
Complexities of JC Polyomavirus Receptor-Dependent and -Independent Mechanisms of Infection


Abstract
:1. Introduction
2. Progressive Multifocal Leukoencephalopathy
3. JC Polyomavirus Genome Organization
4. Capsid Morphology and Assembly
5. JC Polyomavirus Receptor-Dependent Infection
6. Extracellular Vesicles
7. JC Polyomavirus Receptor-Independent Infection
8. JC Polyomavirus(+) Extracellular Vesicle Dissemination to the Brain Parenchyma
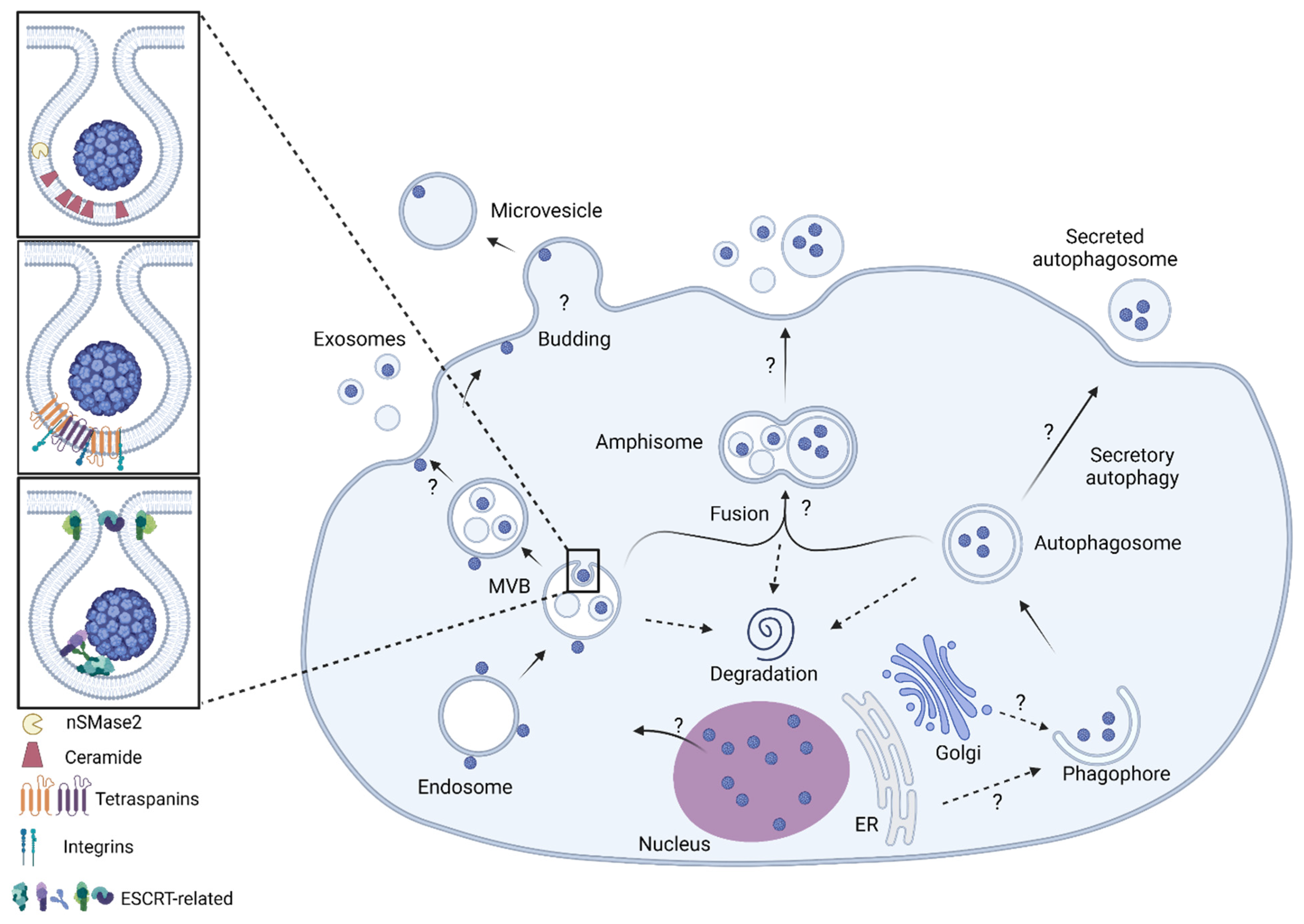
9. Biogenesis of Extracellular Vesicles
10. Virus-EV Biogenesis Pathways
11. JCPyV and EV Purification and Characterization Methods
12. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.W.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Polyomaviridae. J. Gen. Virol. 2017, 98, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Chen, Z.; Bernard, H.U.; Chan, P.K.S.; DeSalle, R.; Dillner, J.; Forslund, O.; Haga, T.; McBride, A.A.; Villa, L.L.; et al. ICTV Virus Taxonomy Profile: Papillomaviridae. J. Gen. Virol. 2018, 99, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.D.; Field, A.M.; Coleman, D.V.; Hulme, B. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet 1971, 19, 1253–1257. [Google Scholar] [CrossRef]
- Padgett, B.L.; Walker, D.L.; ZuRhein, G.M.; Eckroade, R.J.; Dessel, B.H. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet 1971, 19, 1257–1260. [Google Scholar] [CrossRef]
- Boothpur, R.; Brennan, D.C. Human polyoma viruses and disease with emphasis on clinical BK and JC. J. Clin. Virol. 2010, 47, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.S.; Koralnik, I.J. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: Clinical features and pathogenesis. Lancet Neurol. 2010, 9, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 22, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Spurgeon, M.E.; Lambert, P.F. Merkel cell polyomavirus: A newly discovered human virus with oncogenic potential. Virology 2013, 435, 118–130. [Google Scholar] [CrossRef] [Green Version]
- van der Meijden, E.; Janssens, R.W.; Lauber, C.; Bouwes Bavinck, J.N.; Gorbalenya, A.E.; Feltkamp, M.C. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog. 2010, 6, e1001024. [Google Scholar] [CrossRef] [Green Version]
- Kazem, S.; van der Meijden, E.; Kooijman, S.; Rosenberg, A.S.; Hughey, L.C.; Browning, J.C.; Sadler, G.; Busam, K.; Pope, E.; Benoit, T.; et al. Trichodysplasia spinulosa is characterized by active polyomavirus infection. J. Clin. Virol. 2012, 53, 225–230. [Google Scholar]
- Ho, J.; Jedrych, J.J.; Feng, H.; Natalie, A.A.; Grandinetti, L.; Mirvish, E.; Crespo, M.M.; Yadav, D.; Fasanella, K.E.; Proksell, S.; et al. Human polyomavirus 7-associated pruritic rash and viremia in transplant recipients. J. Infect. Dis. 2015, 211, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.D.; Lee, E.E.; Yue, Y.; Stork, J.; Pock, L.; North, J.P.; Vandergriff, T.; Cockerell, C.; Hosler, G.A.; Pastrana, D.V.; et al. Human polyomavirus 6 and 7 are associated with pruritic and dyskeratotic dermatoses. J. Am. Acad. Dermatol. 2017, 76, 932–940.e3. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Chattaraj, S. Entry, infection, replication, and egress of human polyomaviruses: An update. Can. J. Microbiol. 2017, 63, 193–211. [Google Scholar] [CrossRef]
- Handala, L.; Blanchard, E.; Raynal, P.-I.; Roingeard, P.; Morel, V.; Descamps, V.; Castelain, S.; Francois, C.; Duverlie, G.; Brochot, E.; et al. BK Polyomavirus Hijacks Extracellular Vesicles for En Bloc Transmission. J. Virol. 2020, 94, e01834-19. [Google Scholar] [CrossRef] [Green Version]
- Morris-Love, J.; Gee, G.V.; O’Hara, B.A.; Assetta, B.; Atkinson, A.L.; Dugan, A.S.; Haley, S.A.; Atwood, W.J. JC Polyomavirus Uses Extracellular Vesicles To Infect Target Cells. mBio 2019, 10, e00379-19. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, B.A.; Morris-Love, J.; Gee, G.V.; Haley, S.A.; Atwood, W.J. JC Virus infected choroid plexus epithelial cells produce extracellular vesicles that infect glial cells independently of the virus attachment receptor. PLoS Pathog. 2020, 16, e1008371. [Google Scholar] [CrossRef]
- Adang, L.; Berger, J. Progressive Multifocal Leukoencephalopathy. F1000Research 2015, 4, 1424. [Google Scholar] [CrossRef]
- Haley, S.A.; Atwood, W.J. Progressive Multifocal Leukoencephalopathy: Endemic Viruses and Lethal Brain Disease. Annu. Rev. Virol. 2017, 4, 349–367. [Google Scholar] [CrossRef]
- Brooks, B.R.; Walker, D.L. Progressive multifocal leukoencephalopathy. Neurol. Clin. 1984, 2, 299–313. [Google Scholar] [CrossRef]
- Anand, P.; Hotan, G.C.; Vogel, A.; Venna, N.; Mateen, F.J. Progressive multifocal leukoencephalopathy: A 25-year retrospective cohort study. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e618. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.R.; Pall, L.; Lanska, D.; Whiteman, M. Progressive multifocal leukoencephalopathy in patients with HIV infection. J. Neurovirol. 1998, 4, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.L.; Holman, R.C.; Hammett, T.A.; Belay, E.D.; Schonberger, L.B. Progressive multifocal leukoencephalopathy deaths in the USA, 1979-2005. Neuroepidemiology 2010, 35, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Langer-Gould, A.; Atlas, S.W.; Green, A.J.; Bollen, A.W.; Pelletier, D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 2005, 353, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinschmidt-DeMasters, B.K.; Tyler, K.L. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N. Engl. J. Med. 2005, 353, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Van Assche, G.; Van Ranst, M.; Sciot, R.; Dubois, B.; Vermeire, S.; Noman, M.; Verbeeck, J.; Geboes, K.; Rutgeerts, P. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N. Engl. J. Med. 2005, 353, 362–368. [Google Scholar] [CrossRef] [Green Version]
- Chalkley, J.J.; Berger, J.R. Progressive multifocal leukoencephalopathy in multiple sclerosis. Curr. Neurol. Neurosci. Rep. 2013, 13, 408. [Google Scholar] [CrossRef]
- Berger, J.R. Classifying PML risk with disease modifying therapies. Mult. Scler. Relat. Disord. 2017, 12, 59–63. [Google Scholar] [CrossRef]
- Major, E.O.; Nath, A. A link between long-term natalizumab dosing in MS and PML: Putting the puzzle together. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e235. [Google Scholar] [CrossRef] [Green Version]
- Assetta, B.; Atwood, W.J. The biology of JC polyomavirus. Biol. Chem. 2017, 398, 839–855. [Google Scholar] [CrossRef]
- Berger, J.R.; Aksamit, A.J.; Clifford, D.B.; Davis, L.; Koralnik, I.J.; Sejvar, J.J.; Bartt, R.; Major, E.O.; Nath, A. PML diagnostic criteria: Consensus statement from the AAN Neuroinfectious Disease Section. Neurology 2013, 80, 1430–1438. [Google Scholar] [CrossRef] [Green Version]
- White, M.K.; Sariyer, I.K.; Gordon, J.; Delbue, S.; Pietropaolo, V.; Berger, J.R.; Khalili, K. Diagnostic assays for polyomavirus JC and progressive multifocal leukoencephalopathy. Rev. Med. Virol. 2016, 26, 102–114. [Google Scholar] [CrossRef] [Green Version]
- Hammarin, A.L.; Bogdanovic, G.; Svedhem, V.; Pirskanen, R.; Morfeldt, L.; Grandien, M. Analysis of PCR as a tool for detection of JC virus DNA in cerebrospinal fluid for diagnosis of progressive multifocal leukoencephalopathy. J. Clin. Microbiol. 1996, 34, 2929–2932. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.R. The clinical features of PML. Cleve. Clin. J. Med. 2011, 78 (Suppl. S2), S8–S12. [Google Scholar] [CrossRef]
- Williamson, E.M.L.; Berger, J.R. Diagnosis and Treatment of Progressive Multifocal Leukoencephalopathy Associated with Multiple Sclerosis Therapies. Neurotherapeutics 2017, 14, 961–973. [Google Scholar] [CrossRef]
- Tan, K.; Roda, R.; Ostrow, L.; McArthur, J.; Nath, A. PML-IRIS in patients with HIV infection: Clinical manifestations and treatment with steroids. Neurology 2009, 72, 1458–1464. [Google Scholar] [CrossRef]
- Kartau, M.; Sipila, J.O.; Auvinen, E.; Palomaki, M.; Verkkoniemi-Ahola, A. Progressive Multifocal Leukoencephalopathy: Current Insights. Degener. Neurol. Neuro. Dis. 2019, 9, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Fournier, A.; Martin-Blondel, G.; Lechapt-Zalcman, E.; Dina, J.; Kazemi, A.; Verdon, R.; Mortier, E.; De La Blanchardière, A. Immune Reconstitution Inflammatory Syndrome Unmasking or Worsening AIDS-Related Progressive Multifocal Leukoencephalopathy: A Literature Review. Front. Immunol. 2017, 8, 577. [Google Scholar] [CrossRef] [Green Version]
- Ferenczy, M.W.; Marshall, L.J.; Nelson, C.D.; Atwood, W.J.; Nath, A.; Khalili, K.; Major, E.O. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin. Microbiol. Rev. 2012, 25, 471–506. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, A.L.; Atwood, W.J. Fifty Years of JC Polyomavirus: A Brief Overview and Remaining Questions. Viruses 2020, 12, 969. [Google Scholar] [CrossRef]
- Daniel, A.M.; Swenson, J.J.; Mayreddy, R.P.; Khalili, K.; Frisque, R.J. Sequences within the early and late promoters of archetype JC virus restrict viral DNA replication and infectivity. Virology 1996, 216, 90–101. [Google Scholar] [CrossRef]
- Gosert, R.; Kardas, P.; Major, E.O.; Hirsch, H.H. Rearranged JC virus noncoding control regions found in progressive multifocal leukoencephalopathy patient samples increase virus early gene expression and replication rate. J. Virol. 2010, 84, 10448–10456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisque, R.J. Structure and function of JC virus T’ proteins. J. Neurovirol. 2001, 7, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Agostini, H.T.; Ryschkewitsch, C.F.; Singer, E.J.; Stoner, G.L. JC virus regulatory region rearrangements and genotypes in progressive multifocal leukoencephalopathy: Two independent aspects of virus variation. J. Gen. Virol. 1997, 78, 659–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agostini, H.T.; Ryschkewitsch, C.F.; Mory, R.; Singer, E.J.; Stoner, G.L. JC virus (JCV) genotypes in brain tissue from patients with progressive multifocal leukoencephalopathy (PML) and in urine from controls without PML: Increased frequency of JCV type 2 in PML. J. Infect. Dis. 1997, 176, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Yogo, Y.; Zhong, S.; Shibuya, A.; Kitamura, T.; Homma, Y. Transcriptional control region rearrangements associated with the evolution of JC polyomavirus. Virology 2008, 380, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Sugimoto, C.; Kato, A.; Ebihara, H.; Suzuki, M.; Taguchi, F.; Kawabe, K.; Yogo, Y. Persistent JC virus (JCV) infection is demonstrated by continuous shedding of the same JCV strains. J. Clin. Microbiol. 1997, 35, 1255–1257. [Google Scholar] [CrossRef] [Green Version]
- Ikegaya, H.; Iwase, H.; Yogo, Y. Detection of identical JC virus DNA sequences in both human kidneys. Arch. Virol. 2004, 149, 1215–1220. [Google Scholar] [CrossRef]
- Yogo, Y.; Kitamura, T.; Sugimoto, C.; Ueki, T.; Aso, Y.; Hara, K.; Taguchi, F. Isolation of a possible archetypal JC virus DNA sequence from nonimmunocompromised individuals. J. Virol. 1990, 64, 3139–3143. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.T.; Frisque, R.J. Detection of archetype and rearranged variants of JC virus in multiple tissues from a pediatric PML patient. J. Med. Virol. 1997, 52, 243–252. [Google Scholar] [CrossRef]
- Kato, A.; Kitamura, T.; Takasaka, T.; Tominaga, T.; Ishikawa, A.; Zheng, H.Y.; Yogo, Y. Detection of the archetypal regulatory region of JC virus from the tonsil tissue of patients with tonsillitis and tonsilar hypertrophy. J. Neurovirol. 2004, 10, 244–249. [Google Scholar] [CrossRef]
- Van Loy, T.; Thys, K.; Ryschkewitsch, C.; Lagatie, O.; Monaco, M.C.; Major, E.O.; Tritsmans, L.; Stuyver, L.J. JC virus quasispecies analysis reveals a complex viral population underlying progressive multifocal leukoencephalopathy and supports viral dissemination via the hematogenous route. J. Virol. 2015, 89, 1340–1347. [Google Scholar] [CrossRef] [Green Version]
- Marzocchetti, A.; Wuthrich, C.; Tan, C.S.; Tompkins, T.; Bernal-Cano, F.; Bhargava, P.; Ropper, A.H.; Koralnik, I.J. Rearrangement of the JC virus regulatory region sequence in the bone marrow of a patient with rheumatoid arthritis and progressive multifocal leukoencephalopathy. J. Neurovirol. 2008, 14, 455–458. [Google Scholar] [CrossRef] [Green Version]
- Monaco, M.C.; Atwood, W.J.; Gravell, M.; Tornatore, C.S.; Major, E.O. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: Implications for viral latency. J. Virol. 1996, 70, 7004–7012. [Google Scholar] [CrossRef] [Green Version]
- Cubitt, C.L.; Cui, X.; Agostini, H.T.; Nerurkar, V.R.; Scheirich, I.; Yanagihara, R.; Ryschkewitsch, C.F.; Stoner, G.L.; Cubitt, C.L. Predicted amino acid sequences for 100 JCV strains. J. Neurovirol. 2001, 7, 339–344. [Google Scholar]
- Atwood, W.J. Genotypes, archetypes, and tandem repeats in the molecular epidemiology and pathogenesis of JC virus induced disease. J. Neurovirol. 2003, 9, 519–521. [Google Scholar] [CrossRef]
- Agostini, H.T.; Deckhut, A.; Jobes, D.V.; Girones, R.; Schlunck, G.; Prost, M.G.; Frias, C.; Pérez-Trallero, E.; Ryschkewitsch, C.F.; Stoner, G.L. Genotypes of JC virus in East, Central and Southwest Europe. J. Gen. Virol. 2001, 82 Pt 5, 1221–1331. [Google Scholar] [CrossRef] [Green Version]
- Jobes, D.V.; Chima, S.C.; Ryschkewitsch, C.F.; Stoner, G.L. Phylogenetic analysis of 22 complete genomes of the human polyomavirus JC virus. J. Gen. Virol. 1998, 79, 2491–2498. [Google Scholar] [CrossRef] [Green Version]
- Agostini, H.T.; Ryschkewitsch, C.F.; Stoner, G.L. Genotype profile of human polyomavirus JC excreted in urine of immunocompetent individuals. J. Clin. Microbiol. 1996, 34, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Dubois, V.; Moret, H.; Lafon, M.E.; Brodard, V.; Icart, J.; Ruffault, A.; Guist’Hau, O.; Buffet-Janvresse, C.; Abbed, K.; Dussaix, E.; et al. JC virus genotypes in France: Molecular epidemiology and potential significance for progressive multifocal leukoencephalopathy. J. Infect. Dis. 2001, 183, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Sugimoto, C.; Zheng, H.Y.; Kitamura, T.; Yogo, Y. Lack of disease-specific amino acid changes in the viral proteins of JC virus isolates from the brain with progressive multifocal leukoencephalopathy. Arch. Virol. 2000, 145, 2173–2182. [Google Scholar] [CrossRef]
- Zheng, H.Y.; Yasuda, Y.; Kato, S.; Kitamura, T.; Yogo, Y. Stability of JC virus coding sequences in a case of progressive multifocal leukoencephalopathy in which the viral control region was rearranged markedly. Arch. Pathol. Lab. Med. 2004, 128, 275–278. [Google Scholar] [CrossRef] [PubMed]
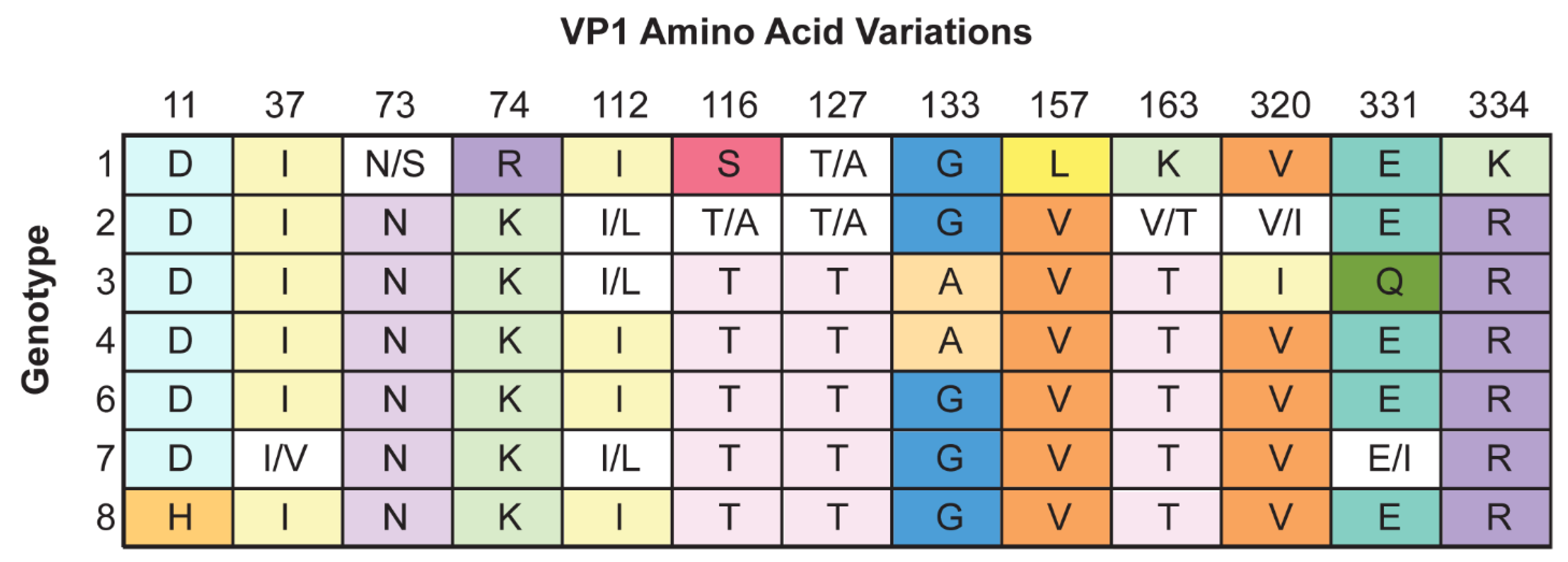
- Gorelik, L.; Reid, C.; Testa, M.; Brickelmaier, M.; Bossolasco, S.; Pazzi, A.; Bestetti, A.; Carmillo, P.; Wilson, E.; McAuliffe, M.; et al. Progressive multifocal leukoencephalopathy (PML) development is associated with mutations in JC virus capsid protein VP1 that change its receptor specificity. J. Infect. Dis. 2011, 204, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Sunyaev, S.R.; Lugovskoy, A.; Simon, K.; Gorelik, L. Adaptive mutations in the JC virus protein capsid are associated with progressive multifocal leukoencephalopathy (PML). PLoS Genet. 2009, 5, e1000368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.Y.; Ikegaya, H.; Takasaka, T.; Matsushima-Ohno, T.; Sakurai, M.; Kanazawa, I.; Kishida, S.; Nagashima, K.; Kitamura, T.; Yogo, Y. Characterization of the VP1 loop mutations widespread among JC polyomavirus isolates associated with progressive multifocal leukoencephalopathy. Biochem. Biophys. Res. Commun. 2005, 333, 996–1002. [Google Scholar] [CrossRef]
- Stoner, G.L.; Agostini, H.T.; Ryschkewitsch, C.F.; Baumhefner, R.W.; Tourtellotte, W.W. Characterization of JC virus DNA amplified from urine of chronic progressive multiple sclerosis patients. Mult. Scler. 1996, 1, 193–199. [Google Scholar] [CrossRef]
- Ferrante, P.; Delbue, S.; Pagani, E.; Mancuso, R.; Marzocchetti, A.; Borghi, E.; Maserati, R.; Bestetti, A.; Cinque, P. Analysis of JC virus genotype distribution and transcriptional control region rearrangements in human immunodeficiency virus-positive progressive multifocal leukoencephalopathy patients with and without highly active antiretroviral treatment. J. Neurovirol. 2003, 9 (Suppl. S1), 42–46. [Google Scholar] [CrossRef]
- Stehle, T.; Gamblin, S.J.; Yan, Y.; Harrison, S.C. The structure of simian virus 40 refined at 3.1 A resolution. Structure 1996, 4, 165–182. [Google Scholar] [CrossRef] [Green Version]
- Liddington, R.C.; Yan, Y.; Moulai, J.; Sahli, R.; Benjamin, T.L.; Harrison, S.C. Structure of simian virus 40 at 3.8-A resolution. Nature 1991, 354, 278–284. [Google Scholar] [CrossRef]
- Yan, Y.; Stehle, T.; Liddington, R.C.; Zhao, H.; Harrison, S.C. Structure determination of simian virus 40 and murine polyomavirus by a combination of 30-fold and 5-fold electron-density averaging. Structure 1996, 4, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Salunke, D.M.; Caspar, D.L.; Garcea, R.L. Self-assembly of purified polyomavirus capsid protein VP1. Cell 1986, 46, 895–904. [Google Scholar] [CrossRef]
- Chen, X.S.; Stehle, T.; Harrison, S.C. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J. 1998, 17, 3233–3240. [Google Scholar] [CrossRef]
- Kawano, M.A.; Inoue, T.; Tsukamoto, H.; Takaya, T.; Enomoto, T.; Takahashi, R.U.; Yokoyama, N.; Yamamoto, N.; Nakanishi, A.; Imai, T.; et al. The VP2/VP3 minor capsid protein of simian virus 40 promotes the in vitro assembly of the major capsid protein VP1 into particles. J. Biol. Chem. 2006, 281, 10164–10173. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, S.D.; Stehle, T.; Garcea, R. Glycan receptors of the Polyomaviridae: Structure, function, and pathogenesis. Curr. Opin. Virol. 2014, 7, 73–78. [Google Scholar] [CrossRef]
- Stroh, L.J.; Rustmeier, N.H.; Blaum, B.S.; Botsch, J.; Rossler, P.; Wedekink, F.; Lipkin, W.I.; Mishra, N.; Stehle, T. Structural Basis and Evolution of Glycan Receptor Specificities within the Polyomavirus Family. mBio 2020, 11, e00745-20. [Google Scholar] [CrossRef]
- Stroh, L.J.; Stehle, T. Glycan Engagement by Viruses: Receptor Switches and Specificity. Annu. Rev. Virol. 2014, 1, 285–306. [Google Scholar] [CrossRef]
- Neu, U.; Maginnis, M.S.; Palma, A.S.; Stroh, L.J.; Nelson, C.D.; Feizi, T.; Atwood, W.J.; Stehle, T. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell. Host Microbe. 2010, 8, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Stroh, L.J.; Maginnis, M.S.; Blaum, B.S.; Nelson, C.D.; Neu, U.; Gee, G.V.; O’Hara, B.A.; Motamedi, N.; DiMaio, D.; Atwood, W.J.; et al. The Greater Affinity of JC Polyomavirus Capsid for alpha2,6-Linked Lactoseries Tetrasaccharide c than for Other Sialylated Glycans Is a Major Determinant of Infectivity. J. Virol. 2015, 89, 6364–6375. [Google Scholar] [CrossRef] [Green Version]
- Reid, C.E.; Li, H.; Sur, G.; Carmillo, P.; Bushnell, S.; Tizard, R.; McAuliffe, M.; Tonkin, C.; Simon, K.; Goelz, S.; et al. Sequencing and analysis of JC virus DNA from natalizumab-treated PML patients. J. Infect. Dis. 2011, 204, 237–244. [Google Scholar] [CrossRef]
- Maginnis, M.S.; Stroh, L.J.; Gee, G.V.; O’Hara, B.A.; Derdowski, A.; Stehle, T.; Atwood, W.J. Progressive multifocal leukoencephalopathy-associated mutations in the JC polyomavirus capsid disrupt lactoseries tetrasaccharide c binding. mBio 2013, 4, e00247-13. [Google Scholar] [CrossRef] [Green Version]
- Haley, S.A.; O’Hara, B.A.; Nelson, C.D.; Brittingham, F.L.; Henriksen, K.J.; Stopa, E.G.; Atwood, W.J. Human polyomavirus receptor distribution in brain parenchyma contrasts with receptor distribution in kidney and choroid plexus. Am. J. Pathol. 2015, 185, 2246–2258. [Google Scholar] [CrossRef] [Green Version]
- Komagome, R.; Sawa, H.; Suzuki, T.; Suzuki, Y.; Tanaka, S.; Atwood, W.J.; Nagashima, K. Oligosaccharides as receptors for JC virus. J. Virol. 2002, 76, 12992–13000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayberry, C.L.; Bond, A.C.; Wilczek, M.P.; Mehmood, K.; Maginnis, M.S. Sending mixed signals: Polyomavirus entry and trafficking. Curr. Opin. Virol. 2021, 47, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Sipione, S.; Monyror, J.; Galleguillos, D.; Steinberg, N.; Kadam, V. Gangliosides in the Brain: Physiology, Pathophysiology and Therapeutic Applications. Front. Neurosci. 2020, 14, 572965. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, E.M.; Pastrana, D.V.; Schowalter, R.M.; Ray, U.; Gao, W.; Ho, M.; Pauly, G.T.; Sigano, D.M.; Kaynor, C.; Cahir-McFarland, E.; et al. Infectious Entry and Neutralization of Pathogenic JC Polyomaviruses. Cell. Rep. 2017, 21, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Haley, S.A.; O’Hara, B.A.; Atwood, W.J. Adipocyte Plasma Membrane Protein (APMAP) promotes JC Virus (JCPyV) infection in human glial cells. Virology 2020, 548, 17–24. [Google Scholar] [CrossRef]
- Mosser, S.; Alattia, J.R.; Dimitrov, M.; Matz, A.; Pascual, J.; Schneider, B.L.; Fraering, P.C. The adipocyte differentiation protein APMAP is an endogenous suppressor of Abeta production in the brain. Hum. Mol. Genet. 2015, 24, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Ilhan, A.; Gartner, W.; Nabokikh, A.; Daneva, T.; Majdic, O.; Cohen, G.; Böhmig, G.A.; Base, W.; Hörl, W.H.; Wagner, L. Localization and characterization of the novel protein encoded by C20orf3. Biochem. J. 2008, 414, 485–495. [Google Scholar] [CrossRef]
- Elphick, G.F.; Querbes, W.; Jordan, J.A.; Gee, G.V.; Eash, S.; Manley, K.; Dugan, A.; Stanifer, M.; Bhatnagar, A.; Kroeze, W.K.; et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science 2004, 306, 1380–1383. [Google Scholar] [CrossRef] [Green Version]
- Assetta, B.; Maginnis, M.S.; Gracia Ahufinger, I.; Haley, S.A.; Gee, G.V.; Nelson, C.D.; O’Hara, B.A.; Ramdial, S.A.A.; Atwood, W.J. 5-HT2 receptors facilitate JC polyomavirus entry. J. Virol. 2013, 87, 13490–13498. [Google Scholar] [CrossRef] [Green Version]
- Assetta, B.; Morris-Love, J.; Gee, G.V.; Atkinson, A.L.; O’Hara, B.A.; Maginnis, M.S.; Haley, S.A.; Atwood, W.J. Genetic and Functional Dissection of the Role of Individual 5-HT2 Receptors as Entry Receptors for JC Polyomavirus. Cell Rep. 2019, 27, 1960–1966.e6. [Google Scholar] [CrossRef] [Green Version]
- DuShane, J.K.; Wilczek, M.P.; Mayberry, C.L.; Maginnis, M.S. ERK Is a Critical Regulator of JC Polyomavirus Infection. J. Virol. 2018, 92, e01529-17. [Google Scholar] [CrossRef] [Green Version]
- Mayberry, C.L.; Soucy, A.N.; Lajoie, C.R.; DuShane, J.K.; Maginnis, M.S. JC Polyomavirus Entry by Clathrin-Mediated Endocytosis Is Driven by beta-Arrestin. J. Virol. 2019, 93, e01948-18. [Google Scholar] [CrossRef] [Green Version]
- Mayberry, C.L.; Wilczek, M.P.; Fong, T.M.; Nichols, S.L.; Maginnis, M.S. GRK2 mediates beta-arrestin interactions with 5-HT2 receptors for JC polyomavirus endocytosis. J. Virol. 2021, 95, e02139-20. [Google Scholar] [CrossRef]
- Maginnis, M.S.; Nelson, C.D.; Atwood, W.J. JC polyomavirus attachment, entry, and trafficking: Unlocking the keys to a fatal infection. J. Neurovirol. 2015, 21, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Maginnis, M.S.; Haley, S.A.; Gee, G.V.; Atwood, W.J. Role of N-linked glycosylation of the 5-HT2A receptor in JC virus infection. J. Virol. 2010, 84, 9677–9684. [Google Scholar] [CrossRef] [Green Version]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell. Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles. 2013, 2, 20389. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Higginbotham, J.N.; Zhang, Q.; Coffey, R.J. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell. Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzan, E.; Tine, M.; Casara, A.; Biondini, D.; Semenzato, U.; Cocconcelli, E.; Balestro, E.; Damin, M.; Radu, C.; Turato, G.; et al. Critical Review of the Evolution of Extracellular Vesicles’ Knowledge: From 1946 to Today. Int. J. Mol. Sci. 2021, 22, 6417. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.F.; Jeong, S.-H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altan-Bonnet, N. Extracellular vesicles are the Trojan horses of viral infection. Curr. Opin. Microbiol. 2016, 32, 77–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altan-Bonnet, N.; Perales, C.; Domingo, E. Extracellular vesicles: Vehicles of en bloc viral transmission. Virus Res. 2019, 265, 143–149. [Google Scholar] [CrossRef]
- Kerviel, A.; Zhang, M.; Altan-Bonnet, N. A New Infectious Unit: Extracellular Vesicles Carrying Virus Populations. Ann. Rev. Cell. Dev. Biol. 2021, 37, 171–197. [Google Scholar] [CrossRef]
- Bird, S.W.; Maynard, N.D.; Covert, M.W.; Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 2014, 111, 13081–13086. [Google Scholar] [CrossRef] [Green Version]
- van der Grein, S.G.; Defourny, K.A.Y.; Rabouw, H.H.; Galiveti, C.R.; Langereis, M.A.; Wauben, M.H.M.; Arkesteijn, G.; Van Kuppeveld, F.J.M.; Hoen, E.N.M.N. Picornavirus infection induces temporal release of multiple extracellular vesicle subsets that differ in molecular composition and infectious potential. PLoS Pathog. 2019, 15, e1007594. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, J.; Li, S.; Dang, W.; Xin, S.; Long, S.; Zhang, W.; Cao, P.; Lu, J. Extracellular Vesicles Regulated by Viruses and Antiviral Strategies. Front. Cell Dev. Biol. 2021, 9, 722020. [Google Scholar] [CrossRef]
- Sadeghipour, S.; Mathias, R.A. Herpesviruses hijack host exosomes for viral pathogenesis. Semin. Cell Dev. Biol. 2017, 67, 91–100. [Google Scholar] [CrossRef]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippe, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [Green Version]
- Dogrammatzis, C.; Deschamps, T.; Kalamvoki, M. Biogenesis of Extracellular Vesicles during Herpes Simplex Virus 1 Infection: Role of the CD63 Tetraspanin. J. Virol. 2019, 93, e01102-18. [Google Scholar] [CrossRef] [Green Version]
- Bello-Morales, R.; Praena, B.; de la Nuez, C.; Rejas, M.T.; Guerra, M.; Galan-Ganga, M.; Izquierdo, M.; Calvo, V.; Krummenacher, C.; López-Guerrero, J.A. Role of Microvesicles in the Spread of Herpes Simplex Virus 1 in Oligodendrocytic Cells. J. Virol. 2018, 92, e00088-18. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. beta-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e14. [Google Scholar] [CrossRef]
- Saribas, A.S.; Cicalese, S.; Ahooyi, T.M.; Khalili, K.; Amini, S.; Sariyer, I.K. HIV-1 Nef is released in extracellular vesicles derived from astrocytes: Evidence for Nef-mediated neurotoxicity. Cell. Death Dis. 2017, 8, e2542. [Google Scholar] [CrossRef] [Green Version]
- Rahimian, P.; He, J.J. Exosome-associated release, uptake, and neurotoxicity of HIV-1 Tat protein. J. Neurovirol. 2016, 22, 774–788. [Google Scholar] [CrossRef] [Green Version]
- Muratori, C.; Cavallin, L.E.; Kratzel, K.; Tinari, A.; De Milito, A.; Fais, S.; D’Aloja, P.; Federico, M.; Vullo, V.; Fomina, A.; et al. Massive secretion by T cells is caused by HIV Nef in infected cells and by Nef transfer to bystander cells. Cell Host Microbe 2009, 6, 218–230. [Google Scholar] [CrossRef] [Green Version]
- McNamara, R.P.; Costantini, L.M.; Myers, T.A.; Schouest, B.; Maness, N.J.; Griffith, J.D.; Damania, B.A.; MacLean, A.G.; Dittmer, D.P. Nef Secretion into Extracellular Vesicles or Exosomes Is Conserved across Human and Simian Immunodeficiency Viruses. mBio 2018, 9, e02344-17. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.; Prasad, A. Exosomes Derived from HIV-1 Infected DCs Mediate Viral trans-Infection via Fibronectin and Galectin-3. Sci. Rep. 2017, 7, 14787. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.K.; Kaminski, R.; Mullen, B.; Gordon, J.; Burdo, T.H.; Cheung, J.Y.; Feldman, A.M.; Madesh, M.; Khalili, K. HIV-1 Nef-induced cardiotoxicity through dysregulation of autophagy. Sci. Rep. 2017, 7, 8572. [Google Scholar] [CrossRef] [PubMed]
- Atwood, W.J.; Amemiya, K.; Traub, R.; Harms, J.; Major, E.O. Interaction of the human polyomavirus, JCV, with human B-lymphocytes. Virology 1992, 190, 716–723. [Google Scholar] [CrossRef]
- Buckley, M.W.; McGavern, D.B. Immune dynamics in the CNS and its barriers during homeostasis and disease. Immunol. Rev. 2022, 306, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Falcao, A.M.; Marques, F.; Novais, A.; Sousa, N.; Palha, J.A.; Sousa, J.C. The path from the choroid plexus to the subventricular zone: Go with the flow! Front. Cell Neurosci. 2012, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, B.A.; Gee, G.V.; Atwood, W.J.; Haley, S.A. Susceptibility of Primary Human Choroid Plexus Epithelial Cells and Meningeal Cells to Infection by JC Virus. J. Virol. 2018, 92, e00105-18. [Google Scholar] [CrossRef] [Green Version]
- Balusu, S.; Van Wonterghem, E.; De Rycke, R.; Raemdonck, K.; Stremersch, S.; Gevaert, K.; Brkic, M.; Demeestere, D.; Vanhooren, V.; Hendrix, A.; et al. Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol. Med. 2016, 8, 1162–1183. [Google Scholar] [CrossRef]
- Vandendriessche, C.; Balusu, S.; Van Cauwenberghe, C.; Brkic, M.; Pauwels, M.; Plehiers, N.; Bruggeman, A.; Dujardin, P.; Van Imschoot, G.; Van Wonterghem, E.; et al. Importance of extracellular vesicle secretion at the blood-cerebrospinal fluid interface in the pathogenesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 143. [Google Scholar] [CrossRef]
- Lepko, T.; Pusch, M.; Muller, T.; Schulte, D.; Ehses, J.; Kiebler, M.; Hasler, J.; Huttner, H.B.; Vandenbroucke, R.E.; VandenDriessche, C.; et al. Choroid plexus-derived miR-204 regulates the number of quiescent neural stem cells in the adult brain. EMBO J. 2019, 38, e100481. [Google Scholar] [CrossRef]
- Corbridge, S.M.; Rice, R.C.; Bean, L.A.; Wuthrich, C.; Dang, X.; Nicholson, D.A.; Koralnik, I.J. JC virus infection of meningeal and choroid plexus cells in patients with progressive multifocal leukoencephalopathy. J. Neurovirol. 2019, 25, 520–524. [Google Scholar] [CrossRef]
- O’Hara, B.A.; Gee, G.V.; Haley, S.A.; Morris-Love, J.; Nyblade, C.; Nieves, C.; Hanson, B.A.; Dang, X.; Turner, T.J.; Chavin, J.M.; et al. Teriflunomide Inhibits JCPyV Infection and Spread in Glial Cells and Choroid Plexus Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 9809. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Stoorvogel, W.; Kleijmeer, M.J.; Geuze, H.J.; Raposo, G. The biogenesis and functions of exosomes. Traffic 2002, 3, 321–330. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Ann. Rev. Cell. Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. J. Lipid Res. 2018, 59, 1325–1340. [Google Scholar] [CrossRef] [Green Version]
- Rojas, C.; Sala, M.; Thomas, A.G.; Datta Chaudhuri, A.; Yoo, S.W.; Li, Z.; Dash, R.P.; Rais, R.; Haughey, N.J.; Nencka, R.; et al. A novel and potent brain penetrant inhibitor of extracellular vesicle release. Br. J. Pharmacol. 2019, 176, 3857–3870. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Canals, D.; Hannun, Y.A. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal. 2009, 21, 836–846. [Google Scholar] [CrossRef] [Green Version]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Zoller, M. Tetraspanins: Push and pull in suppressing and promoting metastasis. Nat. Rev. Cancer 2009, 9, 40–55. [Google Scholar] [CrossRef]
- Andreu, Z.; Yanez-Mo, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef] [Green Version]
- van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [Green Version]
- Snead, W.T.; Hayden, C.C.; Gadok, A.K.; Zhao, C.; Lafer, E.M.; Rangamani, P.; Stachowiak, J.C. Membrane fission by protein crowding. Proc. Natl. Acad. Sci. USA. 2017, 114, E3258–E3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachowiak, J.C.; Schmid, E.M.; Ryan, C.J.; Ann, H.S.; Sasaki, D.Y.; Sherman, M.B.; Geissler, P.L.; Fletcher, D.A.; Hayden, C.C. Membrane bending by protein-protein crowding. Nat. Cell. Biol. 2012, 14, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juan, T.; Furthauer, M. Biogenesis and function of ESCRT-dependent extracellular vesicles. Semin. Cell. Dev. Biol. 2018, 74, 66–77. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, O.; Teis, D. The ESCRT machinery. Curr. Biol. 2012, 22, R116–R120. [Google Scholar] [CrossRef] [Green Version]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H. ESCRTs are everywhere. EMBO J. 2015, 34, 2398–2407. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Schekman, R. Cell biology. Unconventional secretion, unconventional solutions. Science 2013, 340, 559–561. [Google Scholar] [CrossRef]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell. Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Camfield, R.; Gorski, S.M. The interplay between exosomes and autophagy—partners in crime. J. Cell Sci. 2018, 131, jcs215210. [Google Scholar] [CrossRef] [Green Version]
- Claude-Taupin, A.; Jia, J.; Mudd, M.; Deretic, V. Autophagy’s secret life: Secretion instead of degradation. Essays Biochem. 2017, 61, 637–647. [Google Scholar]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Kenny, S.J.; Ge, L.; Xu, K.; Schekman, R. Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. Elife 2015, 2, e11205. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y. Nonredundant Roles of GRASP55 and GRASP65 in the Golgi Apparatus and Beyond. Trends Biochem. Sci. 2020, 45, 1065–1079. [Google Scholar] [CrossRef]
- Chen, Y.D.; Fang, Y.T.; Cheng, Y.L.; Lin, C.F.; Hsu, L.J.; Wang, S.Y.; Anderson, R.; Chang, C.-P.; Lin, Y.S. Exophagy of annexin A2 via RAB11, RAB8A and RAB27A in IFN-gamma-stimulated lung epithelial cells. Sci. Rep. 2017, 7, 5676. [Google Scholar] [CrossRef]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Ejlerskov, P.; Rasmussen, I.; Nielsen, T.T.; Bergstrom, A.L.; Tohyama, Y.; Jensen, P.H.; Vilhardt, F. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J. Biol. Chem. 2013, 288, 17313–17335. [Google Scholar] [CrossRef] [Green Version]
- Pleet, M.L.; Branscome, H.; DeMarino, C.; Pinto, D.O.; Zadeh, M.A.; Rodriguez, M.; Sariyer, I.K.; El-Hage, N.; Kashanchi, F. Autophagy, EVs, and Infections: A Perfect Question for a Perfect Time. Front. Cell. Infect. Microbiol. 2018, 8, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, K.; Hurley, J.H.; Freed, E.O. Beyond Tsg101: The role of Alix in ’ESCRTing’ HIV-1. Nat. Rev. Microbiol. 2007, 5, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Sette, P.; Mu, R.; Dussupt, V.; Jiang, J.; Snyder, G.; Smith, P.; Xiao, T.S.; Bouamr, F. The Phe105 loop of Alix Bro1 domain plays a key role in HIV-1 release. Structure 2011, 19, 1485–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandon, R.; AuCoin, D.P.; Mocarski, E.S. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J. Virol. 2009, 83, 10797–10807. [Google Scholar] [CrossRef] [Green Version]
- Gordon-Alonso, M.; Yanez-Mo, M.; Barreiro, O.; Alvarez, S.; Munoz-Fernandez, M.A.; Valenzuela-Fernandez, A.; Sánchez-Madrid, F. Tetraspanins CD9 and CD81 modulate HIV-1-induced membrane fusion. J. Immunol. 2006, 177, 5129–5137. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Sun, E.; Bujny, M.V.; Kim, D.; Davidson, M.W.; Zhuang, X. Dual function of CD81 in influenza virus uncoating and budding. PLoS Pathog. 2013, 9, e1003701. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, F.S.Y.; Teixeira, F.M.E.; Sato, M.N.; Oliveira, L. Delivery of microRNAs by Extracellular Vesicles in Viral Infections: Could the News be Packaged? Cells 2019, 8, 611. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Woodson, M.; Neupane, B.; Bai, F.; Sherman, M.B.; Choi, K.H.; Neelakanta, G.; Sultana, H. Exosomes serve as novel modes of tick-borne flavivirus transmission from arthropod to human cells and facilitates dissemination of viral RNA and proteins to the vertebrate neuronal cells. PLoS Pathog. 2018, 14, e1006764. [Google Scholar] [CrossRef] [Green Version]
- Urbanelli, L.; Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Porcellati, S.; Emiliani, C. The Role of Extracellular Vesicles in Viral Infection and Transmission. Vaccines 2019, 7, 102. [Google Scholar] [CrossRef] [Green Version]
- Stenovec, M.; Lasic, E.; Dominkus, P.P.; Bobnar, S.T.; Zorec, R.; Lenassi, M.; Kreft, M. Slow Release of HIV-1 Protein Nef from Vesicle-like Structures Is Inhibited by Cytosolic Calcium Elevation in Single Human Microglia. Mol. Neurobiol. 2019, 56, 102–118. [Google Scholar] [CrossRef]
- Hu, G.; Niu, F.; Liao, K.; Periyasamy, P.; Sil, S.; Liu, J.; Dravid, S.M.; Buch, S. HIV-1 Tat-Induced Astrocytic Extracellular Vesicle miR-7 Impairs Synaptic Architecture. J. Neuroimmune. Pharmacol. 2020, 15, 538–553. [Google Scholar] [CrossRef]
- Morris-Love, J.; O’Hara, B.A.; Gee, G.V.; Dugan, A.S.; O’Rourke, R.S.; Armstead, B.E.; Assetta, B.; Haley, S.A.; Atwood, W.J. Biogenesis of JC polyomavirus associated extracellular vesicles. J. Extracell. Biol. 2022, 1, e43. [Google Scholar] [CrossRef]
- Wharton, K.A.; Quigley, C., Jr.; Themeles, M.; Dunstan, R.W.; Doyle, K.; Cahir-McFarland, E.; Wei, J.; Buko, A.; Reid, C.E.; Sun, C.; et al. JC Polyomavirus Abundance and Distribution in Progressive Multifocal Leukoencephalopathy (PML) Brain Tissue Implicates Myelin Sheath in Intracerebral Dissemination of Infection. PLoS ONE 2016, 11, e0155897. [Google Scholar] [CrossRef]
- Gee, G.V.; O’Hara, B.A.; Derdowski, A.; Atwood, W.J. Pseudovirus mimics cell entry and trafficking of the human polyomavirus JCPyV. Virus Res. 2013, 178, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Buck, C.B.; Thompson, C.D. Production of papillomavirus-based gene transfer vectors. Curr. Protoc. Cell Biol. 2007, 37, 26.1. [Google Scholar] [CrossRef]
- Shen, P.S.; Enderlein, D.; Nelson, C.D.; Carter, W.S.; Kawano, M.; Xing, L.; Swenson, R.D.; Olson, N.H.; Baker, T.S.; Cheng, R.H.; et al. The structure of avian polyomavirus reveals variably sized capsids, non-conserved inter-capsomere interactions, and a possible location of the minor capsid protein VP4. Virology 2011, 411, 142–152. [Google Scholar] [CrossRef]
- Gupta, S.; Rawat, S.; Arora, V.; Kottarath, S.K.; Dinda, A.K.; Vaishnav, P.K.; Nayak, B.; Mohanty, S. An improvised one-step sucrose cushion ultracentrifugation method for exosome isolation from culture supernatants of mesenchymal stem cells. Stem. Cell. Res. Ther. 2018, 9, 180. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Liangsupree, T.; Multia, E.; Riekkola, M.L. Modern isolation and separation techniques for extracellular vesicles. J. Chromatogr. A 2021, 1636, 461773. [Google Scholar] [CrossRef]
- Royo, F.; Thery, C.; Falcon-Perez, J.M.; Nieuwland, R.; Witwer, K.W. Methods for Separation and Characterization of Extracellular Vesicles: Results of a Worldwide Survey Performed by the ISEV Rigor and Standardization Subcommittee. Cells 2020, 9, 1955. [Google Scholar] [CrossRef]
- Witwer, K.W.; Soekmadji, C.; Hill, A.F.; Wauben, M.H.; Buzas, E.I.; Di Vizio, D.; Falcon-Perez, J.M.; Gardiner, C.; Hochberg, F.; Kurochkin, I.V.; et al. Updating the MISEV minimal requirements for extracellular vesicle studies: Building bridges to reproducibility. J. Extracell. Vesicles 2017, 6, 1396823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coumans, F.A.W.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.F.; et al. Methodological Guidelines to Study Extracellular Vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef] [PubMed]
- Chitoiu, L.; Dobranici, A.; Gherghiceanu, M.; Dinescu, S.; Costache, M. Multi-Omics Data Integration in Extracellular Vesicle Biology-Utopia or Future Reality? Int. J. Mol. Sci. 2020, 21, 8550. [Google Scholar] [CrossRef] [PubMed]
- Sass, S.; Buettner, F.; Mueller, N.S.; Theis, F.J. A modular framework for gene set analysis integrating multilevel omics data. Nucleic. Acids. Res. 2013, 41, 9622–9633. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef]
- McNamara, R.P.; Dittmer, D.P. Modern Techniques for the Isolation of Extracellular Vesicles and Viruses. J. Neuroimmune Pharmacol. 2020, 15, 459–472. [Google Scholar] [CrossRef]
- Liu, C.K.; Atwood, W.J. Propagation and Assay of the JC Virus. In SV40 Protocols; Humana Press: Totowa, NJ, USA, 2000; Volume 165, pp. 9–17. [Google Scholar]
- Scribano, S.; Guerrini, M.; Arvia, R.; Guasti, D.; Nardini, P.; Romagnoli, P.; Giannecchini, S. Archetype JC polyomavirus DNA associated with extracellular vesicles circulates in human plasma samples. J. Clin. Virol. 2020, 128, 104435. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Pentamer | VLP | PsV | Virus |
|---|---|---|---|---|
| LSTc | Type 1 [76], 3 [77] | --- | Type 1, 3 [77] | Type 1 [76,77], 3 [77] |
| Gangliosides | Type 1, 3 [77] | Type 1 [81], 2 & 3 [84], 3 [62] | Type 1 & 3 [77], 2 & 3 [62,84] | Type 1, 3 [77] |
| GAGs | --- | Type 2, 3 [84] | Type 2, 3 [84] | --- |
| 5HT2A/B/C | --- | --- | Type 1 [90] | Type 1 [88,89,90,95] |
| APMAP | --- | --- | --- | Type 1 [85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morris-Love, J.; Atwood, W.J. Complexities of JC Polyomavirus Receptor-Dependent and -Independent Mechanisms of Infection. Viruses 2022, 14, 1130. https://doi.org/10.3390/v14061130
Morris-Love J, Atwood WJ. Complexities of JC Polyomavirus Receptor-Dependent and -Independent Mechanisms of Infection. Viruses. 2022; 14(6):1130. https://doi.org/10.3390/v14061130
Chicago/Turabian StyleMorris-Love, Jenna, and Walter J. Atwood. 2022. "Complexities of JC Polyomavirus Receptor-Dependent and -Independent Mechanisms of Infection" Viruses 14, no. 6: 1130. https://doi.org/10.3390/v14061130