
Different Mutation Tolerance of Lentiviral (HIV-1) and Deltaretroviral (BLV and HTLV) Protease Precursors


Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression Constructs and Mutagenesis
2.2. Transformation and Expression of MBP-Fused Proteases
2.3. Expression and Purification of the Cleavage Site Mutant BLV PR
2.4. Western Blot
2.5. Enzyme Assay Using Oligopeptide Substrate
3. Results and Discussion
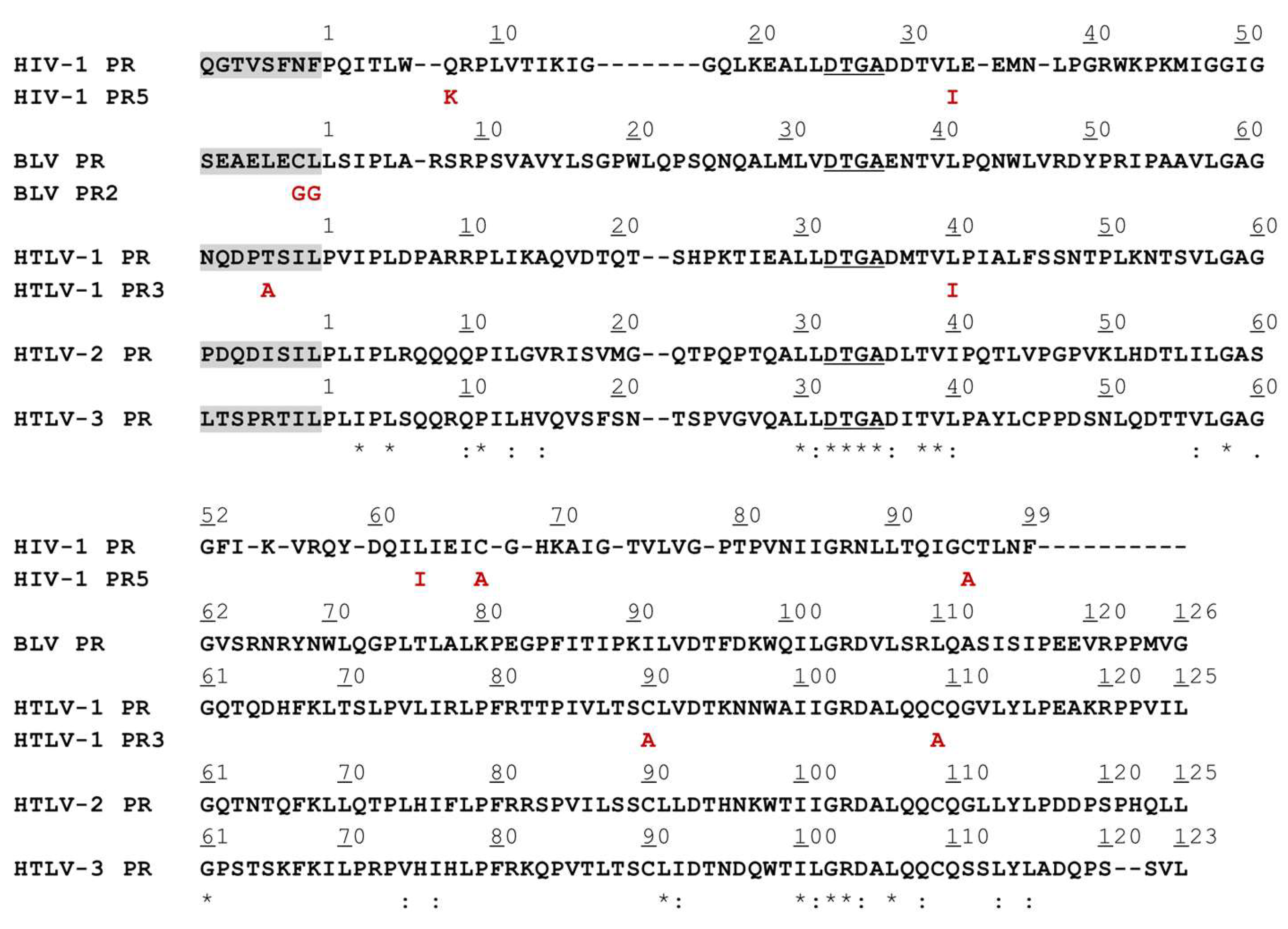
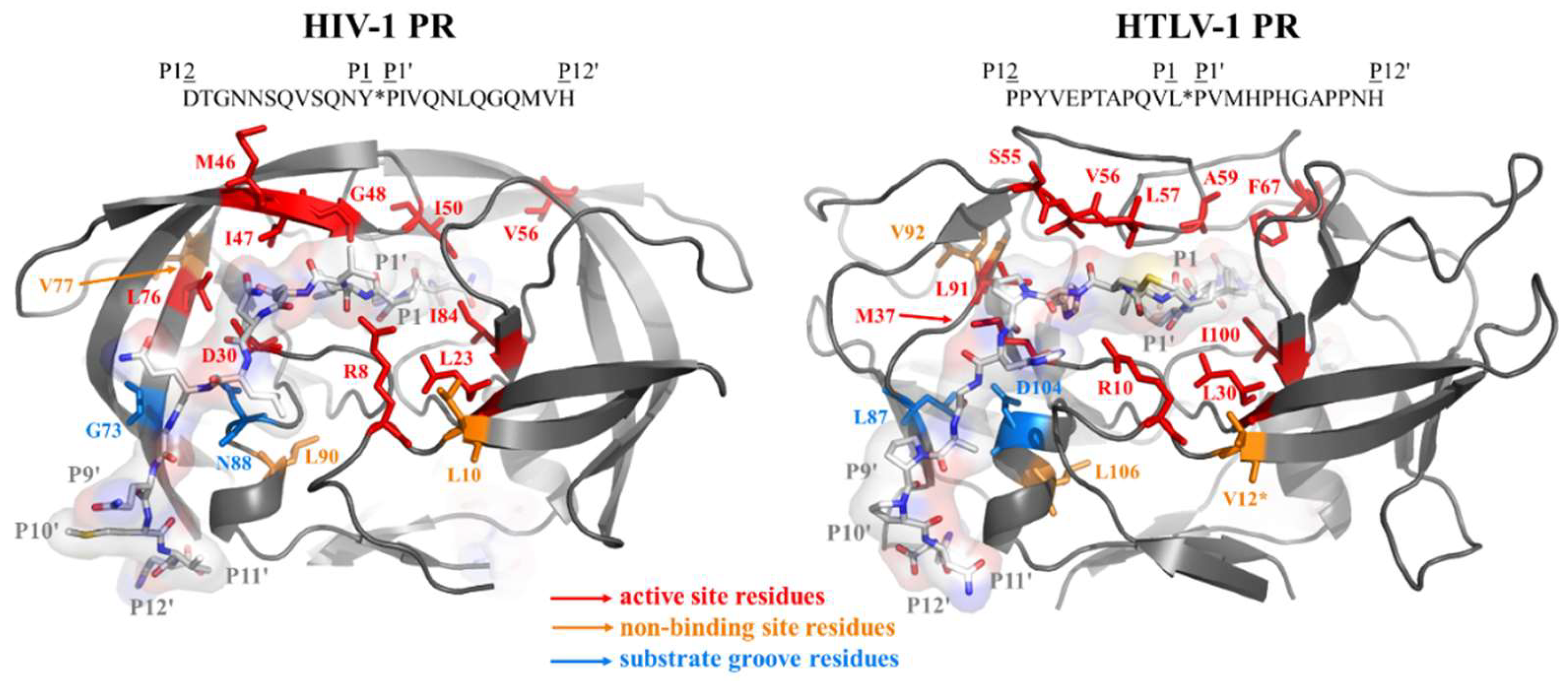
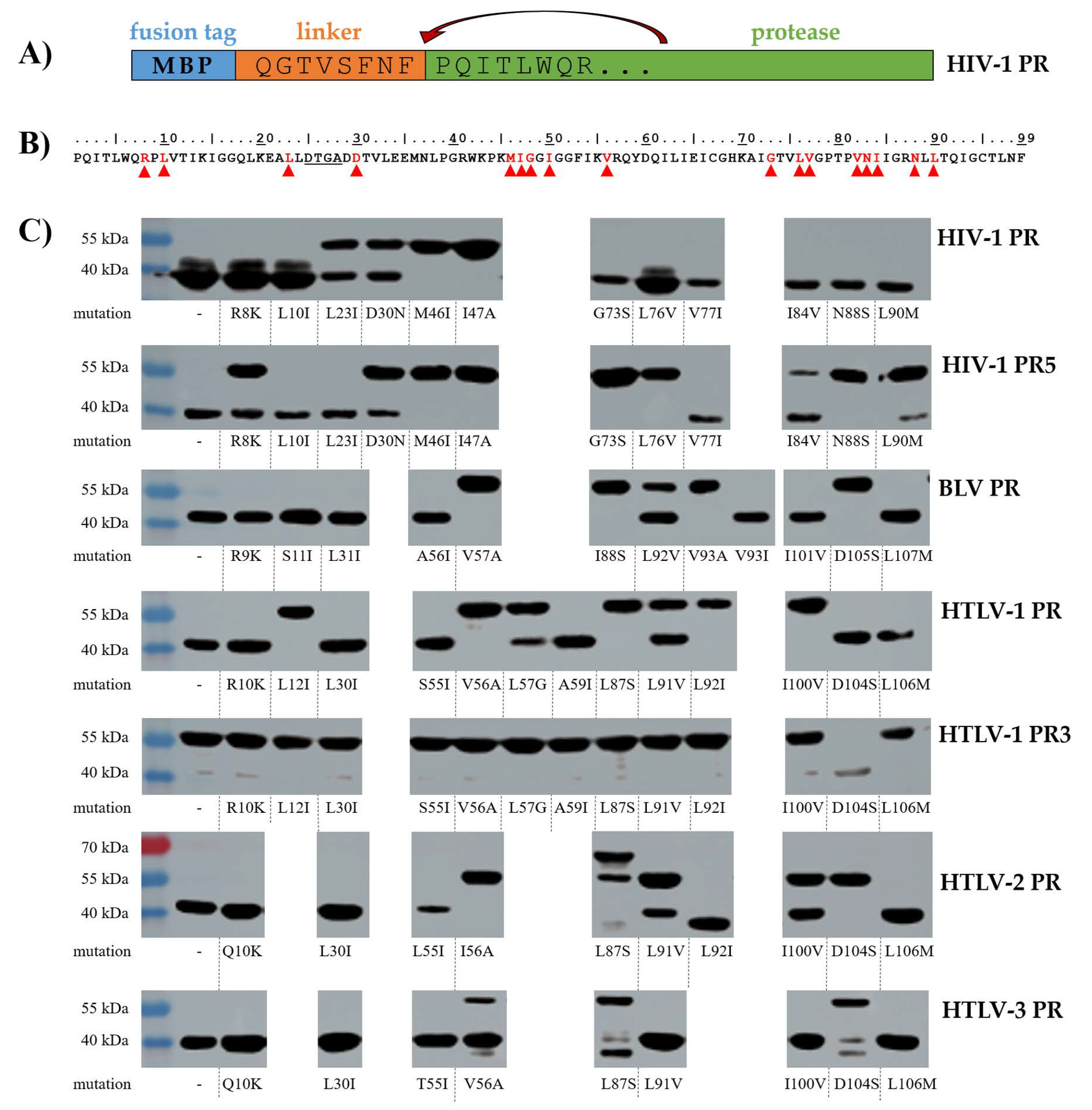
3.1. Mutation Design
3.2. Studies on Autoproteolysis
3.2.1. HIV-1 PR
3.2.2. HIV-1 PR5
3.2.3. BLV PR
3.2.4. HTLV-1 PR
3.2.5. HTLV-1 PR3
3.2.6. HTLV-2 PR
3.2.7. HTLV-3 PR
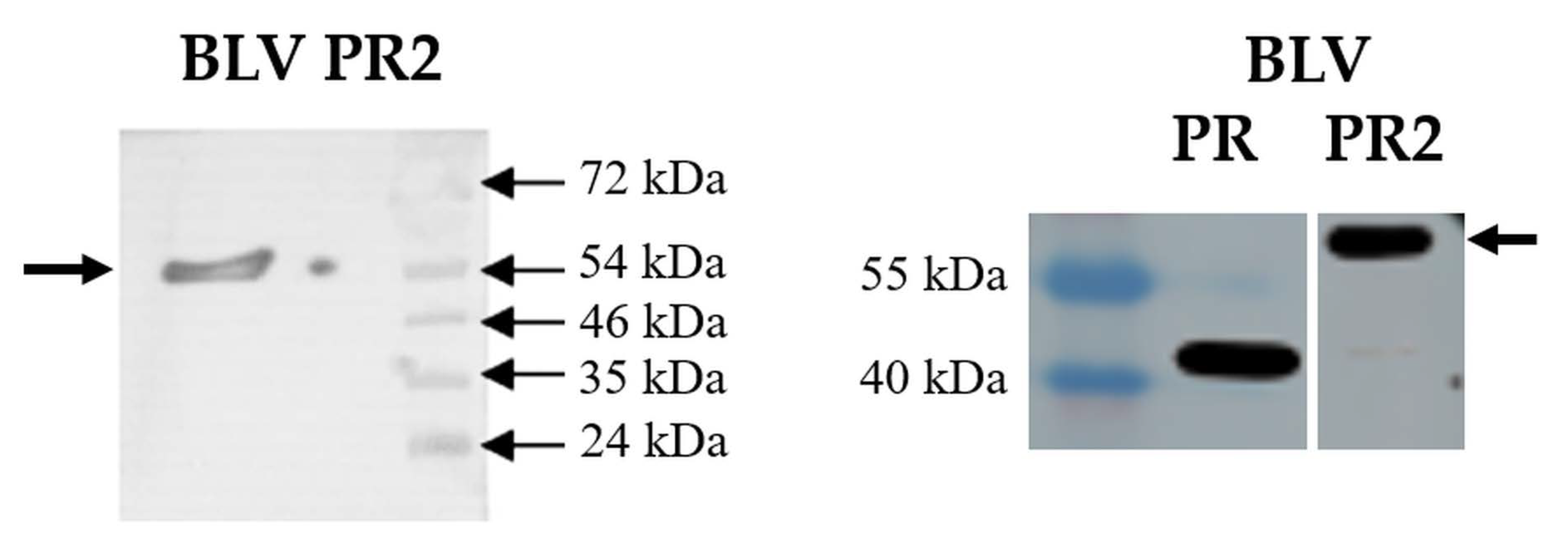
3.3. Autoproteolysis, Kinetic Analysis and Inhibition of BLV PR2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. (Eds.) Retroviruses; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. Available online: https://www.ncbi.nlm.nih.gov/books/NBK19376/ (accessed on 10 April 2022).
- Ghysdael, J.; Bruck, C.; Kettmann, R.; Burny, A. Bovine leukemia virus. Curr. Top. Microbiol. Immunol. 1984, 112, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Olaya-Galán, N.N.; Corredor-Figueroa, A.P.; Guzmán-Garzón, T.C.; Ríos-Hernandez, K.S.; Salas-Cárdenas, S.P.; Patarroyo, M.A.; Gutierrez, M.F. Bovine leukaemia virus DNA in fresh milk and raw beef for human consumption. Epidemiol. Infect. 2017, 145, 3125–3130. [Google Scholar] [CrossRef] [PubMed]
- Olaya-Galán, N.N.; Blume, S.; Tong, K.; Shen, H.; Gutierrez, M.F.; Buehring, G.C. In vitro Susceptibility of Human Cell Lines Infection by Bovine Leukemia Virus. Front. Microbiol. 2022, 13, 793348. [Google Scholar] [CrossRef] [PubMed]
- Afzal, S.; Fiaz, K.; Noor, A.; Sindhu, A.S.; Hanif, A.; Bibi, A.; Asad, M.; Nawaz, S.; Zafar, S.; Ayub, S.; et al. Interrelated Oncogenic Viruses and Breast Cancer. Front. Mol. Biosci. 2022, 9, 781111. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS Data 2021. Available online: https://www.unaids.org/en/resources/documents/2021/2021_unaids_data (accessed on 30 May 2022).
- ECDC. Geographical Distribution of Areas with a High Prevalence of HTLV-1 Infection. ECDC: Stockholm, Sweden. Available online: www.ecdc.europa.eu/en/publications-data/geographical-distribution-areas-high-prevalence-htlv-1-infection (accessed on 30 May 2022).
- De Thé, G.; Bomford, R. An HTLV-I vaccine: Why, how, for whom? AIDS Res. Hum. Retrovir. 1993, 9, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Edlich, R.F.; Arnette, J.A.; Williams, F.M. Global epidemic of human T-cell lymphotropic virus type-I (HTLV-I). J. Emerg. Med. 2000, 18, 109–119. [Google Scholar] [CrossRef]
- Gessain, A.; Cassar, O. Epidemiological aspects and world distribution of HTLV-1 infection. Front. Microbiol. 2012, 3, 1–23. [Google Scholar] [CrossRef]
- WHO. Human T-Lymphotropic Virus Type1: Technical Report February World Health Organization; World Health Organization: Geneva, Switzerland; ISBN 978–92–4-002022. Available online: https://www.who.int/publications/i/item/9789240020221 (accessed on 15 April 2022).
- Nicolás, D.; Ambrosioni, J.; Paredes, R.; Marcos, M.Á.; Manzardo, C.; Moreno, A.; Miró, J.M. Infection with human retroviruses other than HIV-1: HIV-2, HTLV-1, HTLV-2, HTLV-3 and HTLV. Expert Rev. Anti. Infect. Ther. 2015, 13, 947–963. [Google Scholar] [CrossRef]
- Murphy, E.L.; Cassar, O.; Gessain, A. Estimating the number of HTLV-2 infected persons in the world. Retrovirology 2015, 12. [Google Scholar] [CrossRef]
- Marawan, M.A.; Alouffi, A.; El Tokhy, S.; Badawy, S.; Shirani, I.; Dawood, A.; Guo, A.; Almutairi, M.M.; Alshammari, F.A.; Selim, A. Bovine Leukaemia Virus: Current Epidemiological Circumstance and Future Prospective. Viruses 2021, 13, 2167. [Google Scholar] [CrossRef]
- Bell, N.M.; Lever, A.M. HIV Gag polyprotein: Processing and early viral particle assembly. Trends Microbiol. 2013, 21, 136–144. [Google Scholar] [CrossRef]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers 2015, 1, 15035. [Google Scholar] [CrossRef]
- Smyth, R.P.; Davenport, M.P.; Mak, J. The origin of genetic diversity in HIV-1. Virus Res. 2012, 169, 415–429. [Google Scholar] [CrossRef]
- Craigie, R.; Bushman, F.D. HIV DNA integration. Cold Spring Harb. Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Genet. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Nejmeddine, M.; Bangham, C.R. The HTLV-1 Virological Synapse. Viruses 2010, 2, 1427–1447. [Google Scholar] [CrossRef]
- Wattel, E.; Vartanian, J.P.; Pannetier, C.; Wain-Hobson, S. Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J. Virol. 1995, 69, 2863–2868. [Google Scholar] [CrossRef]
- Wattel, E.; Cavrois, M.; Gessain, A.; Wain-Hobson, S. Clonal expansion of infected cells: A way of life for HTLV-I. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 13, S92–S99. [Google Scholar] [CrossRef]
- Bagossi, P.; Bander, P.; Bozóki, B.; Tözsér, J. Discovery and significance of new human T-lymphotropic viruses: HTLV-3 and HTLV-4. Expert Rev. Anti-Infect. Ther. 2009, 7, 1235–1249. [Google Scholar] [CrossRef]
- Gessain, A.; Rua, R.; Betsem, E.; Turpin, J.; Mahieux, R. HTLV-3/4 and simian foamy retroviruses in humans: Discovery, epidemiology, cross-species transmission and molecular virology. Virology 2013, 435, 187–199. [Google Scholar] [CrossRef]
- Weber, I.T.; Agniswamy, J. HIV-1 Protease: Structural Perspectives on Drug Resistance. Viruses 2009, 1, 1110–1136. [Google Scholar] [CrossRef]
- Weber, I.T.; Kneller, D.W.; Wong-Sam, A. Highly resistant HIV-1 proteases and strategies for their inhibition. Futur. Med. Chem. 2015, 7, 1023–1038. [Google Scholar] [CrossRef]
- Weber, I.T.; Wang, Y.-F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [Google Scholar] [CrossRef]
- Humphris-Narayanan, E.; Akiva, E.; Varela, R.; Conchúir, S.Ó.; Kortemme, T. Prediction of mutational tolerance in HIV-1 protease and reverse transcriptase using flexible backbone protein design. PLoS Comput. Biol. 2012, 8, e1002639. [Google Scholar] [CrossRef]
- Áy, É.; Müller, V.; Mezei, M.; Pocskay, Á.; Koroknai, A.; Müller, D.; Győri, Z.; Marschalkó, M.; Tóth, B.; Kárpáti, S.; et al. Transmitted drug resistance in newly diagnosed and treatment-naïve HIV type 1-infected patients in Hungary. J. Glob. Antimicrob. Resist. 2020, 20, 124–130. [Google Scholar] [CrossRef]
- Tözsér, J. Comparative studies on retroviral proteases: Substrate specificity. Viruses 2010, 2, 147–165. [Google Scholar] [CrossRef]
- Shuker, S.B.; Mariani, V.L.; Herger, B.E.; Dennison, K.J. Understanding HTLV-I protease. Chem. Biol. 2003, 10, 373–380. [Google Scholar] [CrossRef]
- Sperka, T.; Miklóssy, G.; Tie, Y.; Bagossi, P.; Zahuczky, G.; Boross, P.; Matúz, K.; Harrison, R.W.; Weber, I.T.; Tözsér, J. Bovine leukemia virus protease: Comparison with human T-lymphotropic virus and human immunodeficiency virus proteases. J. Gen. Virol. 2007, 88, 2052–2063. [Google Scholar] [CrossRef]
- Kádas, J.; Weber, I.T.; Bagossi, P.; Miklóssy, G.; Boross, P.; Oroszlan, S.; Tözsér, J. Narrow substrate specificity and sensitivity toward ligand-binding site mutations of human T-cell leukemia virus type 1 protease. J. Biol. Chem. 2004, 279, 27148–27157. [Google Scholar] [CrossRef] [Green Version]
- Kassay, N.; Mótyán, J.A.; Matúz, K.; Golda, M.; Tőzsér, J. Biochemical Characterization, Specificity and Inhibition Studies of HTLV-1, HTLV-2, and HTLV-3 Proteases. Life 2021, 11, 127. [Google Scholar] [CrossRef] [PubMed]
- Louis, J.M.; McDonald, R.A.; Nashed, N.T.; Wondrak, E.M.; Jerina, D.M.; Oroszlan, S.; Mora, P.T. Autoprocessing of the HIV-1 protease using purified wild-type and mutated fusion proteins expressed at high levels in Escherichia coli. Eur. J. Biochem. 1991, 199, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Louis, J.M.; Oroszlan, S.; Tözsér, J. Stabilization from autoproteolysis and kinetic characterization of the human T-cell leukemia virus type 1 proteinase. J. Biol. Chem. 1999, 274, 6660–6666. [Google Scholar] [CrossRef] [PubMed]
- Zahuczky, G.; Boross, P.; Bagossi, P.; Emri, G.; Copeland, T.D.; Oroszlan, S.; Louis, J.M.; Tözsér, J. Cloning of the bovine leukemia virus proteinase in Escherichia coli and comparison of its specificity to that of human T-cell leukemia virus proteinase. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1478, 1–8. [Google Scholar] [CrossRef]
- Kádas, J.; Boross, P.; Weber, I.T.; Bagossi, P.; Matúz, K.; Tözsér, J. C-terminal residues of mature human T-lymphotropic virus type 1 protease are critical for dimerization and catalytic activity. Biochem. J. 2008, 416, 357–364. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef]
- Shafer, R.W. Rationale and uses of a public HIV drug-resistance database. J. Infect. Dis. 2006, 194, S51–S58. [Google Scholar] [CrossRef]
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Günthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D.D. 2019 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2019, 27, 111–121. [Google Scholar]
- Laco, G.S. HIV-1 protease substrate-groove: Role in substrate recognition and inhibitor resistance. Biochimie 2015, 118, 90–103. [Google Scholar] [CrossRef]
- Laco, G.S. Retroviral proteases: Correlating substrate recognition with both selected and native inhibitor resistance. J. Mol. Biochem. 2017, 6, 45–63. [Google Scholar]
- Eizert, H.; Bander, P.; Bagossi, P.; Sperka, T.; Miklóssy, G.; Boross, P.; Weber, I.T.; Tözsér, J. Amino Acid Preferences of Retroviral Proteases for Amino-Terminal Positions in a Type 1 Cleavage Site. J. Virol. 2008, 82, 10111–10117. [Google Scholar] [CrossRef]
- Mótyán, J.A.; Miczi, M.; Tőzsér, J. Dimer Interface Organization is a Main Determinant of Intermonomeric Interactions and Correlates with Evolutionary Relationships of Retroviral and Retroviral-Like Ddi1 and Ddi2 Proteases. Int. J. Mol. Sci. 2020, 21, 1352. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, B.; Louis, J.M.; Reed, C.C.; Adomat, J.M.; Krouse, J.; Wang, Y.F.; Harrison, R.W.; Weber, I.T. Structural and kinetic analysis of drug resistant mutants of HIV-1 protease. Eur. J. Biochem. 1999, 263, 238–245. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Schock, H.B.; Hall, D.; Chen, E.; Kuo, L.C. Three-dimensional structure of a mutant HIV-1 protease displaying cross-resistance to all protease inhibitors in clinical trials. J. Biol. Chem. 1995, 270, 21433–21436. [Google Scholar] [CrossRef]
- Piana, S.; Carloni, P.; Rothlisberger, U. Drug resistance in HIV-1 protease: Flexibility-assisted mechanism of compensatory mutations. Protein Sci. 2002, 11, 2393–2402. [Google Scholar] [CrossRef]
- Zennou, V.; Mammano, F.; Paulous, S.; Mathez, D.; Clavel, F. Loss of viral fitness associated with multiple Gag and Gag-Pol processing defects in human immunodeficiency virus type 1 variants selected for resistance to protease inhibitors in vivo. J. Virol. 1998, 72, 3300–3306. [Google Scholar] [CrossRef]
- Kagan, R.M.; Shenderovich, M.D.; Heseltine, P.N.; Ramnarayan, K. Structural analysis of an HIV-1 protease I47A mutant resistant to the protease inhibitor lopinavir. Protein Sci. 2005, 14, 1870–1878. [Google Scholar] [CrossRef]
- Sasková, K.G.; Kozísek, M.; Lepsík, M.; Brynda, J.; Rezácová, P.; Václavíková, J.; Kagan, R.M.; Machala, L.; Konvalinka, J. Enzymatic and structural analysis of the I47A mutation contributing to the reduced susceptibility to HIV protease inhibitor lopinavir. Protein Sci. 2008, 17, 1555–1564. [Google Scholar] [CrossRef]
- Kozísek, M.; Bray, J.; Rezácová, P.; Sasková, K.; Brynda, J.; Pokorná, J.; Mammano, F.; Rulísek, L.; Konvalinka, J. Molecular analysis of the HIV-1 resistance development: Enzymatic activities, crystal structures, and thermodynamics of nelfinavir-resistant HIV protease mutants. J. Mol. Biol. 2007, 374, 1005–1016. [Google Scholar] [CrossRef]
- Tözsér, J.; Zahuczky, G.; Bagossi, P.; Louis, J.M.; Copeland, T.D.; Oroszlan, S.; Harrison, R.W.; Weber, I.T. Comparison of the substrate specificity of the human T-cell leukemia virus and human immunodeficiency virus proteinases. Eur. J. Biochem. 2000, 267, 6287–6295. [Google Scholar] [CrossRef]
- Bagossi, P.; Kádas, J.; Miklóssy, G.; Boross, P.; Weber, I.T.; Tözsér, J. Development of a microtiter plate fluorescent assay for inhibition studies on the HTLV-1 and HIV-1 proteinases. J. Virol. Methods 2004, 119, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Fehér, A.; Weber, I.T.; Bagossi, P.; Boross, P.; Mahalingam, B.; Louis, J.M.; Copeland, T.D.; Torshin, I.Y.; Harrison, R.W.; Tözsér, J. Effect of sequence polymorphism and drug resistance on two HIV-1 Gag processing sites. Eur. J. Biochem. 2002, 269, 4114–4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miklóssy, G.; Tözsér, J.; Kádas, J.; Ishima, R.; Louis, J.M.; Bagossi, P. Novel macromolecular inhibitors of human immunodeficiency virus-1 protease. Protein Eng. Des. Sel. 2008, 21, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Matúz, K.; Mótyán, J.; Li, M.; Wlodawer, A.; Tőzsér, J. Inhibition of XMRV and HIV-1 proteases by pepstatin A and acetyl-pepstatin. FEBS J. 2012, 279, 3276–3286. [Google Scholar] [CrossRef]
- Tóth, F.; Kádas, J.; Mótyán, J.A.; Tőzsér, J. Effect of internal cleavage site mutations in human immunodeficiency virus type 1 capsid protein on its structure and function. FEBS Open Bio. 2016, 6, 847–859. [Google Scholar] [CrossRef]
- Miczi, M.; Diós, Á.; Bozóki, B.; Tőzsér, J.; Mótyán, J.A. Development of a Bio-Layer Interferometry-Based Protease Assay Using HIV-1 Protease as a Model. Viruses 2021, 13, 1183. [Google Scholar] [CrossRef]
- Bagossi, P.; Sperka, T.; Fehér, A.; Kádas, J.; Zahuczky, G.; Miklóssy, G.; Boross, P.; Tözsér, J. Amino acid preferences for a critical substrate binding subsite of retroviral proteases in type 1 cleavage sites. J. Virol. 2005, 79, 4213–4218. [Google Scholar] [CrossRef]
- Awahara, C.; Oku, D.; Furuta, S.; Kobayashi, K.; Teruya, K.; Akaji, K.; Hattori, Y. The Effects of Side-Chain Configurations of a Retro-Inverso-Type Inhibitor on the Human T-Cell Leukemia Virus (HTLV)-1 Protease. Molecules 2022, 27, 1646. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protease | Plasmid | Cloning Site | Protease Mutation | N-terminal Auto- Proteolytic Sequence | Reference |
|---|---|---|---|---|---|
| BLV PR | pMalc2x | EcoRI/SalI | no | SEAELECL↓LSIPLARS | [38] |
| BLV PR2 | pMalc2 | EcoRI/SalI | no | SEAELEGG↓LSIPLARS | this study |
| HIV-1 PR | pMalc5x | NdeI/BamHI | no | QGTVSFNF↓PQITLWQR | [36] |
| HIV-1 PR5 | pMalc5x | NdeI/BamHI | Q7K, L33I, L63I, C67A, C95A | QGTVSFNF↓PQITLWQR | this study |
| HTLV-1 PR | pMalc2x | EcoRI/BamHI | no | NQDPTSIL↓PVIPLDPA | [34] |
| HTLV_1 PR3 | pMalc5x | NdeI/BamHI | L40I, C90A, C109A | NQDPASIL↓PVIPLDPA | this study |
| HTLV-2 | pMalc2x | EcoRI/BamHI | no | PDQDISIL↓PLIPLRQQ | [35] |
| HTLV-3 | pMalc2x | EcoRI/BamHI | no | LTSPRTIL↓PLIPLSQQ | [35] |
| HIV-1 PR | HIV-1 PR5 | BLV PR | HTLV-1 PR | HTLV-1 PR3 | HTLV-2 PR | HTLV-3 PR | Modified Position |
|---|---|---|---|---|---|---|---|
| wt | wt | wt 1,* | wt 2,* | wt | wt 3,* | wt 3,* | - |
| R8K | R8K | R9K | R10K | R10K | Q10K | Q10K | S3, S1 |
| L10I | L10I | S11I | L12I | L12I | I12 (wt) 3,* | I12 (wt) 3,* | - |
| L23I | L23I | L31I | L30I | L30I | L30I | L30I | S1 |
| D30 (wt) | D30 (wt) | N38D 1 | M37D 2 | n.d. | L37D 3 | I37D 3 | S4, S2 |
| D30N | D30N | N38 (wt) 1,* | M37N 2 | n.d. | L37N 3 | I37N 3 | |
| n.d. | n.d. | n.d. | M37A 2 | n.d. | n.d. | n.d. | |
| M46I | M46I | A56I | S55I | S55I | L55I | T55I | S4 |
| I47A | I47A | V57A | V56A | V56A | I56A | V56A | S4, S2 |
| I47 (wt) | I47 (wt) | V57I 1 | n.d. | n.d. | n.d. | n.d. | |
| G48 (wt) | G48 (wt) | L58G 1 | L57G | L57G | L57G 3 | L57G 3 | S4, S3, S2 |
| I50 (wt) | I50 (wt) | A60I 1 | A59I | A59I | A59I 3 | A59I 3 | S3, S2, S1 |
| V56 (wt) | V56 (wt) | Y68 (wt) 1,* | F67Q 2 | n.d. | F67Q 3 | F67Q 3 | S4 |
| G73S | G73S | I88S | L87S | L87S | L87S | L87S | S-groove |
| L76V | L76V | L92V | L91V | L91V | L91V | L91V | S4 |
| V77I | V77I | V93I | V92I | V92I | L92I | I92 (wt) 3,* | - |
| n.d. | n.d. | V93A | n.d. | n.d. | n.d. | n.d. | |
| P82 (wt) | P82 (wt) | K98P 1 | n.d. | n.d. | n.d. | n.d. | S1 |
| V83 (wt) | V83 (wt) | W99V 1 | n.d. | n.d. | n.d. | n.d. | S1 |
| I84V | I84V | I101V | I100V | I100V | I100V | I100V | S2, S1 |
| N88S | N88S | D105S | D104S | D104S | D104S | D104S | S-groove |
| L90M | L90M | L107M | L106M | L106M | L106M | L106M | - |
| 12 | 12 | 18 | 17 | 13 | 15 | 14 | Total number of mutants |
| 2/12 14.3% | 5/12 41.7% | 7/18 38.9% | 9/17 52.9% | 12/13 92.3% | 6/15 40.0% | 3/14 21.4% | Number and percentage of non-processing mutants |
| KM (mM) | kcat (s−1) | kcat/KM (mM−1s−1) | Folding Efficiency | Ki (nM) | ||
|---|---|---|---|---|---|---|
| BLV | PR | 0.011 1 | 0.27 1 | 24.5 1 | 100% 1 | 13 1 |
| PR-N38D | 0.18 1 | 0.023 1 | 0.13 1 | 21% 1 | - | |
| PR2 | 0.014 ± 0.002 | 0.058 ± 0.002 | 4.14 ± 0.61 | 100% | 3.93 | |
| HIV-1 | PR5 | 0.16 3 | 3.6 3 | 22.5 3 | 100% 3 | 11.215 2 |
| HTLV-1 | PR3 | 0.063 3 | 10.0 3 | 158.7 3 | 100% 3 | 298 2 |
| 0.051 ± 0.005 4 | 7.68±0.17 4 | 150.6 4 | - | - | ||
| HTLV-2 | PR | 0.168 ± 0.078 # | 11.27 ± 1.932 # | 67.2 5 | - | 37 5 |
| HTLV-3 | PR | 0.144 ± 0.066 # | 4.344 ± 0.985 # | 30.1 5 | - | 214 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mótyán, J.A.; Kassay, N.; Matúz, K.; Tőzsér, J. Different Mutation Tolerance of Lentiviral (HIV-1) and Deltaretroviral (BLV and HTLV) Protease Precursors. Viruses 2022, 14, 1888. https://doi.org/10.3390/v14091888
Mótyán JA, Kassay N, Matúz K, Tőzsér J. Different Mutation Tolerance of Lentiviral (HIV-1) and Deltaretroviral (BLV and HTLV) Protease Precursors. Viruses. 2022; 14(9):1888. https://doi.org/10.3390/v14091888
Chicago/Turabian StyleMótyán, János András, Norbert Kassay, Krisztina Matúz, and József Tőzsér. 2022. "Different Mutation Tolerance of Lentiviral (HIV-1) and Deltaretroviral (BLV and HTLV) Protease Precursors" Viruses 14, no. 9: 1888. https://doi.org/10.3390/v14091888