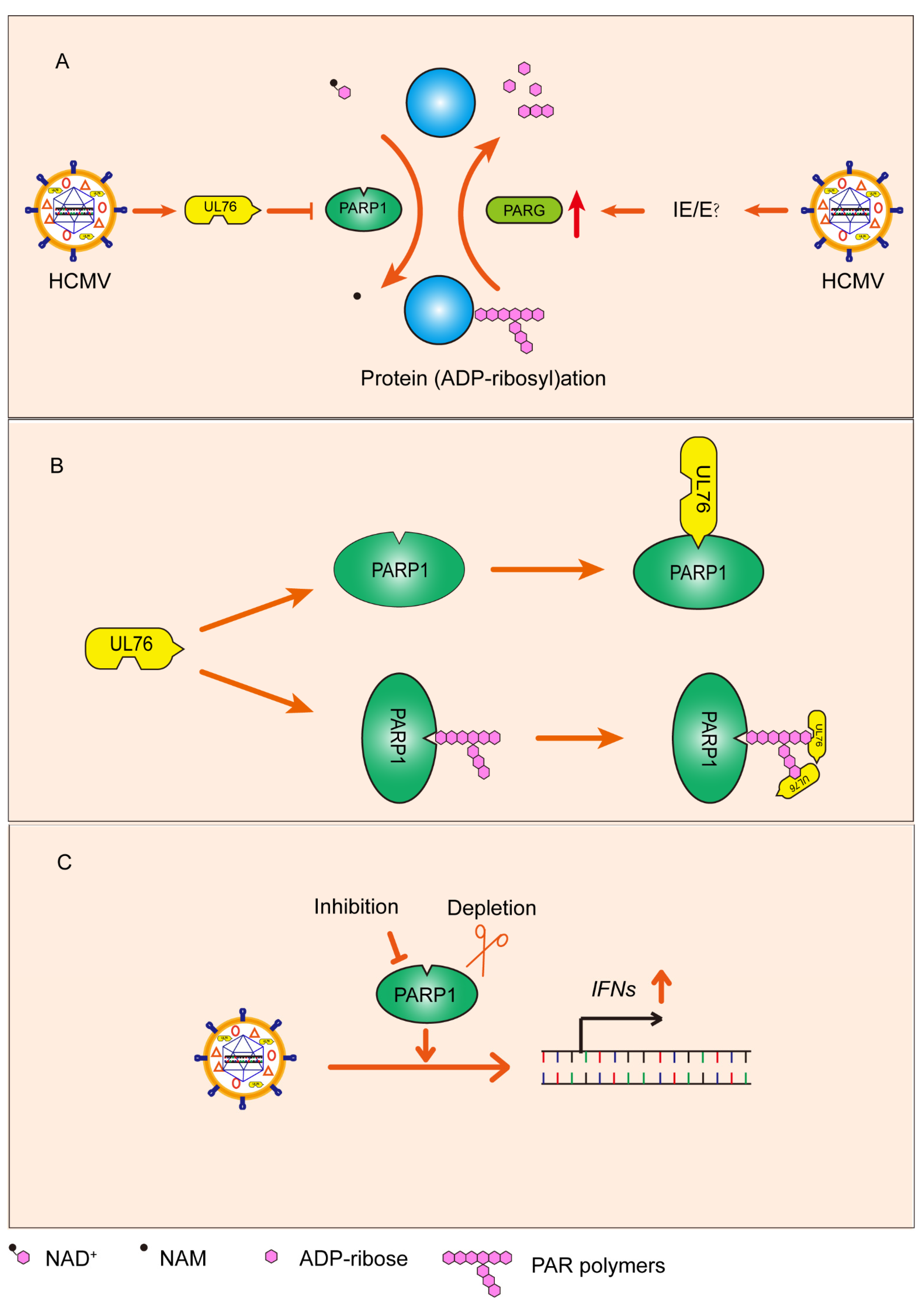
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kumar, R.; Cruz, L.; Sandhu, P.K.; Buchkovich, N.J. UL88 Mediates the Incorporation of a Subset of Proteins into the Virion Tegument. J. Virol. 2020, 94, e00474-20. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.S.M. Human Cytomegalovirus; Springer: Berlin/Heidelberg, Germany, 2008; Volume 325, pp. 1–476. [Google Scholar]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Stinski, F.S.M. Sequence of Protein Synthesis in Cells Infected by Human Cytomegalovirus: Early and Late Virus-Induced Polypeptides. J. Virol. 1978, 26, 686–701. [Google Scholar] [CrossRef]
- Clement, M.; Humphreys, I.R. Cytokine-Mediated Induction and Regulation of Tissue Damage During Cytomegalovirus Infection. Front Immunol 2019, 10, 78. [Google Scholar] [CrossRef]
- Wujcicka, W.; Paradowska, E.; Studzinska, M.; Wilczynski, J.; Nowakowska, D. Toll-like receptors genes polymorphisms and the occurrence of HCMV infection among pregnant women. Virol. J. 2017, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, M.; Shenk, T. Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 2821–2826. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.H.; Rosenke, K.; Czornak, K.; Fortunato, E.A. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J. Virol. 2007, 81, 1934–1950. [Google Scholar] [CrossRef]
- Qian, Z.; Xuan, B.; Gualberto, N.; Yu, D. The human cytomegalovirus protein pUL38 suppresses endoplasmic reticulum stress-mediated cell death independently of its ability to induce mTORC1 activation. J. Virol. 2011, 85, 9103–9113. [Google Scholar] [CrossRef]
- Fliss, P.M.; Brune, W. Prevention of cellular suicide by cytomegaloviruses. Viruses 2012, 4, 1928–1949. [Google Scholar] [CrossRef]
- Brune, W.; Andoniou, C.E. Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus. Viruses 2017, 9, 249. [Google Scholar] [CrossRef] [Green Version]
- Knizewski, L.; Kinch, L.; Grishin, N.V.; Rychlewski, L.; Ginalski, K. Human herpesvirus 1 UL24 gene encodes a potential PD-(D/E)XK endonuclease. J. Virol. 2006, 80, 2575–2577. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.K.; Duh, C.Y.; Wu, C.W. Human cytomegalovirus UL76 encodes a novel virion-associated protein that is able to inhibit viral replication. J. Virol. 2004, 78, 9750–9762. [Google Scholar] [CrossRef]
- Lin, S.R.; Jiang, M.J.; Wang, H.H.; Hu, C.H.; Hsu, M.S.; Hsi, E.; Duh, C.Y.; Wang, S.K. Human cytomegalovirus UL76 elicits novel aggresome formation via interaction with S5a of the ubiquitin proteasome system. J. Virol. 2013, 87, 11562–11578. [Google Scholar] [CrossRef] [PubMed]
- Siew, V.K.; Duh, C.Y.; Wang, S.K. Human cytomegalovirus UL76 induces chromosome aberrations. J. Biomed. Sci. 2009, 16, 1–10. [Google Scholar] [CrossRef]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 12396–12401. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.; Cassie, C.; Hong, L.; Rong, H.; David, P.; Viktor, S.; Hua, Z.; Fenyong, L. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef]
- Burkle, A. Poly(ADP-ribose). The most elaborate metabolite of NAD+. FEBS J. 2005, 272, 4576–4589. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Teloni, F.; Altmeyer, M. Readers of poly(ADP-ribose): Designed to be fit for purpose. Nucleic Acids Res. 2016, 44, 993–1006. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Vyas, A.; Kassab, M.A.; Singh, A.K.; Yu, X. The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 2017, 45, 8129–8141. [Google Scholar] [CrossRef]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.A.; Umanah, G.K.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagne, J.P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Fouquerel, E.; Ame, J.C.; Leonhardt, H.; Schreiber, V. PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res. 2011, 39, 5045–5056. [Google Scholar] [CrossRef]
- Meyer-Ficca, M.L.; Meyer, R.G.; Coyle, D.L.; Jacobson, E.L.; Jacobson, M.K. Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp. Cell Res. 2004, 297, 521–532. [Google Scholar] [CrossRef]
- Shang, J.; Smith, M.R.; Anmangandla, A.; Lin, H. NAD+-consuming enzymes in immune defense against viral infection. Biochem. J. 2021, 478, 4071–4092. [Google Scholar] [CrossRef]
- Malgras, M.; Garcia, M.; Jousselin, C.; Bodet, C.; Leveque, N. The Antiviral Activities of Poly-ADP-Ribose Polymerases. Viruses 2021, 13, 582. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zheng, C. When PARPs Meet Antiviral Innate Immunity. Trends Microbiol. 2021, 29, 776–778. [Google Scholar] [CrossRef]
- Ha, H.C.; Juluri, K.; Zhou, Y.; Leung, S.; Hermankova, M.; Snyder, S.H. Poly(ADP-ribose) polymerase-1 is required for efficient HIV-1 integration. Proc. Natl. Acad. Sci. USA 2001, 98, 3364–3368. [Google Scholar] [CrossRef]
- Na, T.Y.; Ka, N.L.; Rhee, H.; Kyeong, D.; Kim, M.H.; Seong, J.K.; Park, Y.N.; Lee, M.O. Interaction of hepatitis B virus X protein with PARP1 results in inhibition of DNA repair in hepatocellular carcinoma. Oncogene 2016, 35, 5435–5445. [Google Scholar] [CrossRef]
- Nebenzahl-Sharon, K.; Sharf, R.; Amer, J.; Shalata, H.; Khoury-Haddad, H.; Sohn, S.-Y.; Ayoub, N.; Hearing, P.; Kleinberger, T. An Interaction with PARP-1 and Inhibition of Parylation Contribute to Attenuation of DNA Damage Signaling by the Adenovirus E4orf4 Protein. J. Virol. 2019, 93, e02253-18. [Google Scholar] [CrossRef] [PubMed]
- Grady, S.L.; Hwang, J.; Vastag, L.; Rabinowitz, J.D.; Shenk, T. Herpes simplex virus 1 infection activates poly(ADP-ribose) polymerase and triggers the degradation of poly(ADP-ribose) glycohydrolase. J. Virol. 2012, 86, 8259–8268. [Google Scholar] [CrossRef] [PubMed]
- Tempera, I.; Deng, Z.; Atanasiu, C.; Chen, C.J.; D’Erme, M.; Lieberman, P.M. Regulation of Epstein-Barr virus OriP replication by poly(ADP-ribose) polymerase 1. J. Virol. 2010, 84, 4988–4997. [Google Scholar] [CrossRef] [PubMed]
- Ohsaki, E.; Ueda, K.; Sakakibara, S.; Do, E.; Yada, K.; Yamanishi, K. Poly(ADP-ribose) polymerase 1 binds to Kaposi’s sarcoma-associated herpesvirus (KSHV) terminal repeat sequence and modulates KSHV replication in latency. J. Virol. 2004, 78, 9936–9946. [Google Scholar] [CrossRef]
- Gwack, Y.; Nakamura, H.; Lee, S.H.; Souvlis, J.; Yustein, J.T.; Gygi, S.; Kung, H.J.; Jung, J.U. Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication. Mol. Cell Biol. 2003, 23, 8282–8294. [Google Scholar] [CrossRef]
- Chung, W.-C.; Park, J.-H.; Kang, H.-R.; Song, M.J. Downregulation of Poly(ADP-Ribose) Polymerase 1 by a Viral Processivity Factor Facilitates Lytic Replication of Gammaherpesvirus. J. Virol. 2015, 89, 9676–9682. [Google Scholar] [CrossRef]
- Chung, W.C.; Lee, S.; Kim, Y.; Seo, J.B.; Song, M.J. Kaposi’s sarcoma-associated herpesvirus processivity factor (PF-8) recruits cellular E3 ubiquitin ligase CHFR to promote PARP1 degradation and lytic replication. PLoS Pathog. 2021, 17, e1009261. [Google Scholar] [CrossRef]
- Noh, C.W.; Cho, H.J.; Kang, H.R.; Jin, H.Y.; Lee, S.; Deng, H.; Wu, T.T.; Arumugaswami, V.; Sun, R.; Song, M.J. The virion-associated open reading frame 49 of murine gammaherpesvirus 68 promotes viral replication both in vitro and in vivo as a derepressor of RTA. J. Virol. 2012, 86, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.C.; Kim, J.; Kim, B.C.; Kang, H.R.; Son, J.; Ki, H.; Hwang, K.Y.; Song, M.J. Structure-based mechanism of action of a viral poly(ADP-ribose) polymerase 1-interacting protein facilitating virus replication. IUCrJ. 2018, 5, 866–879. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, M.; Chang, B.; Zhou, Y.; Wu, X.; Ma, M.; Liu, S.; Cao, Y.; Zheng, M.; Dang, Y.; et al. Cytoplasmic PARP1 links the genome instability to the inhibition of antiviral immunity through PARylating cGAS. Mol. Cell 2022, 82, 2032–2049.e7. [Google Scholar] [CrossRef]
- Schuhwerk, H.; Bruhn, C.; Siniuk, K.; Min, W.; Erener, S.; Grigaravicius, P.; Kruger, A.; Ferrari, E.; Zubel, T.; Lazaro, D.; et al. Kinetics of poly(ADP-ribosyl)ation, but not PARP1 itself, determines the cell fate in response to DNA damage in vitro and in vivo. Nucleic Acids Res. 2017, 45, 11174–11192. [Google Scholar] [CrossRef] [PubMed]
- Yingshan, C.; Yongxuan, Y.; Kaitao, Z.; Canyu, L.; Yifei, Y.; Hao, S.; Dan, H.; Yi, Z.; Yuan, Z.; Jizheng, C.; et al. DNA repair factor poly(ADP-Ribose) polymerase 1 is a proviral factor in Hepatitis B virus covalently closed circular DNA formation. J. Virol. 2022, 96, 1–17. [Google Scholar] [CrossRef]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef]
- Lupey-Green, L.N.; Moquin, S.A.; Martin, K.A.; McDevitt, S.M.; Hulse, M.; Caruso, L.B.; Pomerantz, R.T.; Miranda, J.L.; Tempera, I. PARP1 restricts Epstein Barr Virus lytic reactivation by binding the BZLF1 promoter. Virology 2017, 507, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Nobre, L.V.; Nightingale, K.; Ravenhill, B.J.; Antrobus, R.; Soday, L.; Nichols, J.; Davies, J.A.; Seirafian, S.; Wang, E.C.; Davison, A.J.; et al. Human cytomegalovirus interactome analysis identifies degradation hubs, domain associations and viral protein functions. Elife 2019, 8, e49894. [Google Scholar] [CrossRef] [PubMed]
- Glover, J.M.; Williams, R.S.; Lee, M.S. Interactions between BRCT repeats and phosphoproteins: Tangled up in two. TRENDS Biochem. Sci. 2004, 29, 579–585. [Google Scholar] [CrossRef]
- Gibbs-Seymour, I.; Fontana, P.; Rack, J.G.M.; Ahel, I. HPF1/C4orf27 Is a PARP-1-Interacting Protein that Regulates PARP-1 ADP-Ribosylation Activity. Mol. Cell 2016, 62, 432–442. [Google Scholar] [CrossRef]
- Kong, X.; Wakida, N.M.; Yokomori, K. Application of Laser Microirradiation in the Investigations of Cellular Responses to DNA Damage. Front. Phys. 2021, 8, 66. [Google Scholar] [CrossRef]
- Barkauskaite, E.; Jankevicius, G.; Ladurner, A.G.; Ahel, I.; Timinszky, G. The recognition and removal of cellular poly(ADP-ribose) signals. FEBS J. 2013, 280, 3491–3507. [Google Scholar] [CrossRef]
- Barkauskaite, E.; Jankevicius, G.; Ahel, I. Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Mol. Cell 2015, 58, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Altmeyer, M.; Messner, S.; Hassa, P.O.; Fey, M.; Hottiger, M.O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009, 37, 3723–3738. [Google Scholar] [CrossRef]
- Singatulina, A.S.; Hamon, L.; Sukhanova, M.V.; Desforges, B.; Joshi, V.; Bouhss, A.; Lavrik, O.I.; Pastre, D. PARP-1 Activation Directs FUS to DNA Damage Sites to Form PARG-Reversible Compartments Enriched in Damaged DNA. Cell Rep. 2019, 27, 1809–1821.e5. [Google Scholar] [CrossRef] [PubMed]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gomez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Lin, W.L.; Chen, Z.; Liu, H.W. PARP-1-dependent recruitment of cold-inducible RNA-binding protein promotes double-strand break repair and genome stability. Proc. Natl. Acad Sci. USA 2018, 115, E1759–E1768. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Virag, L. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett. 2012, 586, 3771–3777. [Google Scholar] [CrossRef] [PubMed]
- Rosado, M.M.; Bennici, E.; Novelli, F.; Pioli, C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 2013, 139, 428–437. [Google Scholar] [CrossRef]
- Kim, C.; Wang, X.D.; Yu, Y. PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. Elife 2020, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Li, G.L.; Ding, G.X.; Zeng, L.; Ming, S.L.; Fu, P.F.; Wang, Q.; Yang, G.Y.; Wang, J.; Chu, B.B. Inhibition of PARP1 Dampens Pseudorabies Virus Infection through DNA Damage-Induced Antiviral Innate Immunity. J. Virol. 2021, 95, e0076021. [Google Scholar] [CrossRef]
- Lukovac, S.; Belzer, C.; Pellis, L.; Keijser, B.J.; de Vos, W.M.; Montijn, R.C.; Roeselers, G. Herpes Simplex Virus 1 Manipulates Host Cell Antiviral and Proviral DNA Damage Responses. mBio 2021, 12, e03552-20. [Google Scholar] [CrossRef]
- Shah, G.A.; O’Shea, C.C. Viral and Cellular Genomes Activate Distinct DNA Damage Responses. Cell 2015, 162, 987–1002. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.; Reuven, N.; Mohni, K.N.; Schumacher, A.J.; Weller, S.K. Structure of the herpes simplex virus 1 genome: Manipulation of nicks and gaps can abrogate infectivity and alter the cellular DNA damage response. J. Virol. 2014, 88, 10146–10156. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus DNA Replication and the Host DNA Damage Response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, J.A.; Dremel, S.E.; DeLuca, N.A. Replication-Coupled Recruitment of Viral and Cellular Factors to Herpes Simplex Virus Type 1 Replication Forks for the Maintenance and Expression of Viral Genomes. PLoS Pathog. 2017, 13, e1006166. [Google Scholar] [CrossRef] [PubMed]
- Manska, S.; Rossetto, C.C. Identification of cellular proteins associated with human cytomegalovirus (HCMV) DNA replication suggests novel cellular and viral interactions. Virology 2022, 566, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, J.A.; DeLuca, N.A. Selective recruitment of nuclear factors to productively replicating herpes simplex virus genomes. PLoS Pathog. 2015, 11, e1004939. [Google Scholar] [CrossRef]
- Wang, Y.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) signals to mitochondrial AIF: A key event in parthanatos. Exp. Neurol 2009, 218, 193–202. [Google Scholar] [CrossRef]
- Burns, D.M.; Ying, W.; Kauppinen, T.M.; Zhu, K.; Swanson, R.A. Selective down-regulation of nuclear poly(ADP-ribose) glycohydrolase. PLoS ONE 2009, 4, e4896. [Google Scholar] [CrossRef]
- Matthias, K.; Domagoj, V. Cell death pathways: Intricate connections and disease implications. EMBO J. 2021, 40, e106700. [Google Scholar] [CrossRef]
- Costa, H.; Nascimento, R.; Sinclair, J.; Parkhouse, R.M. Human cytomegalovirus gene UL76 induces IL-8 expression through activation of the DNA damage response. PLoS Pathog. 2013, 9, e1003609. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef]
- Ryckman, B.J.; Jarvis, M.A.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 2006, 80, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Albright, E.R.; Mickelson, C.K.; Kalejta, R.F. Human Cytomegalovirus UL138 Protein Inhibits the STING Pathway and Reduces Interferon Beta mRNA Accumulation during Lytic and Latent Infections. mBio 2021, 12, e02267-21. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | Forward | Reverse |
|---|---|---|
| SFB-PARP1 | acgcGTCGACggATGGCGGAGTCTTCGGATAAGC | ggaAGATCTTTACCACAGGGAGGTCTTAAAATTG |
| SFB-PARP2 | ataGGCGCGCCTATGGCGGCGCGGCG | acgcGTCGACTCACCACAGCTGAAGGAAATTAAACTG |
| GFP-PARP1 | acgcGTCGACggATGGCGGAGTCTTCGGATAAGC | ccgCTCGAGTTACCACAGGGAGGTCTTAAAATTG |
| pET30-PARP1 | actGAGCTCcgATGGCGGAGTCTTCGGATAAGC | ccgCTCGAG CCACAGGGAGGTCTTAAAATTG |
| SFB-UL76 | cgGAATTCATGCCGTCCGGGCGT | cgGGATCCCTATAAAGACCGTGTGGGAC |
| myc-UL76 | cgGAATTCggATGCCGTCCGGGCGT | cgGGTACCCTATAAAGACCGTGTGGGAC |
| GFP-UL76 | cccAAGCTTcgATGCCGTCCGGGCGT | cgGGATCCCTATAAAGACCGTGTGG |
| SFB-UL24 | ttGGCGCGCCTATGGCCGCGAGAACG | acgcGTCGACTCATTCGGAGGCGGCT |
| SFB-ORF20 | ttGGCGCGCCTATGGTACGTCCAACCG | acgcGTCGACTCATGGACCTGAACAAGC |
| GFP-IE1 | cgGAATTCtATGGAGTCCTCTGCCA | cgGGATCCTTACTGGTCAGCCTTGCTTC |
| GFP-pp65 | ccgCTCGAGctATGGAGTCGCGCGGTC | cgGGATCCTCAACCTCGGTGCTTTTTGG |
| GFP-UL24 | cccAAGCTTcgATGGCCGCGAGAACG | cgGGATCCTCATTCGGAGGCGGCT |
| GFP-ORF20 | cccAAGCTTcgATGGTACGTCCAACCG | cgGGATCCTCATGGACCTGAACAAGC |
| pGEX4T-UL76 | cgGGATCCATGCCGTCCGGGCGT | cgCTCGAGCTATAAAGACCGTGTGGGAC |
| PARP1 E988K | ATCTGTTACATGAACAATATATCATCTCTCC | TAGACAATGTACTTGTTATATAGTAGAGAGG |
| SFB-PARP1 DN | acgcGTCGACggGTGGATGAAGTGGCGAA | ggaAGATCTTTACCACAGGGAGGTCTTAAAATTG |
| SFB-PARP1 DM | F1:acgcGTCGACggATGGCGGAGTCTTCGGATAAGC R1:GGCCACAACTTCAACAGGTCCATCCACCTCATCGC | F2:GGCGATGAGGTGGATGGACCTGTTGAAGTTGTGG R2:ggaAGATCTTTACCACAGGGAGGTCTTAAAATTG |
| SFB-PARP1 DW | F1:acgcGTCGACggATGGCGGAGTCTTCGGATAAGC R1:CACTGCCTCTTCATCCTGGTTGATACCTTCCTCCTTG | F2:TCAAGGAGGAAGGTATCAACCAGGATGAAGAGGCAG R2:ggaAGATCTTTACCACAGGGAGGTCTTAAAATTG |
| SFB-PARP1 DC | acgcGTCGACggATGGCGGAGTCTTCGGATAAGC | ggaAGATCTTCAGCCAGGATTTACTGTCAGC |
| SFB-PARP1 DZ | F1:acgcGTCGACggATGGCGGAGTCTTCGGATAAGC R1:CACAGCAGCAGGAGCCGATCCATCCACCTCATCGC | F2:CGATGAGGTGGATGGATCGGCTCCTGCTGCT R2:ggaAGATCTTTACCACAGGGAGGTCTTAAAATTG |
| SFB-PARP1 DB | F1:acgcGTCGACggATGGCGGAGTCTTCGGATAAGC R1:GGCCACAACTTCAACAGGGGCTGTGGAGGGCGGA | F2:CCTCCGCCCTCCACAGCCCCTGTTGAAGTTGTGGCCC R2:ggaAGATCTTTACCACAGGGAGGTCTTAAAATTG |
| GFP-UL76 1-150 | cccAAGCTTcgATGCCGTCCGGGCGT | cgGGATCCCTAGACTTGACCGCCACCG |
| GFP-UL76 1-200 | cccAAGCTTcgATGCCGTCCGGGCGT | cgGGATCCCTAAAGGTGTGCAACAGACTCATC |
| GFP-UL76 1-250 | cccAAGCTTcgATGCCGTCCGGGCGT | cgGGATCCCTACGCACCCGGTCCATGA |
| GFP-UL76 1-300 | cccAAGCTTcgATGCCGTCCGGGCGT | cgGGATCCCTATGTAGCAGCGTCCGCG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Guo, J.; Chen, Q. Role of PARP-1 in Human Cytomegalovirus Infection and Functional Partners Encoded by This Virus. Viruses 2022, 14, 2049. https://doi.org/10.3390/v14092049
Zhang W, Guo J, Chen Q. Role of PARP-1 in Human Cytomegalovirus Infection and Functional Partners Encoded by This Virus. Viruses. 2022; 14(9):2049. https://doi.org/10.3390/v14092049
Chicago/Turabian StyleZhang, Wenchang, Jing Guo, and Qiang Chen. 2022. "Role of PARP-1 in Human Cytomegalovirus Infection and Functional Partners Encoded by This Virus" Viruses 14, no. 9: 2049. https://doi.org/10.3390/v14092049