
Genomic Epidemiology of SARS-CoV-2 in Tocantins State and the Diffusion of P.1.7 and AY.99.2 Lineages in Brazil

 ,
,  , ,
, ,  , , , ,
, , , ,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Extraction and Sequencing
2.3. Comparative Genome Analysis
2.4. Phylogenetic Analysis
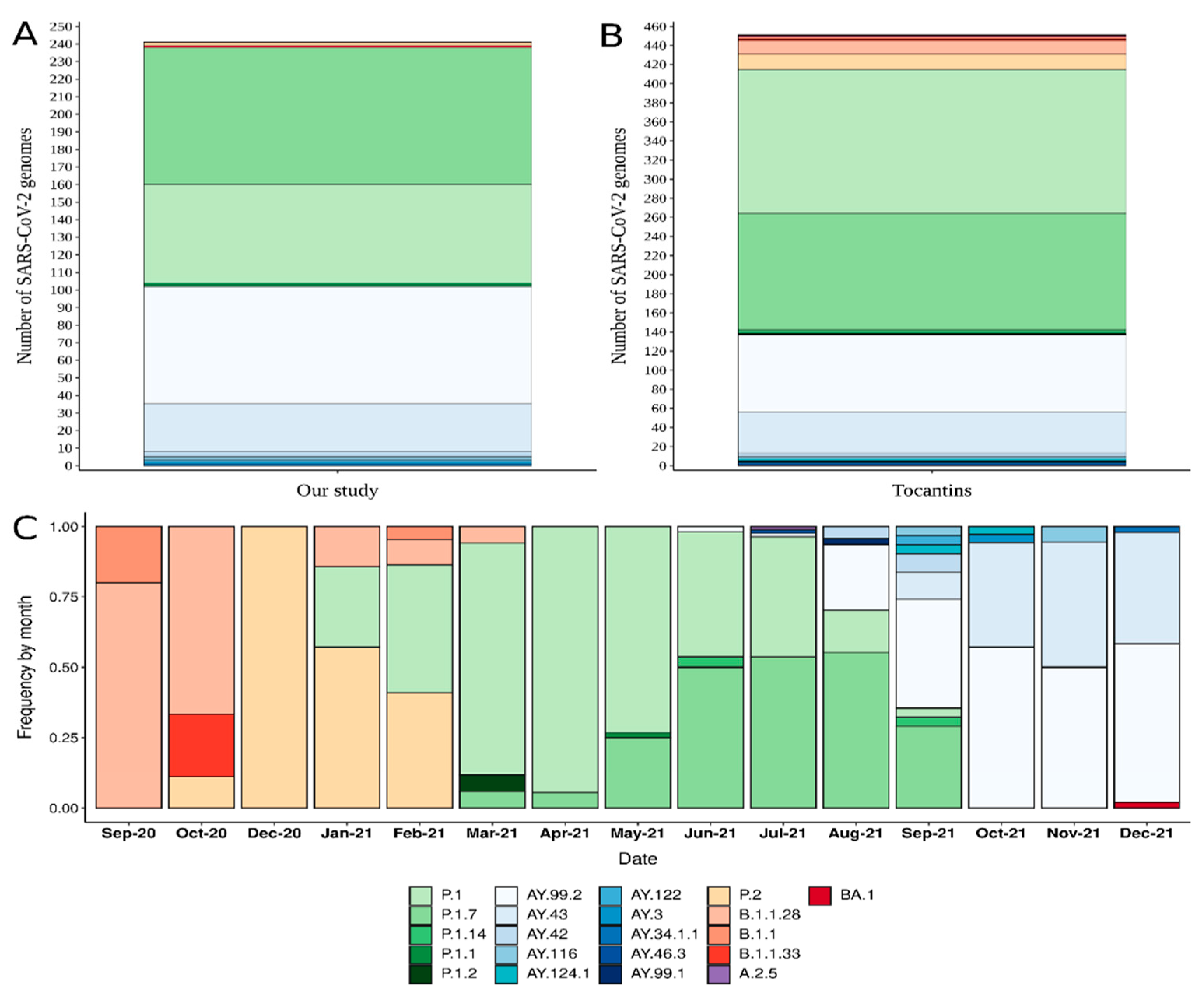
3. Results
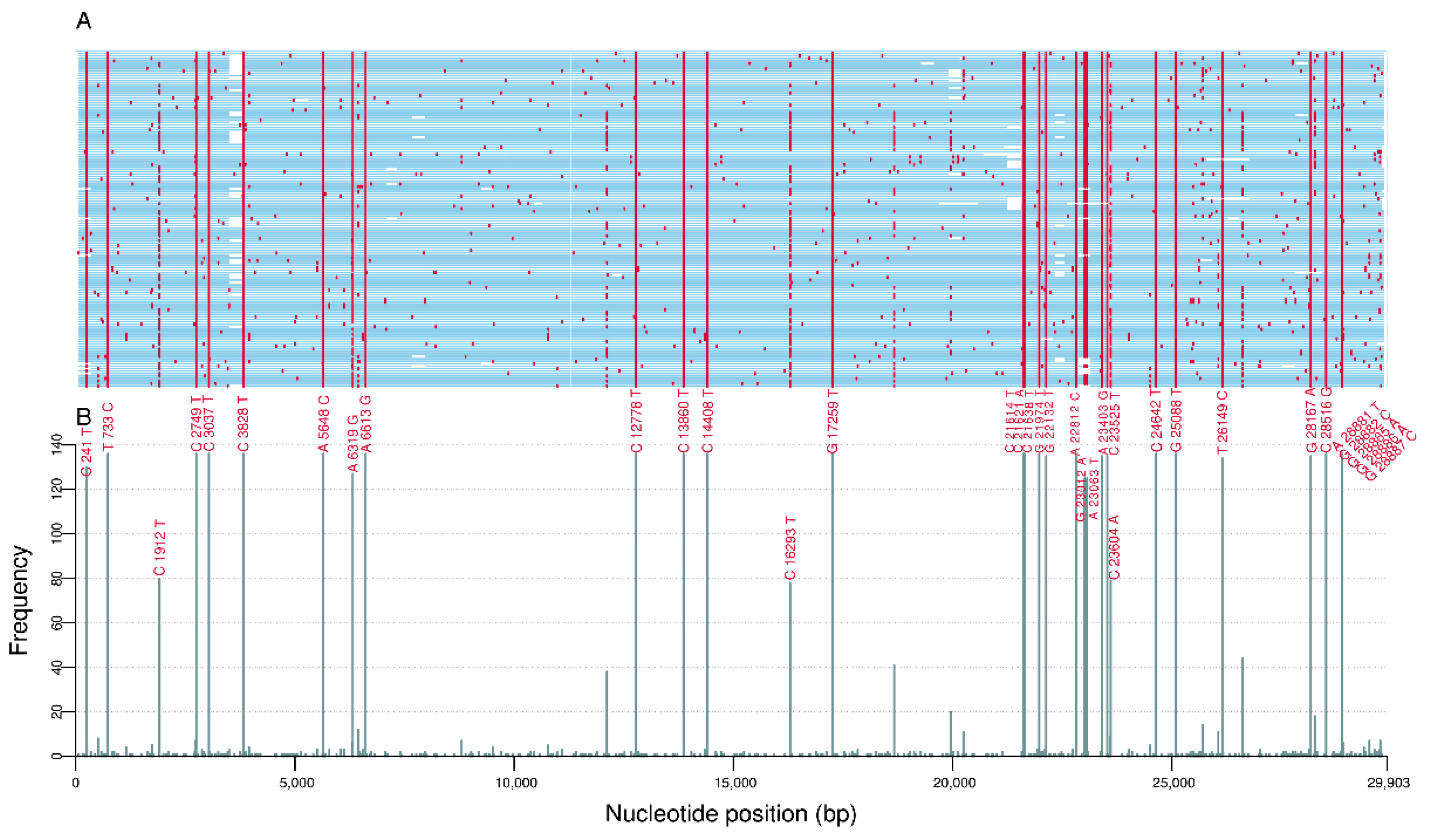
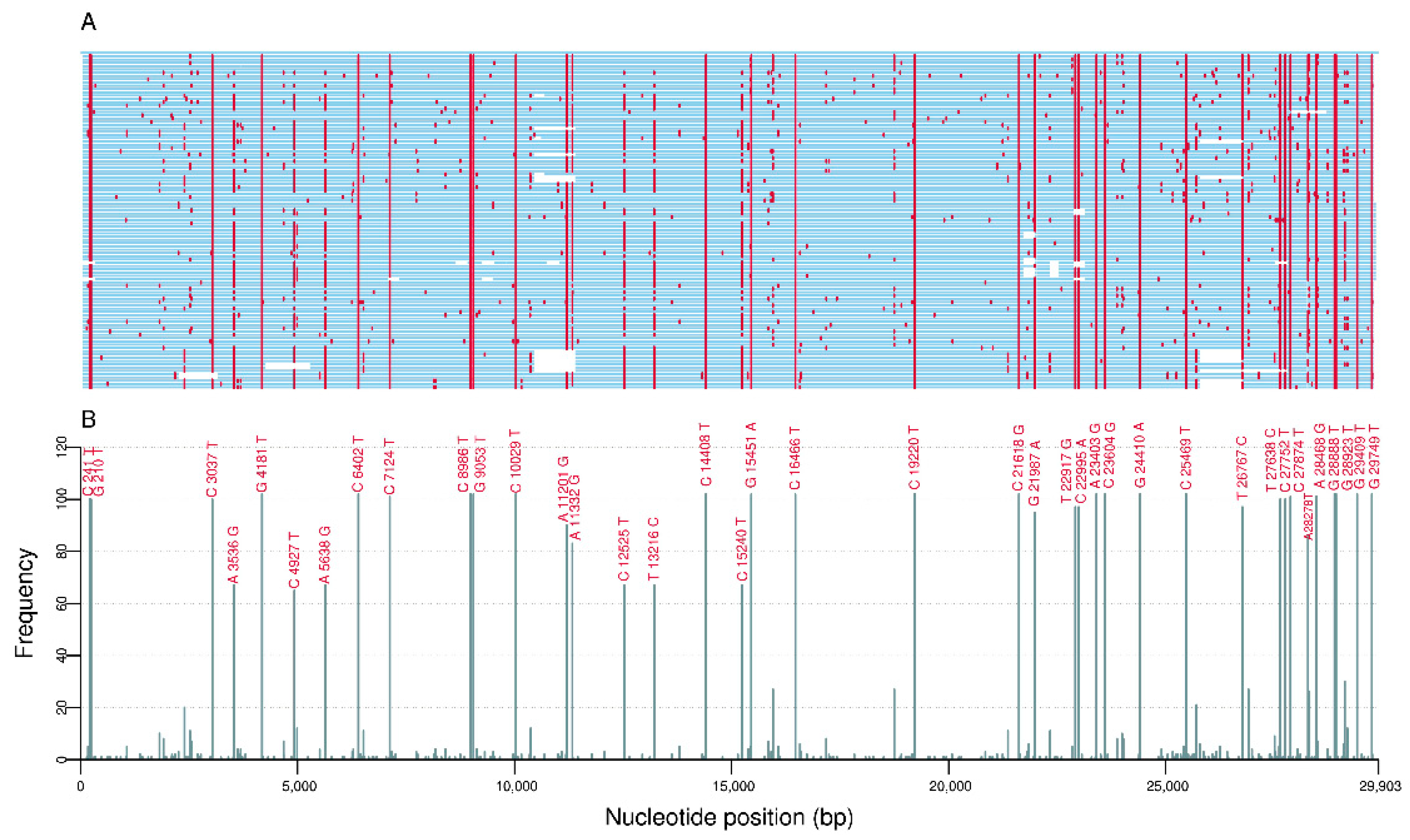
3.1. Comparative Genome Analysis
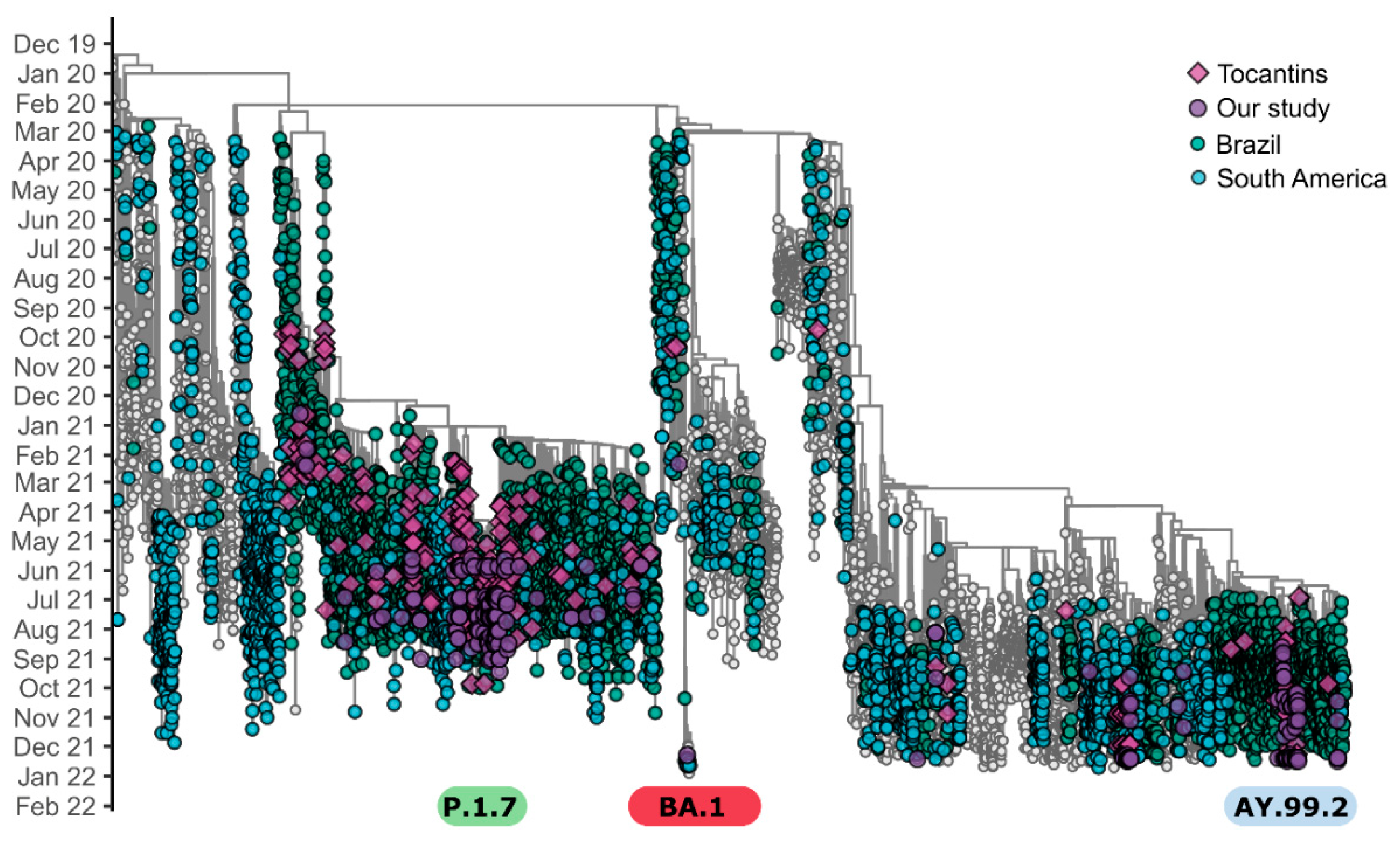
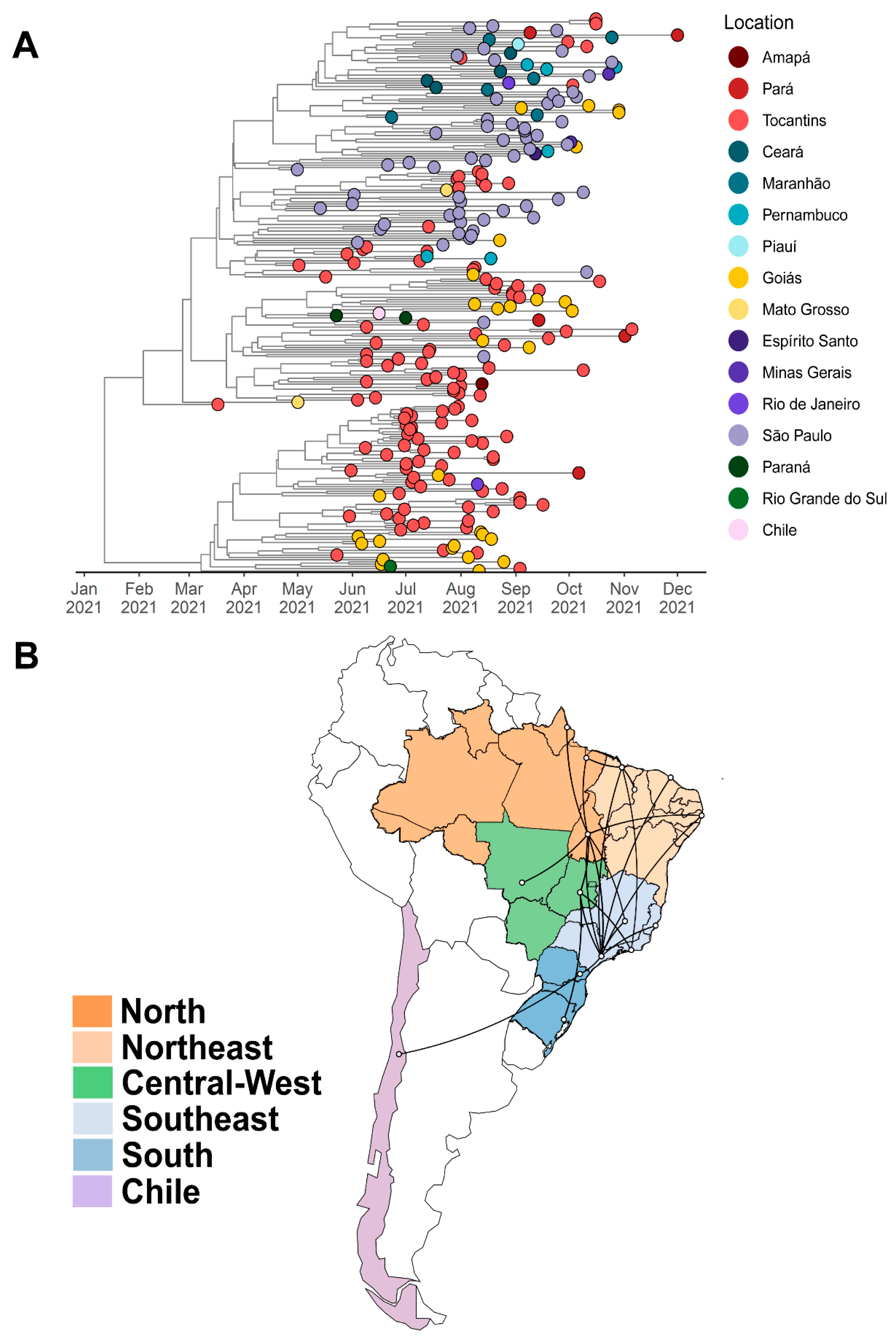
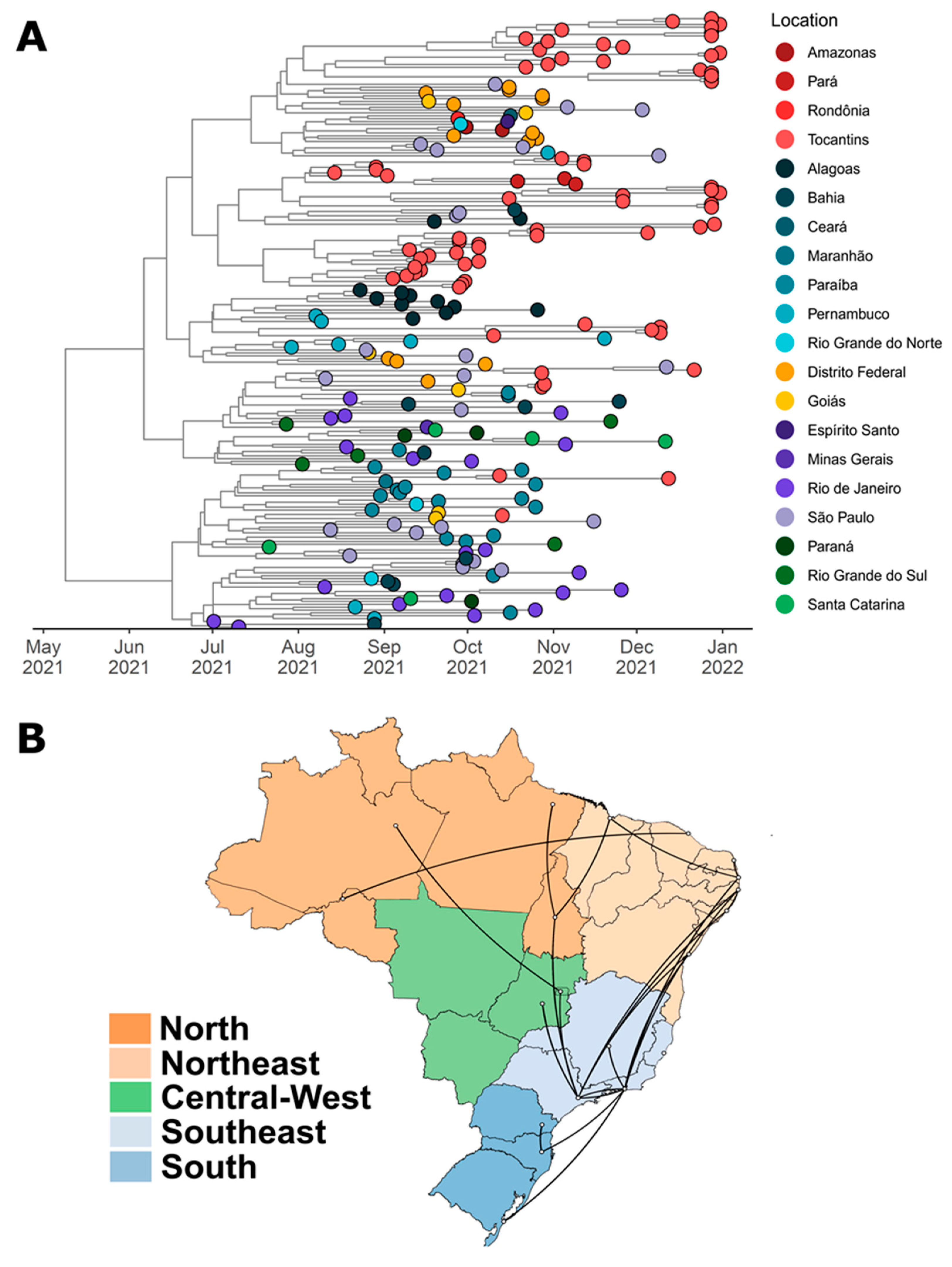
3.2. Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candido, D.S.; Claro, I.M.; de Jesus, J.G.; Souza, W.M.; Moreira, F.R.R.; Dellicour, S.; Mellan, T.A.; Du Plessis, L.; Pereira, R.H.M.; Sales, F.C.S.; et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 2020, 369, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.E.; Mahmud, A.S.; Miller, I.F.; Rajeev, M.; Rasambainarivo, F.; Rice, B.L.; Takahashi, S.; Tatem, A.J.; Wagner, C.E.; Wang, L.F.; et al. Infectious disease in an era of global change. Nat. Rev. Microbiol. 2021, 13, 193–205. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef]
- Long, S.W.; Olsen, R.J.; Christensen, P.A.; Subedi, S.; Olson, R.; Davis, J.J.; Saavedra, M.O.; Yerramilli, P.; Pruitt, L.; Reppond, K.; et al. Sequence Analysis of 20,453 Severe Acute Respiratory Syndrome Coronavirus 2 Genomes from the Houston Metropolitan Area Identifies the Emergence and Widespread Distribution of Multiple Isolates of All Major Variants of Concern. Am. J. Pathol. 2021, 191, 983–992. [Google Scholar] [CrossRef]
- Umair, M.; Ikram, A.; Salman, M.; Khurshid, A.; Alam, M.; Badar, N.; Suleman, R.; Tahir, F.; Sharif, S.; Montgomery, J.; et al. Whole-genome sequencing of SARS-CoV-2 reveals the detection of G614 variant in Pakistan. PLoS ONE 2021, 16, e0248371. [Google Scholar] [CrossRef]
- Souza, U.J.B.; dos Santos, R.N.; Campos, F.S.; Lourenço, K.L.; da Fonseca, F.G.; Spilki, F.R. High Rate of Mutational Events in SARS-CoV-2 Genomes across Brazilian Geographical Regions, February 2020 to June 2021. Viruses 2021, 13, 1806. [Google Scholar] [CrossRef]
- CDC. Emerging SARS-CoV-2 Variants. Available online: https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-emerging-variants.html (accessed on 2 February 2022).
- WHO. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 2 February 2022).
- Drost, S.; Kuepper, B.; Piotrowski, M. Brazilian State of Tocantins: A Hotspot for Cerrado Deforestation. Chain Reaction Research. 2019, pp. 1–15. Available online: https://chainreactionresearch.com/wp-content/uploads/2019/04/Tocantins-Report-2.pdf (accessed on 13 November 2021).
- Esper, F.P.; Cheng, Y.-W.; Adhikari, T.M.; Tu, Z.J.; Li, D.; Li, E.A.; Farkas, D.H.; Procop, G.W.; Ko, J.S.; Chan, T.A.; et al. Genomic Epidemiology of SARS-CoV-2 Infection During the Initial Pandemic Wave and Association with Disease Severity. JAMA Netw. Open 2021, 4, e217746. [Google Scholar] [CrossRef]
- Meredith, L.W.; Hamilton, W.L.; Warne, B.; Houldcroft, C.J.; Hosmillo, M.; Jahun, A.S.; Curran, M.D.; Parmar, S.; Caller, L.G.; Caddy, S.L.; et al. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: A prospective genomic surveillance study. Lancet Infect. Dis. 2020, 20, 1263–1271. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; A Beutler, N.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [Green Version]
- Vairo, F.; Coussoud-Mavoungou, M.P.; Ntoumi, F.; Castilletti, C.; Kitembo, L.; Haider, N.; Carletti, F.; Colavita, F.; Gruber, C.; Iannetta, M.; et al. Chikungunya Outbreak Group Taskforce. Chikungunya Outbreak in the Republic of the Congo, 2019—Epidemiological, Virological and Entomological Findings of a South-North Multidisciplinary Taskforce Investigation. Viruses 2020, 12, 1020. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; du Plessis, L.; Liu, Z.; Hill, V.; Kang, M.; Lin, H.; Sun, J.; François, S.; Kraemer, M.U.; Faria, N.R.; et al. Genomic Epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell 2020, 181, 997–1003.e9. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Bolyen, E.; Dillon, M.R.; Bokulich, N.A.; Ladner, J.T.; Larsen, B.B.; Hepp, C.M.; Lemmer, D.; Sahl, J.W.; Sanchez, A.; Holdgraf, C.; et al. Reproducibly sampling SARS-CoV-2 genomes across time, geography, and viral diversity. F1000Research 2020, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Meth. 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Orme, D.; Freckleton, R.; Thomas, G.; Petzoldt, T.; Fritz, S.; Isaac, N.; Pearse, W. Caper: Comparative analyses of phylogenetics and evolution in R. R package version 05. Proc. R. Soc. B 2012, 2, 458. [Google Scholar]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Ayres, D.L.; Darling, A.; Zwickl, D.J.; Beerli, P.; Holder, M.T.; Lewis, P.O.; Huelsenbeck, J.P.; Ronquist, F.; Swofford, D.L.; Cummings, M.P. BEAGLE: An Application Programming Interface and High-Performance Computing Library for Statistical Phylogenetics. Syst. Biol. 2012, 61, 170–173. [Google Scholar] [CrossRef]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.A.R.; Suchard, M.A. Bayesian analysis of elapsed times in continuous-time Markov chains. Can. J. Stat. 2008, 36, 355–368. [Google Scholar] [CrossRef]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian Population Dynamics Inference: A Coalescent-Based Model for Multiple Loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, V.B.; Caldana, G.D.; de Menezes Mayer, A.; Cybis, G.B.; Neves, C.A.M.; Ferrareze, P.A.G.; Demoliner, M.; de Almeida, P.R.; Gularte, J.S.; Hansen, A.W.; et al. Genomic epidemiology of SARS-CoV-2 in Esteio, Rio Grande do Sul, Brazil. BMC Genom. 2021, 22, 371. [Google Scholar] [CrossRef]
- Slavov, S.N.; Giovanetti, M.; dos Santos Bezerra, R.; Fonseca, V.; Santos, E.V.; Rodrigues, E.S.; Adelino, T.; Xavier, J.; Borges, J.S.; Evaristo, M.; et al. Molecular surveillance of the on-going SARS-COV-2 epidemic in Ribeirao Preto City, Brazil. Infect. Genet. Evol. 2021, 93, 104976. [Google Scholar] [CrossRef]
- Fujino, T.; Nomoto, H.; Kutsuna, S.; Ujiie, M.; Suzuki, T.; Sato, R.; Fujimoto, T.; Kuroda, M.; Wakita, T.; Ohmagari, N. Novel SARS-CoV-2 Variant in Travelers from Brazil to Japan. Emerg. Infect. Dis. 2021, 27, 1243. [Google Scholar] [CrossRef]
- Gularte, J.S.; da Silva, M.S.; Mosena, A.C.S.; Demoliner, M.; Hansen, A.W.; Filippi, M.; Pereira, V.M.d.A.G.; Heldt, F.H.; Weber, M.N.; de Almeida, P.R.; et al. Early introduction.; dispersal and evolution of Delta SARS-CoV-2 in Southern Brazil, late predominance of AY.99.2 and AY.101 related lineages. Virus Res. 2022, 311, 198702. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, C.K.V.; Gräf, T.; Barcia, C.A.L.; Costa, V.F.; de Oliveira, J.L.; Passos, R.D.H.; Bastos, I.N.; de Santana, M.C.B.; Santos, I.M.; de Sousa, K.A.F.; et al. SARS-CoV-2 variant of concern P.1 (Gamma) infection in young and middle-aged patients admitted to the intensive care units of a single hospital in Salvador, Northeast Brazil, February 2021. Int. J. Infect. Dis. 2021, 111, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; Corado, A.L.; Nascimento, F.; Silva, G.; da Costa, C.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, was driven by the persistence of endemic lineages and P.1 emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Sabino, E.C.; Buss, L.F.; Carvalho, M.P.S.; Prete, C.A., Jr.; Crispim, M.A.E.; Fraiji, N.A.; Pereira, R.H.M.; Parag, K.V.; da Silva Peixoto, P.; Kraemer, M.U.G.; et al. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet 2021, 397, 452–455. [Google Scholar] [CrossRef]
- Sonabend, R.; Whittles, L.K.; Imai, N.; Perez-Guzman, P.N.; Knock, E.S.; Rawson, T.; Gaythorpe, K.A.M.; Djaafara, B.; Hinsley, W.; FitzJohn, R.G.; et al. Non-pharmaceutical interventions, vaccination, and the SARS-CoV-2 delta variant in England: A mathematical modelling study. Lancet 2021, 398, 1825–1835. [Google Scholar] [CrossRef]
- Moran, E.F. Roads and Dams: Infrastructure-Driven Transformations in the Brazilian Amazon. Ambiente Soc. 2016, 19, 207–220. [Google Scholar] [CrossRef]
- Naveca, F.; Nascimento, V.; Souza, V.; de Lima Corado, A.; Nascimento, F.; Mejía, M.; Brandão, M.J.; da Silva, A.V.; Benzaken, A.S.; Silva, G.; et al. The SARS-CoV-2 Variant Delta Displaced the Variants Gamma and Gamma Plus in Amazonas, Brazil. Available online: https://virological.org/t/the-sars-cov-2-variant-delta-displaced-the-variants-gamma-and-gamma-plus-in-amazonas-brazil/765 (accessed on 14 November 2021).
- Patané, J.; Viala, V.; Lima, L.; Martins, A.; Barros, C.; Marqueze, E.; Bernardino, J.; Moretti, D.; Slavov, S.; Bezerra, R.; et al. SARS-CoV-2 Delta variant of concern in Brazil—Multiple introductions, communitary transmission, and early signs of local evolution. medRxiv 2021. [Google Scholar] [CrossRef]
- Albuquerque, M.G.; Santos, J.R.; Silveira, A.F.; Bornia Junior, D.L.; Nunes, R.B.; Gandra, T.B.R.; da Silva, A.L.C.; de Miranda, G.B.; Trevisol, A.R. Influence of socio-economic indicators and territorial networks at the spatiotemporal spread dynamics of COVID-19 in Brazil. Soc. Nat. 2021, 33, e59688. [Google Scholar] [CrossRef]
- Castro, R.R.; Santos, R.S.C.; Sousa, G.J.B.; Pinheiro, Y.T.; Martins, R.R.I.M.; Pereira, M.L.D.; Silva, R.A.R. Spatial dynamics of the COVID-19 pandemic in Brazil. Epidemiol. Infect. 2021, 149, e60. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, U.J.B.; dos Santos, R.N.; de Melo, F.L.; Belmok, A.; Galvão, J.D.; de Rezende, T.C.V.; Cardoso, F.D.P.; Carvalho, R.F.; da Silva Oliveira, M.; Ribeiro Junior, J.C.; et al. Genomic Epidemiology of SARS-CoV-2 in Tocantins State and the Diffusion of P.1.7 and AY.99.2 Lineages in Brazil. Viruses 2022, 14, 659. https://doi.org/10.3390/v14040659
de Souza UJB, dos Santos RN, de Melo FL, Belmok A, Galvão JD, de Rezende TCV, Cardoso FDP, Carvalho RF, da Silva Oliveira M, Ribeiro Junior JC, et al. Genomic Epidemiology of SARS-CoV-2 in Tocantins State and the Diffusion of P.1.7 and AY.99.2 Lineages in Brazil. Viruses. 2022; 14(4):659. https://doi.org/10.3390/v14040659
Chicago/Turabian Stylede Souza, Ueric José Borges, Raíssa Nunes dos Santos, Fernando Lucas de Melo, Aline Belmok, Jucimária Dantas Galvão, Tereza Cristina Vieira de Rezende, Franciano Dias Pereira Cardoso, Rogério Fernandes Carvalho, Monike da Silva Oliveira, Jose Carlos Ribeiro Junior, and et al. 2022. "Genomic Epidemiology of SARS-CoV-2 in Tocantins State and the Diffusion of P.1.7 and AY.99.2 Lineages in Brazil" Viruses 14, no. 4: 659. https://doi.org/10.3390/v14040659