
Viral Metagenomics Reveals Diverse Viruses in Tissue Samples of Diseased Pigs
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. Viral Nucleic Acid Extraction
2.3. Library Construction and Bioinformatics Analysis
2.4. Phylogenetic Analysis
2.5. Nucleotide Sequence Accession Number
3. Results
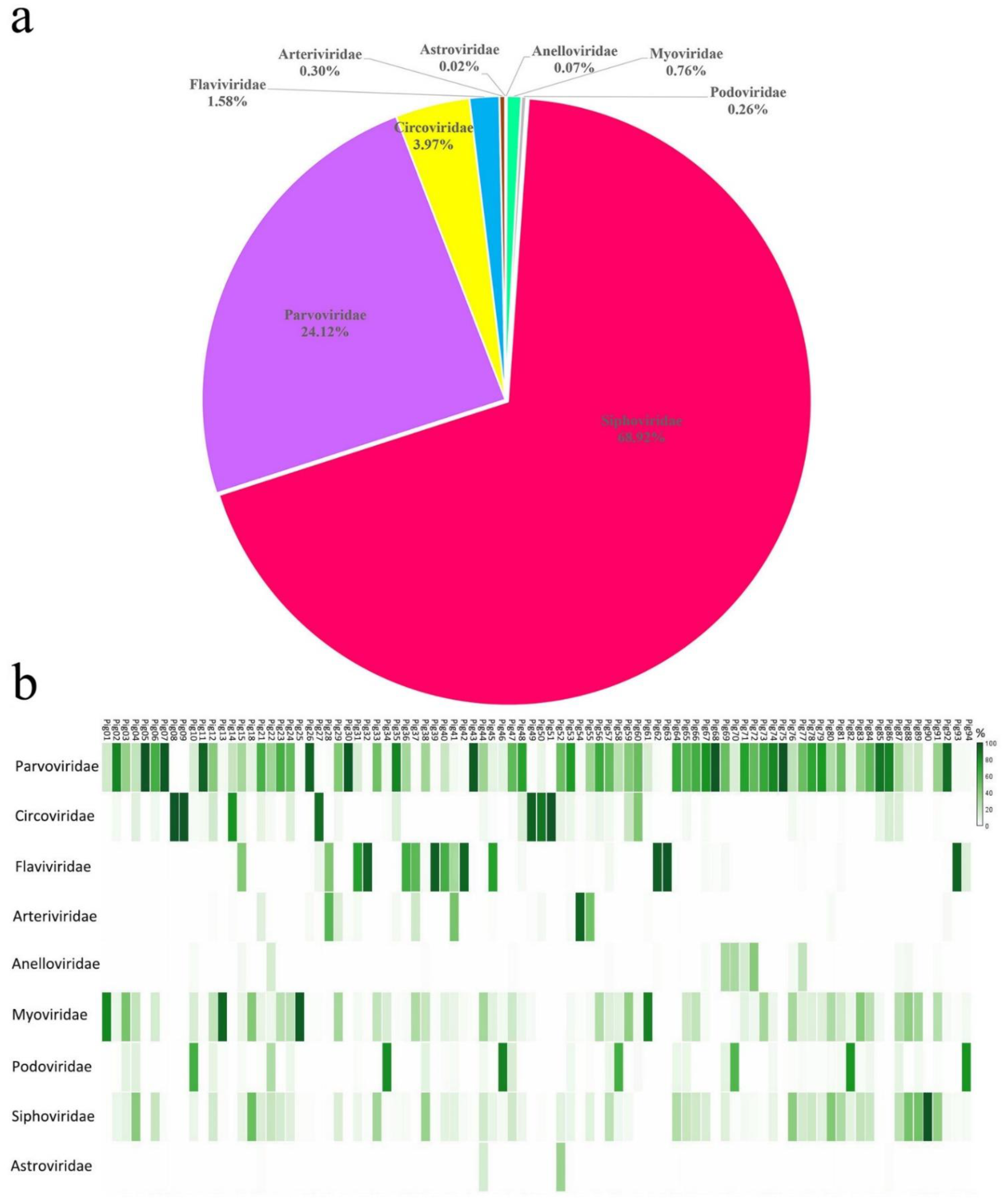
3.1. Viral Metagenomic Overview
3.2. Classical Swine Fever Virus
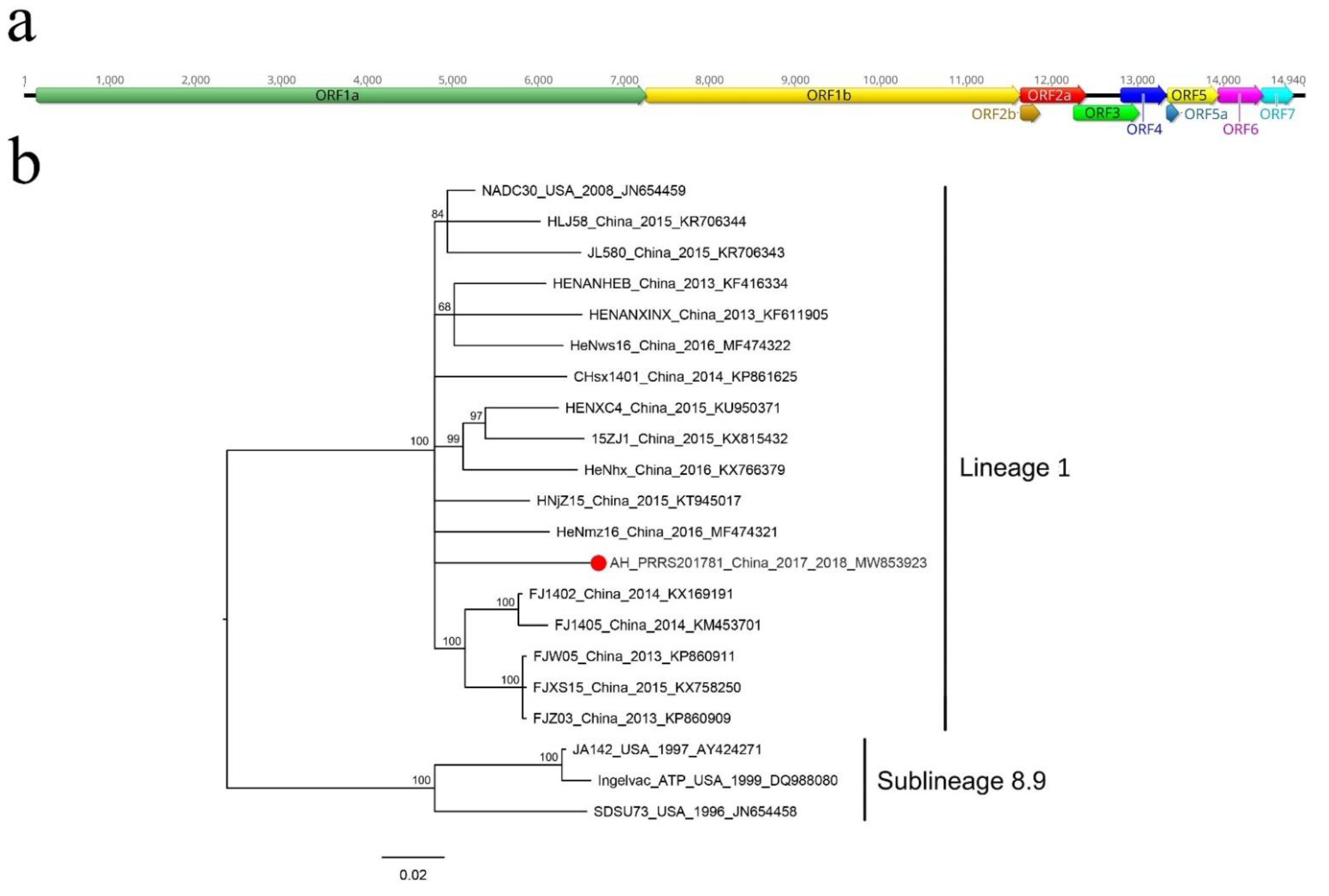
3.3. Porcine Reproductive and Respiratory Syndrome Virus
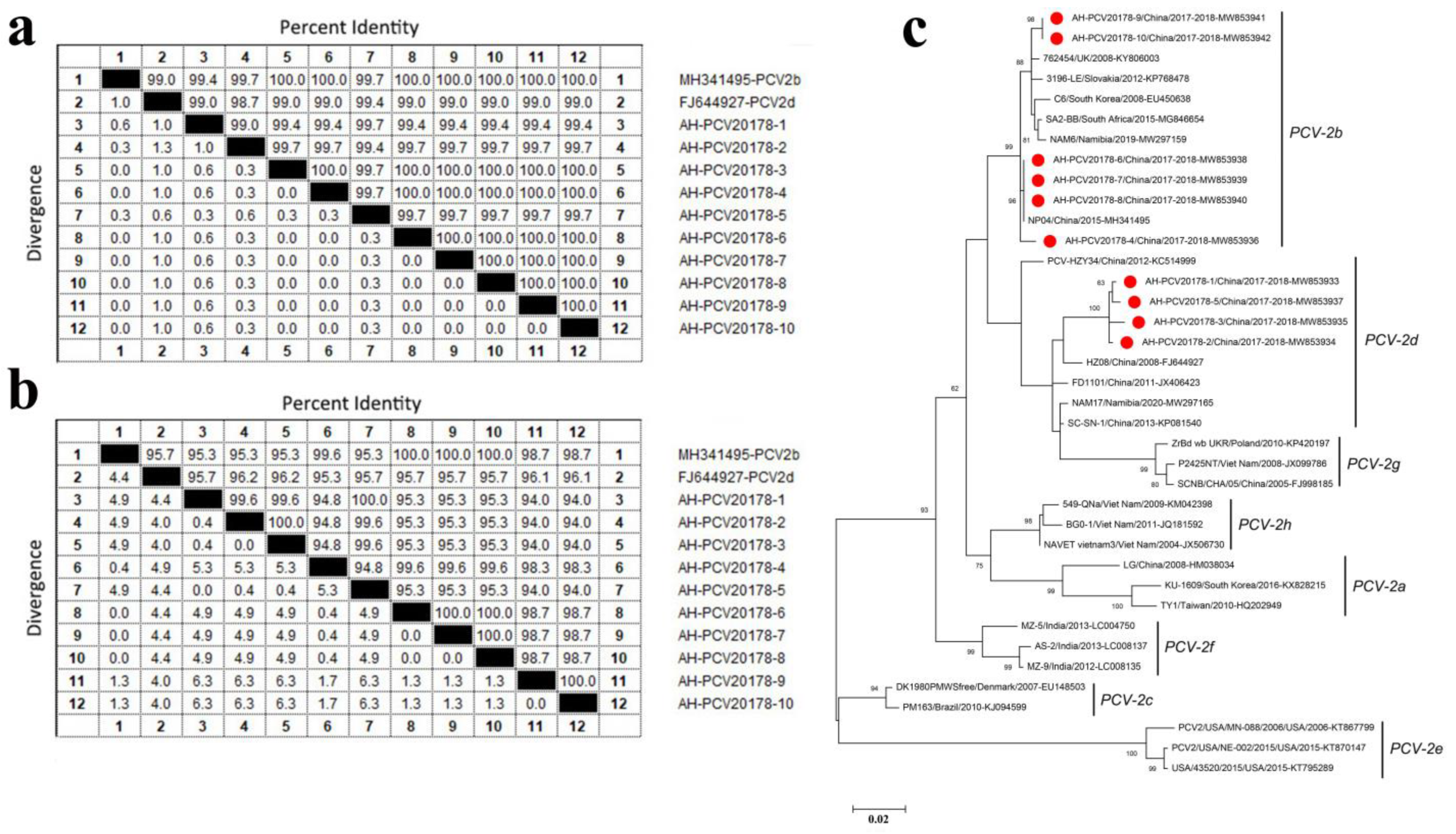
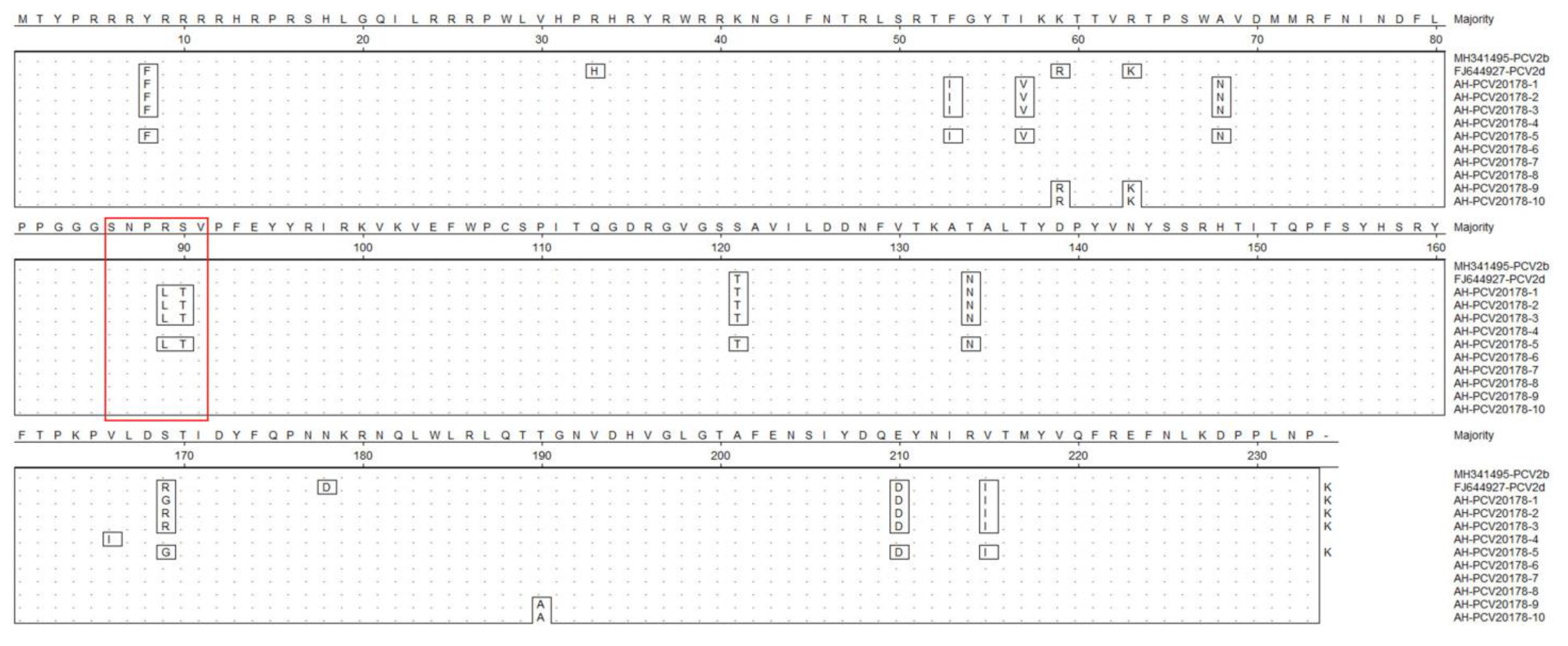
3.4. Porcine Circovirus Type 2
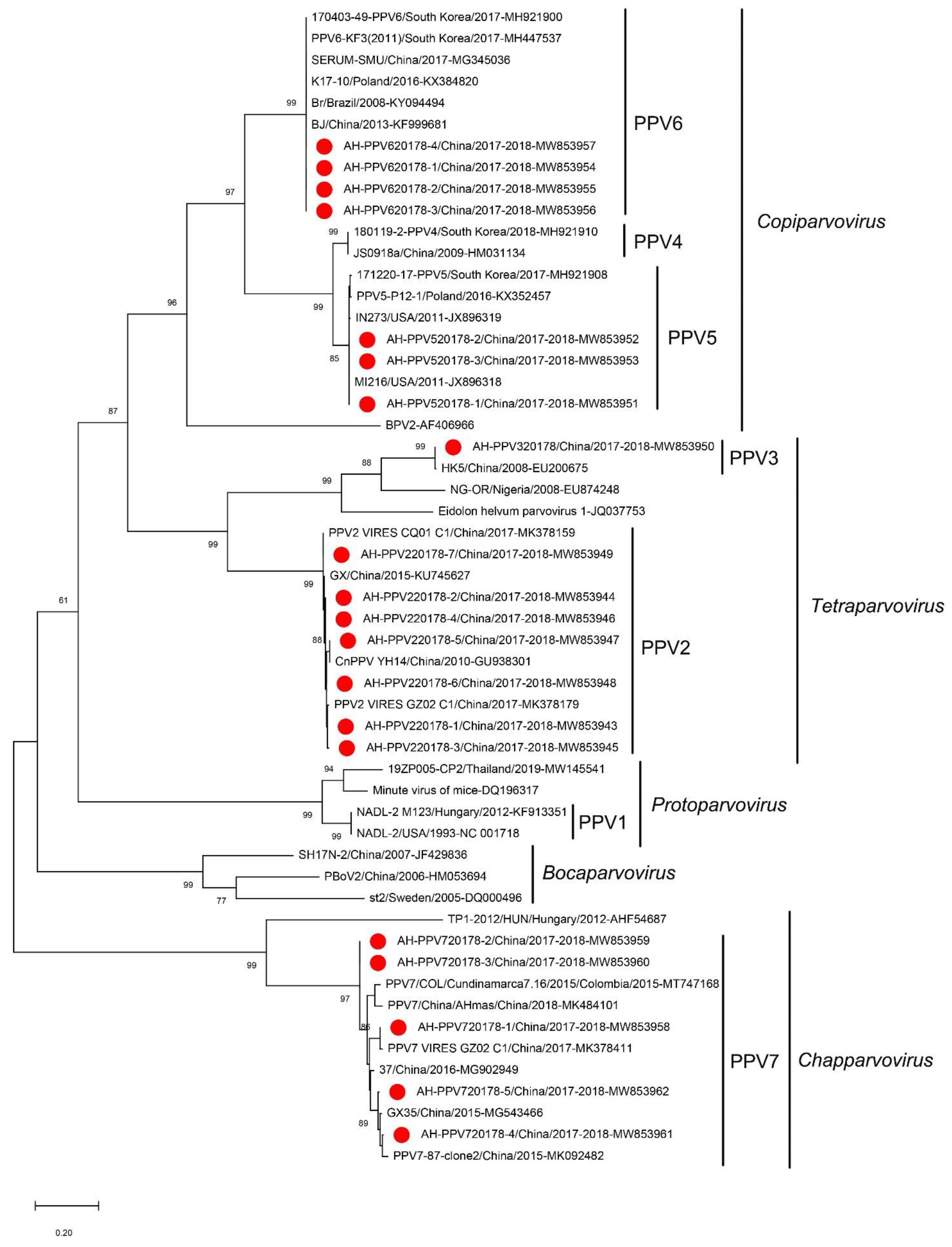
3.5. Porcine Parvovirus
3.6. Other Viruses That Only Obtained Short Fragments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fatima, M.; Luo, Y.; Zhang, L.; Wang, P.-Y.; Song, H.; Fu, Y.; Li, Y.; Sun, Y.; Li, S.; Bao, Y.-J.; et al. Genotyping and Molecular Characterization of Classical Swine Fever Virus Isolated in China during 2016–2018. Viruses 2021, 13, 664. [Google Scholar] [CrossRef] [PubMed]
- Ma, W. Swine Influenza Virus: Current Status and Challenge. Virus Res. 2020, 288, 198118. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Biernacka, K.; Fan, J.; Gerber, P.F.; Stadejek, T.; Opriessnig, T. Circulation of Porcine Parvovirus Types 1 through 6 in Serum Samples Obtained from Six Commercial Polish Pig Farms. Transbound. Emerg. Dis. 2017, 64, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.J.; Halbur, P.G.; Shapiro, M.S.; Govindarajan, S.; Bruna, J.D.; Mushahwar, I.K.; Purcell, R.H.; Emerson, S.U. Genetic and Experimental Evidence for Cross-Species Infection by Swine Hepatitis E Virus. J. Virol. 1998, 72, 9714–9721. [Google Scholar] [CrossRef]
- Chua, K.B.; Bellini, W.J.; Rota, P.A.; Harcourt, B.H.; Tamin, A.; Lam, S.K.; Ksiazek, T.G.; Rollin, P.E.; Zaki, S.R.; Shieh, W.; et al. Nipah Virus: A Recently Emergent Deadly Paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and Pandemic Potential of Swine-Origin H1N1 Influenza Virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef]
- Ricklin, M.E.; García-Nicolás, O.; Brechbühl, D.; Python, S.; Zumkehr, B.; Nougairede, A.; Charrel, R.N.; Posthaus, H.; Oevermann, A.; Summerfield, A. Vector-Free Transmission and Persistence of Japanese Encephalitis Virus in Pigs. Nat. Commun. 2016, 7, 10832. [Google Scholar] [CrossRef]
- Shan, T.; Li, L.; Simmonds, P.; Wang, C.; Moeser, A.; Delwart, E. The Fecal Virome of Pigs on a High-Density Farm. J. Virol. 2011, 85, 11697–11708. [Google Scholar] [CrossRef]
- Blomström, A.-L.; Ye, X.; Fossum, C.; Wallgren, P.; Berg, M. Characterisation of the Virome of Tonsils from Conventional Pigs and from Specific Pathogen-Free Pigs. Viruses 2018, 10, 382. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read de Novo Assembler. Gigascience 2012, 1, 2047-217X. [Google Scholar] [CrossRef]
- Aiyar, A. The Use of CLUSTAL W and CLUSTAL X for Multiple Sequence Alignment. Methods Mol. Biol. 2000, 132, 221–241. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Sepúlveda-Robles, O.; Kameyama, L.; Guarneros, G. High Diversity and Novel Species of Pseudomonas Aeruginosa Bacteriophages. Appl. Environ. Microbiol. 2012, 78, 4510–4515. [Google Scholar] [CrossRef]
- Belák, S.; Karlsson, O.E.; Blomström, A.-L.; Berg, M.; Granberg, F. New Viruses in Veterinary Medicine, Detected by Metagenomic Approaches. Vet. Microbiol. 2013, 165, 95–101. [Google Scholar] [CrossRef]
- Zhang, B.; Tang, C.; Yue, H.; Ren, Y.; Song, Z. Viral Metagenomics Analysis Demonstrates the Diversity of Viral Flora in Piglet Diarrhoeic Faeces in China. J. Gen. Virol. 2014, 95, 1603–1611. [Google Scholar] [CrossRef]
- Franzo, G.; Kekarainen, T.; Llorens, A.; Correa-Fiz, F.; Segalés, J. Exploratory Metagenomic Analyses of Periweaning Failure-to-Thrive Syndrome-Affected Pigs. Vet. Rec. 2019, 184, 25. [Google Scholar] [CrossRef]
- Ganges, L.; Crooke, H.R.; Bohórquez, J.A.; Postel, A.; Sakoda, Y.; Becher, P.; Ruggli, N. Classical Swine Fever Virus: The Past, Present and Future. Virus Res. 2020, 289, 198151. [Google Scholar] [CrossRef]
- Leng, C.; Zhang, H.; Kan, Y.; Yao, L.; Li, M.; Zhai, H.; Li, Z.; Liu, C.; Shi, H.; Ji, J.; et al. Characterisation of Newly Emerged Isolates of Classical Swine Fever Virus in China, 2014–2015. J. Vet. Res. 2017, 61, 1–9. [Google Scholar] [CrossRef]
- Neumann, E.J.; Kliebenstein, J.B.; Johnson, C.D.; Mabry, J.W.; Bush, E.J.; Seitzinger, A.H.; Green, A.L.; Zimmerman, J.J. Assessment of the Economic Impact of Porcine Reproductive and Respiratory Syndrome on Swine Production in the United States. J. Am. Vet. Med. Assoc. 2005, 227, 385–392. [Google Scholar] [CrossRef]
- Firth, A.E.; Zevenhoven-Dobbe, J.C.; Wills, N.M.; Go, Y.Y.; Balasuriya, U.B.R.; Atkins, J.F.; Snijder, E.J.; Posthuma, C.C. Discovery of a Small Arterivirus Gene That Overlaps the GP5 Coding Sequence and Is Important for Virus Production. J. Gen. Virol. 2011, 92, 1097–1106. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, X.X.; Li, R.; Qiao, S.; Zhang, G. The Prevalent Status and Genetic Diversity of Porcine Reproductive and Respiratory Syndrome Virus in China: A Molecular Epidemiological Perspective. Virol. J. 2018, 15, 2. [Google Scholar] [CrossRef]
- Wu, Q.; Li, Z.; Zhang, G.; Niu, J.; Zeng, X.; Sun, B.; Ma, J. Genetic Diversity and Phylogenetic Analysis of Porcine Reproductive and Respiratory Syndrome Virus in Southern China from 2007 to 2014. J. Vet. Sci. 2017, 18, 317–326. [Google Scholar] [CrossRef]
- Tian, K. NADC30-Like Porcine Reproductive and Respiratory Syndrome in China. Open Virol. J. 2017, 11, 59–65. [Google Scholar] [CrossRef]
- Fang, Y.; Schneider, P.; Zhang, W.P.; Faaberg, K.S.; Nelson, E.A.; Rowland, R.R.R. Diversity and Evolution of a Newly Emerged North American Type 1 Porcine Arterivirus: Analysis of Isolates Collected between 1999 and 2004. Arch. Virol. 2007, 152, 1009–1017. [Google Scholar] [CrossRef]
- Zhou, Z.; Wu, J.; Zhang, S.; Hou, B.; Han, T.; Wang, J.; Xu, Q.; Wang, D.; Liu, Y.; Xin, S.; et al. Analysis of Genetic Variation of Two NADC30-like Strains of Porcine Reproductive and Respiratory Syndrome Virus in China. Open Virol. J. 2017, 11, 90–97. [Google Scholar] [CrossRef]
- Opriessnig, T.; Karuppannan, A.K.; Castro, A.M.M.G.; Xiao, C.-T. Porcine Circoviruses: Current Status, Knowledge Gaps and Challenges. Virus Res. 2020, 286, 198044. [Google Scholar] [CrossRef]
- Da Silva, M.S.; Budaszewski, R.F.; Weber, M.N.; Cibulski, S.P.; Paim, W.P.; Mósena, A.C.S.; Canova, R.; Varela, A.P.M.; Mayer, F.Q.; Pereira, C.W.; et al. Liver Virome of Healthy Pigs Reveals Diverse Small SsDNA Viral Genomes. Infect. Genet. Evol. 2020, 81, 104203. [Google Scholar] [CrossRef]
- Liu, Y.; Gong, Q.-L.; Nie, L.-B.; Wang, Q.; Ge, G.-Y.; Li, D.-L.; Ma, B.-Y.; Sheng, C.-Y.; Su, N.; Zong, Y.; et al. Prevalence of Porcine Circovirus 2 throughout China in 2015-2019: A Systematic Review and Meta-Analysis. Microb. Pathog. 2020, 149, 104490. [Google Scholar] [CrossRef]
- He, W.; Zhao, J.; Xing, G.; Li, G.; Wang, R.; Wang, Z.; Zhang, C.; Franzo, G.; Su, S.; Zhou, J. Genetic Analysis and Evolutionary Changes of Porcine Circovirus 2. Mol. Phylogenet. Evol. 2019, 139, 106520. [Google Scholar] [CrossRef]
- Allemandou, A.; Grasland, B.; Hernandez-Nignol, A.-C.; Kéranflec’h, A.; Cariolet, R.; Jestin, A. Modification of PCV-2 Virulence by Substitution of the Genogroup Motif of the Capsid Protein. Vet. Res. 2011, 42, 54. [Google Scholar] [CrossRef] [Green Version]
- Monger, V.R.; Loeffen, W.L.A.; Kus, K.; Stegeman, J.A.; Dukpa, K.; Szymanek, K.; Podgórska, K. Genetic Characterization of Porcine Circovirus Type 2 (PCV2) in Pigs of Bhutan. Transbound. Emerg. Dis. 2017, 64, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Miłek, D.; Woźniak, A.; Guzowska, M.; Stadejek, T. Detection Patterns of Porcine Parvovirus (PPV) and Novel Porcine Parvoviruses 2 through 6 (PPV2-PPV6) in Polish Swine Farms. Viruses 2019, 11, 474. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiao, Y.; Qiu, M.; Li, X.; Li, S.; Lin, H.; Li, X.; Zhu, J.; Chen, N. A Systematic Investigation Unveils High Coinfection Status of Porcine Parvovirus Types 1 through 7 in China from 2016 to 2020. Microbiol. Spectr. 2021, 9, e0129421. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Miller, S.A. Clinical Metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Holmes, E.C. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 2020, 181, 223–227. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Zhang, D.; Ji, Z.; Zhang, Y.; Wang, Y.; Chen, X.; He, Y.; Lu, X.; Li, R.; Guo, Y.; et al. Viral Metagenomics Reveals Diverse Viruses in Tissue Samples of Diseased Pigs. Viruses 2022, 14, 2048. https://doi.org/10.3390/v14092048
Yang S, Zhang D, Ji Z, Zhang Y, Wang Y, Chen X, He Y, Lu X, Li R, Guo Y, et al. Viral Metagenomics Reveals Diverse Viruses in Tissue Samples of Diseased Pigs. Viruses. 2022; 14(9):2048. https://doi.org/10.3390/v14092048
Chicago/Turabian StyleYang, Shixing, Dianqi Zhang, Zexuan Ji, Yuyang Zhang, Yan Wang, Xu Chen, Yumin He, Xiang Lu, Rong Li, Yufei Guo, and et al. 2022. "Viral Metagenomics Reveals Diverse Viruses in Tissue Samples of Diseased Pigs" Viruses 14, no. 9: 2048. https://doi.org/10.3390/v14092048