
Could Phylogenetic Analysis Be Used for Feline Leukemia Virus (FeLV) Classification?

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Recombination Analysis
2.3. Phylogenetic Reconstruction
3. Results
3.1. Dataset Curation
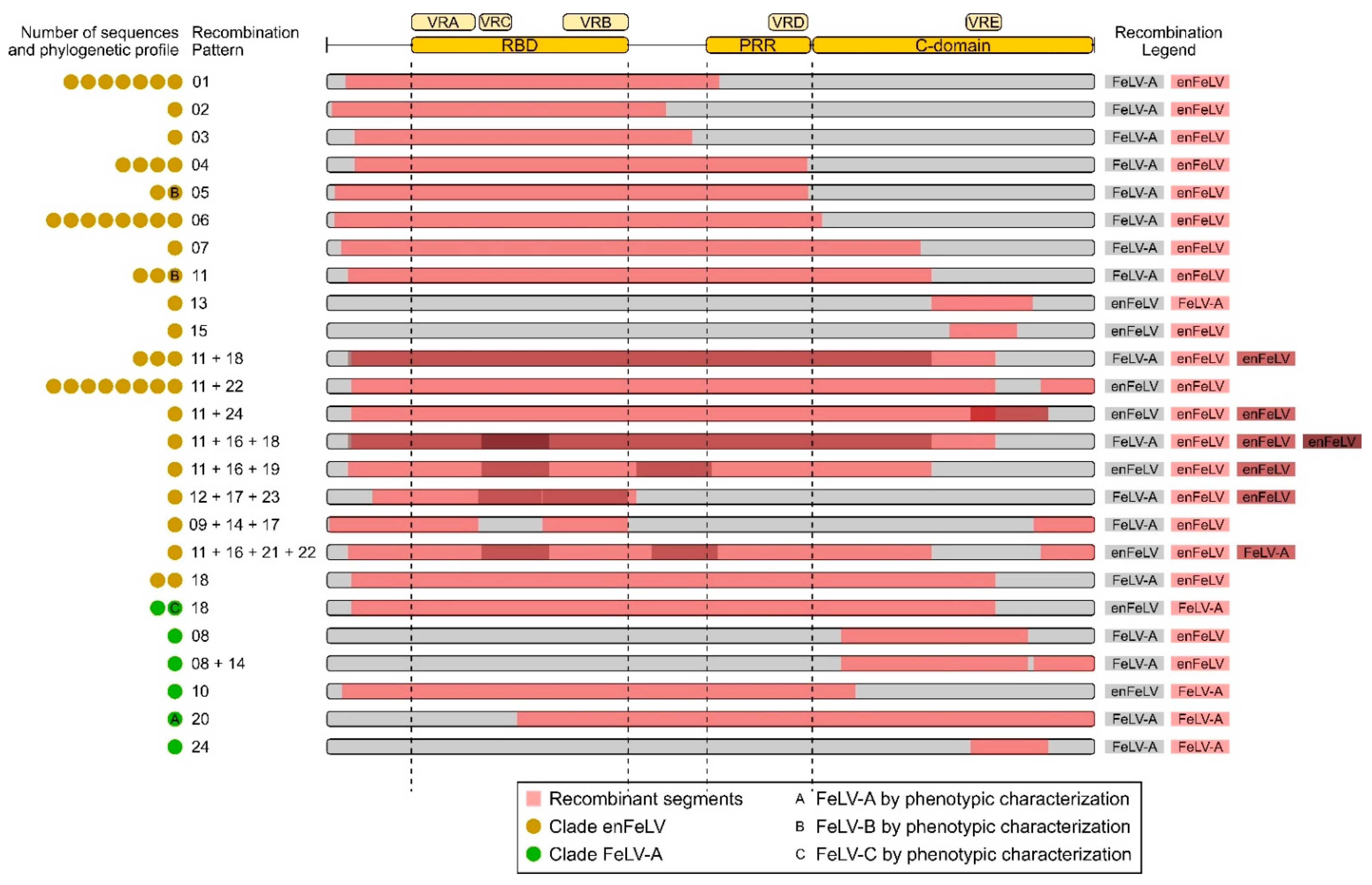
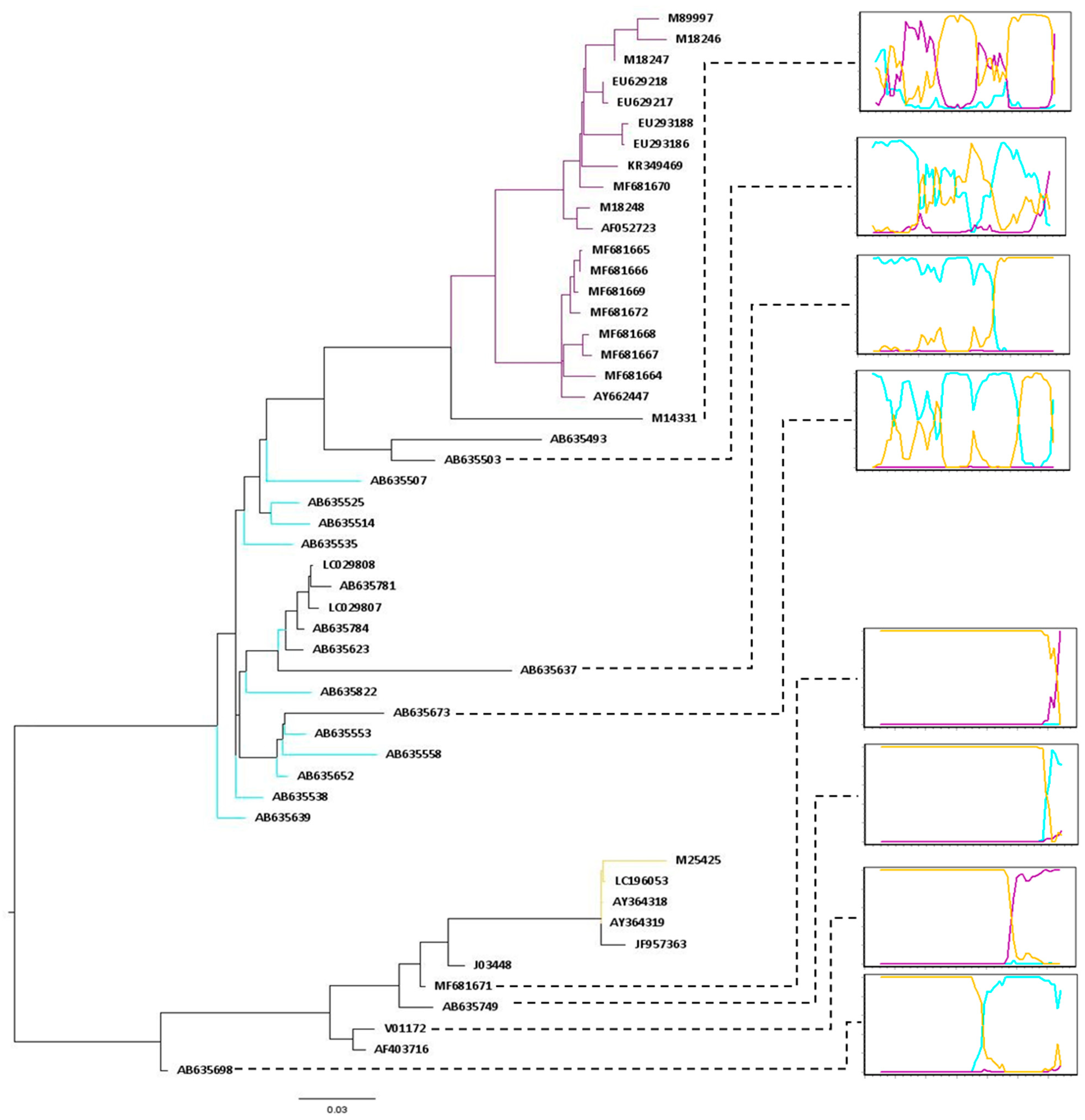
3.2. Different Patterns of Recombination Were Identified
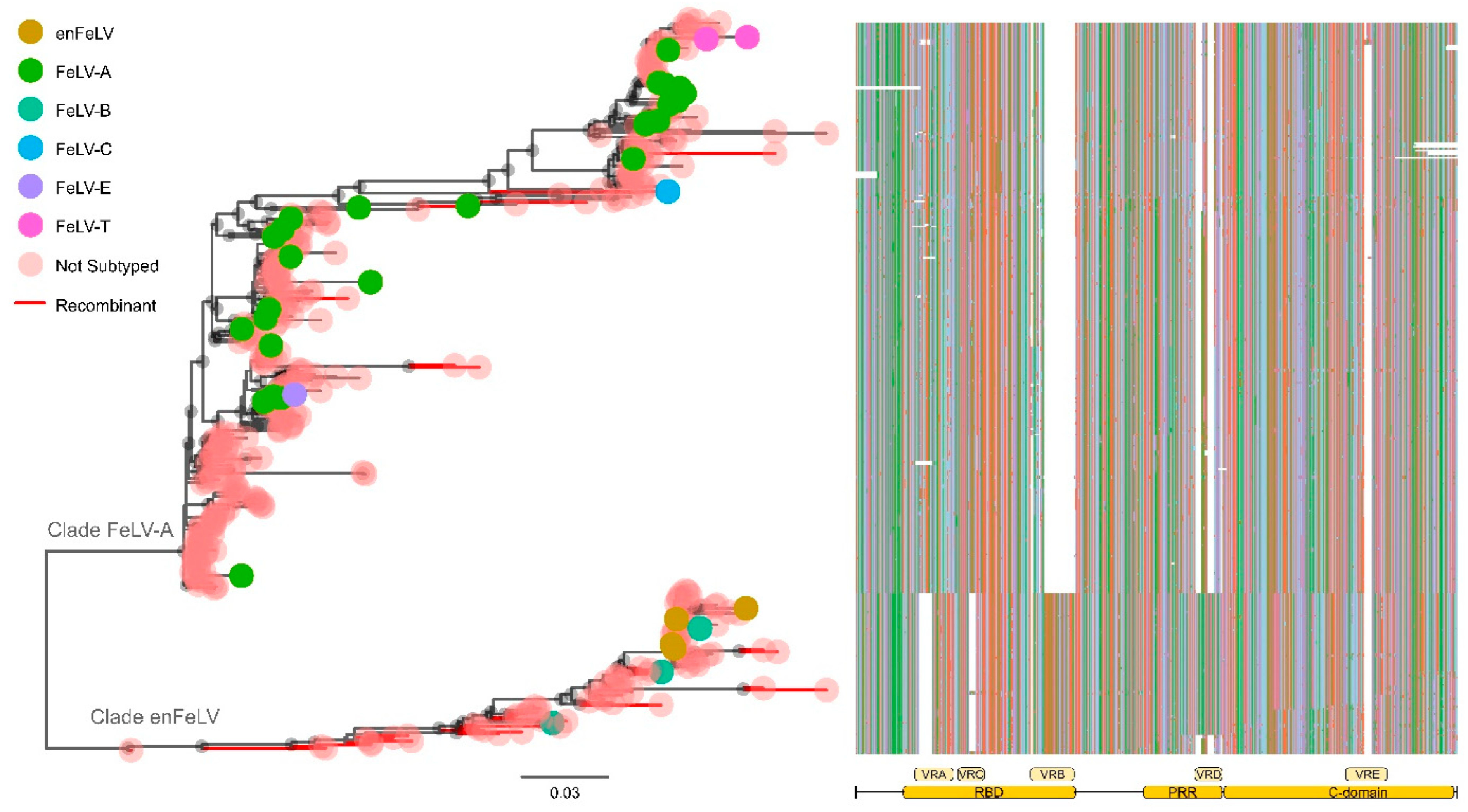
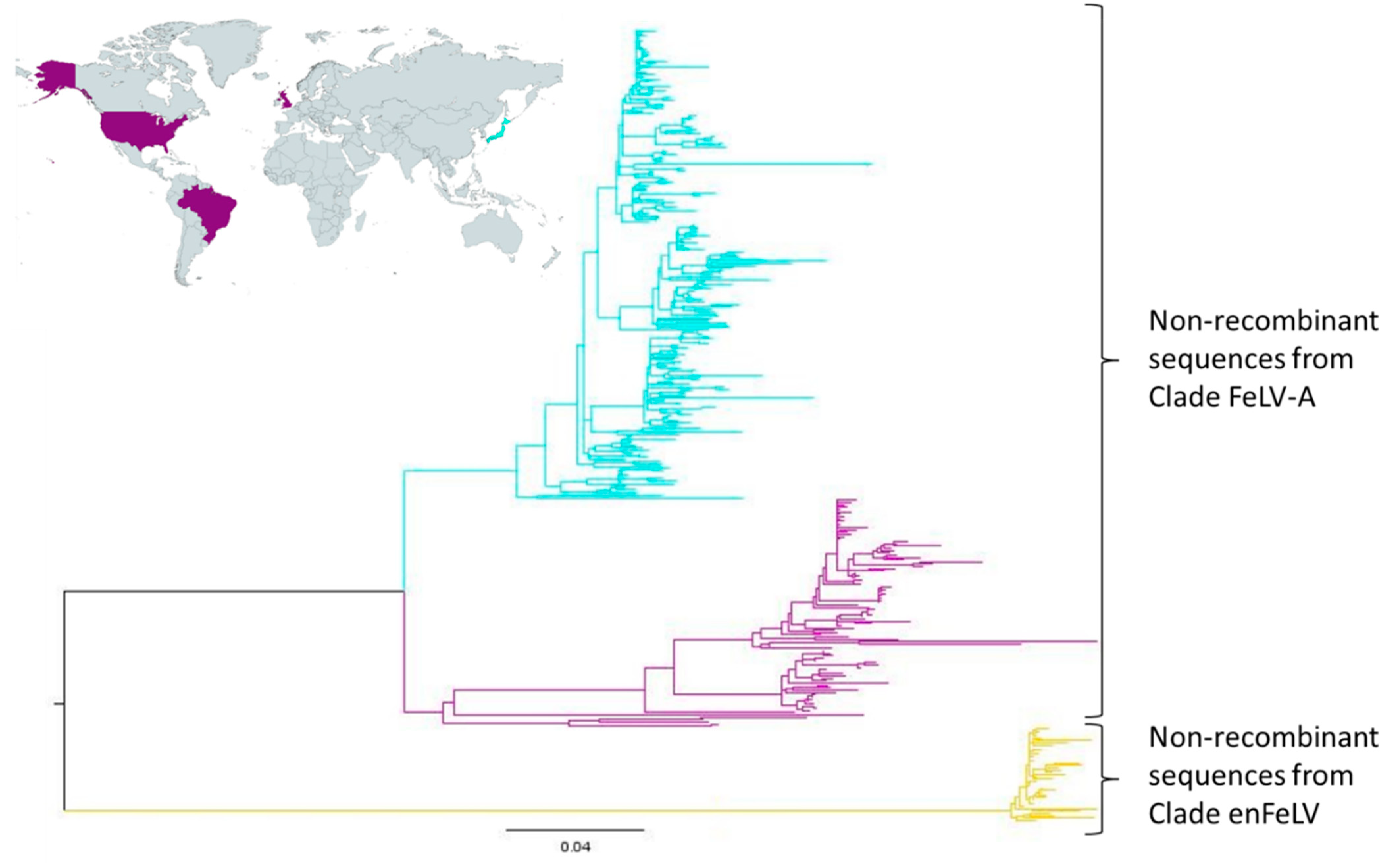
3.3. FeLV SU Gene Can Be Divided into Two Large Clades
3.4. The SU Envelope Gene Has Five Variable Regions; the Pattern of the Variable Regions Is Conserved Intraclade
3.5. The FeLV-A Clade
3.6. The enFeLV Clade
3.7. Geographical Distribution of FeLV-A Clade
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levy, L.S. Advances in understanding molecular determinants in FeLV pathology. Vet. Immunol. Immunopathol. 2008, 123, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, K. Clinical aspects of feline retroviruses: A review. Viruses 2012, 4, 2684–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Kawamura, M.; Odahara, Y.; Anai, Y.; Ochi, H.; Nakagawa, S.; Endo, Y.; Tsujimoto, H.; Nishigaki, K. Phylogenetic and Structural Diversity in the Feline Leukemia Virus Env Gene. PLoS ONE 2013, 8, e61009. [Google Scholar] [CrossRef]
- Powers, J.A.; Chiu, E.S.; Kraberger, S.J.; Roelke-Parker, M.; Lowery, I.; Erbeck, K.; Troyer, R.; Carver, S.; VandeWoude, S. Feline Leukemia Virus (FeLV) Disease Outcomes in a Domestic Cat Breeding Colony: Relationship to Endogenous FeLV and Other Chronic Viral Infections. J. Virol. 2018, 92, e00649-18. [Google Scholar] [CrossRef] [Green Version]
- Vobis, M.; D’Haese, J.; Mehlhorn, H.; Mencke, N. Evidence of horizontal transmission of feline leukemia virus by the cat flea (Ctenocephalides felis). Parasitol. Res. 2003, 91, 467–470. [Google Scholar] [CrossRef]
- Willett, B.J.; Hosie, M.J. Feline leukaemia virus: Half a century since its discovery. Vet. J. 2013, 195, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Addie, D.D.; Toth, S.; Reid, S.; Jarrett, O.; Dennis, J.M.; Callanan, J.J. Long-term impact on a closed household of pet cats of natural infection with feline coronavirus, feline leukaemia virus and feline immunodeficiency virus. Vet. Rec. 2000, 146, 419–424. [Google Scholar] [CrossRef]
- Patel, M.; Carritt, K.; Lane, J.; Jayappa, H.; Stahl, M.; Bourgeois, M. Comparative efficacy of feline leukemia virus (FeLV) inactivated whole-virus vaccine and canarypox virus-vectored vaccine during virulent FeLV challenge and immunosuppression. Clin. Vaccine Immunol. 2015, 22, 798–805. [Google Scholar] [CrossRef] [Green Version]
- Coffin, J.M. Evolution of Retroviruses: Fossils in Our Dna. Proc. Am. Philos. Soc. 2004, 148, 264–280. [Google Scholar]
- Polani, S.; Roca, A.L.; Rosensteel, B.B.; Kolokotronis, S.O.; Bar-Gal, G.K. Evolutionary dynamics of endogenous feline leukemia virus proliferation among species of the domestic cat lineage. Virology 2010, 405, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Koshy, R.; Gallo, R.C.; Wong-Staal, F. Characterization of the endogenous feline leukemia virus-related DNA sequences in cats and attempts to identify exogenous viral sequences in tissues of virus-negative leukemic animals. Virology 1980, 103, 434–445. [Google Scholar] [CrossRef]
- Busch, M.P.; Devi, B.G.; Soe, L.H.; Roy-Burman, P.; Perbal, B.; Marcel, A. Baluda Characterization of the expression of cellular retrovirus genes and oncogenes in feline cells. Hematol. Oncol. 1983, 1, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Cattori, V.; Willi, B.; Lutz, H.; Hofmann-Lehmann, R. Quantification of endogenous and exogenous feline leukemia virus sequences by real-time PCR assays. Vet. Immunol. Immunopathol. 2008, 123, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.B. Interactions between exogenous and endogenous retroviruses. J. Biomed. Sci. 1997, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Cattori, V.; Willi, B.; Meli, M.L.; Gomes-Keller, M.A.; Lutz, H.; Hofmann-Lehmann, R. Copy number polymorphism of endogenous feline leukemia virus-like sequences. Mol. Cell. Probes 2007, 21, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Cattori, V.; Pepin, A.C.; Riond, B.; Meli, M.L.; McDonald, M.; Doherr, M.G.; Lutz, H.; Hofmann-Lehmann, R. Association between endogenous feline leukemia virus loads and exogenous feline leukemia virus infection in domestic cats. Virus Res. 2008, 135, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Chiu, E.S.; Hoover, E.A.; VandeWoude, S. A Retrospective Examination of Feline Leukemia Subgroup Characterization: Viral Interference Assays to Deep Sequencing. Viruses 2018, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauring, A.S.; Anderson, M.M.; Overbaugh, J. Specificity in receptor usage by T-cell-tropic feline leukemia viruses: Implications for the in vivo tropism of immunodeficiency-inducing variants. J. Virol. 2001, 75, 8888–8898. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, I.K.; Spibey, N.; Jarrett, O. The receptor binding site of feline leukemia virus surface glycoprotein is distinct from the site involved in virus neutralization. J. Virol. 1998, 72, 3268–3277. [Google Scholar] [CrossRef] [Green Version]
- Erbeck, K.; Gagne, R.B.; Kraberger, S.; Chiu, E.S.; Roelke-Parker, M.; VandeWoude, S. Feline Leukemia Virus (FeLV) Endogenous and Exogenous Recombination Events Result in Multiple FeLV-B Subtypes during Natural Infection. J. Virol. 2021, 95, 1–10. [Google Scholar] [CrossRef]
- Odelberg, S.J.; Weiss, R.B.; Hata, A.; White, R. Template-switching during DNA synthesis by Thermus aquaticus DNA polymerase I. Nucleic Acids Res. 1995, 23, 2049–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrett, O.; Hardy, W.D.; Golder, M.C.; Hay, D. The frequency of occurrence of feline leukaemia virus subgroups in cats. Int. J. Cancer 1978, 21, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Sarma, P.S.; Log, T. Subgroup classification of feline leukemia and sarcoma viruses by viral interference and neutralization tests. Virology 1973, 54, 160–169. [Google Scholar] [CrossRef]
- Jarrett, O.; Golder, M.C.; Toth, S.; Onions, D.E.; Stewart, M.F. Interaction between feline leukaemia virus subgroups in the pathogenesis of erythroid hypoplasia. Int. J. Cancer 1984, 34, 283–288. [Google Scholar] [CrossRef]
- Anai, Y.; Ochi, H.; Watanabe, S.; Nakagawa, S.; Kawamura, M.; Gojobori, T.; Nishigaki, K. Infectious endogenous retroviruses in cats and emergence of recombinant viruses. J. Virol. 2012, 86, 8634–8644. [Google Scholar] [CrossRef] [Green Version]
- Miyake, A.; Watanabe, S.; Hiratsuka, T.; Ito, J.; Ngo, M.H.; Makundi, I.; Kawasaki, J.; Endo, Y.; Tsujimoto, H.; Nishigakia, K. Novel Feline Leukemia Virus Interference Group Based on the env Gene. J. Virol. 2016, 90, 4832–4837. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree v1.4: Tree Figure Drawing Tool 2009.
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Sarma, P.S.; Log, T. Viral interference in feline leukemia-sarcoma complex. Virology 1971, 44, 352–358. [Google Scholar] [CrossRef]
- Mendoza, R.; Anderson, M.M.; Overbaugh, J. A Putative Thiamine Transport Protein Is a Receptor for Feline Leukemia Virus Subgroup A. J. Virol. 2006, 80, 3378–3385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudra-Ganguly, N.; Ghosh, A.K.; Roy-Burman, P. Retrovirus receptor PiT-1 of the Felis catus. Biochim. Biophys. Acta-Gene Struct. Expr. 1998, 1443, 407–413. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Vile, R.G.; Simpson, G.; O’Hara, B.; Collins, M.K.; Weiss, R.A. Feline leukemia virus subgroup B uses the same cell surface receptor as gibbon ape leukemia virus. J. Virol. 1992, 66, 1219–1222. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.M.; Lauring, A.S.; Robertson, S.; Dirks, C.; Overbaugh, J. Feline Pit2 Functions as a Receptor for Subgroup B Feline Leukemia Viruses. J. Virol. 2001, 75, 10563–10572. [Google Scholar] [CrossRef] [Green Version]
- Tailor, C.S.; Willett, B.J.; Kabat, D. A Putative Cell Surface Receptor for Anemia-Inducing Feline Leukemia Virus Subgroup C Is a Member of a Transporter Superfamily. J. Virol. 1999, 73, 6500–6505. [Google Scholar] [CrossRef] [Green Version]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Riedel, N.; Hoover, E.A.; Gasper, P.W.; Nicolson, M.O.; Mullins, J.I. Molecular analysis and pathogenesis of the feline aplastic anemia retrovirus, feline leukemia virus C-Sarma. J. Virol. 1986, 60, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.M.; Lauring, A.S.; Burns, C.C.; Overbaugh, J. Identification of a Cellular Cofactor Required for Infection by Feline Leukemia Virus. Science 2000, 287, 1828–1830. [Google Scholar] [CrossRef] [PubMed]
- Hoover, E.A.; Mullins, J.I.; Quackenbush, S.L.; Gasper, P.W. Experimental transmission and pathogenesis of immunodeficiency syndrome in cats. Blood 1987, 70, 1880–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.A.; Cunningham, M.W.; Roca, A.L.; Troyer, J.L.; Johnson, W.E.; O’Brien, S.J. Genetic characterization of feline leukemia virus from Florida panthers. Emerg. Infect. Dis. 2008, 14, 252. [Google Scholar] [CrossRef] [PubMed]
- Ortega, C.; Valencia, A.C.; Duque-Valencia, J.; Ruiz-Saenz, J. Prevalence and Genomic Diversity of Feline Leukemia Virus in Privately Owned and Shelter Cats in Aburrá Valley, Colombia. Viruses 2020, 12, 464. [Google Scholar] [CrossRef] [Green Version]
- Rey, M.A.; Prasad, R.; Tailor, C.S. The C domain in the surface envelope glycoprotein of subgroup C feline leukemia virus is a second receptor-binding domain. Virology 2008, 370, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Shojima, T.; Nakata, R.; Miyazawa, T. Host cell range of T-lymphotropic feline leukemia virus in vitro. Biochem. Biophys. Res. Commun. 2006, 345, 1466–1470. [Google Scholar] [CrossRef]
- Stewart, M.A.; Warnock, M.; Wheeler, A.; Wilkie, N.; Mullins, J.I.; Onions, D.E.; Neil, J.C. Nucleotide sequences of a feline leukemia virus subgroup A envelope gene and long terminal repeat and evidence for the recombinational origin of subgroup B viruses. J. Virol. 1986, 58, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Roy-Burman, P. Endogenousenv elements: Partners in generation of pathogenic feline leukemia viruses. Virus Genes 1995, 11, 147–161. [Google Scholar] [CrossRef]
- Pandey, R.; Ghosh, A.K.; Kumar, D.V.; Bachman, B.A.; Shibata, D.; Roy-Burman, P. Recombination between feline leukemia virus subgroup B or C and endogenous env elements alters the in vitro biological activities of the viruses. J. Virol. 1991, 65, 6495–6508. [Google Scholar] [CrossRef] [Green Version]
- Stewart, H.; Jarrett, O.; Hosie, M.J.; Willett, B.J. Are endogenous feline leukemia viruses really endogenous? Vet. Immunol. Immunopathol. 2011, 143, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cano-Ortiz, L.; Tochetto, C.; Roehe, P.M.; Franco, A.C.; Junqueira, D.M. Could Phylogenetic Analysis Be Used for Feline Leukemia Virus (FeLV) Classification? Viruses 2022, 14, 249. https://doi.org/10.3390/v14020249
Cano-Ortiz L, Tochetto C, Roehe PM, Franco AC, Junqueira DM. Could Phylogenetic Analysis Be Used for Feline Leukemia Virus (FeLV) Classification? Viruses. 2022; 14(2):249. https://doi.org/10.3390/v14020249
Chicago/Turabian StyleCano-Ortiz, Lucía, Caroline Tochetto, Paulo Michel Roehe, Ana Cláudia Franco, and Dennis Maletich Junqueira. 2022. "Could Phylogenetic Analysis Be Used for Feline Leukemia Virus (FeLV) Classification?" Viruses 14, no. 2: 249. https://doi.org/10.3390/v14020249