
Tracking SARS-CoV-2 Spike Protein Mutations in the United States (January 2020—March 2021) Using a Statistical Learning Strategy

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
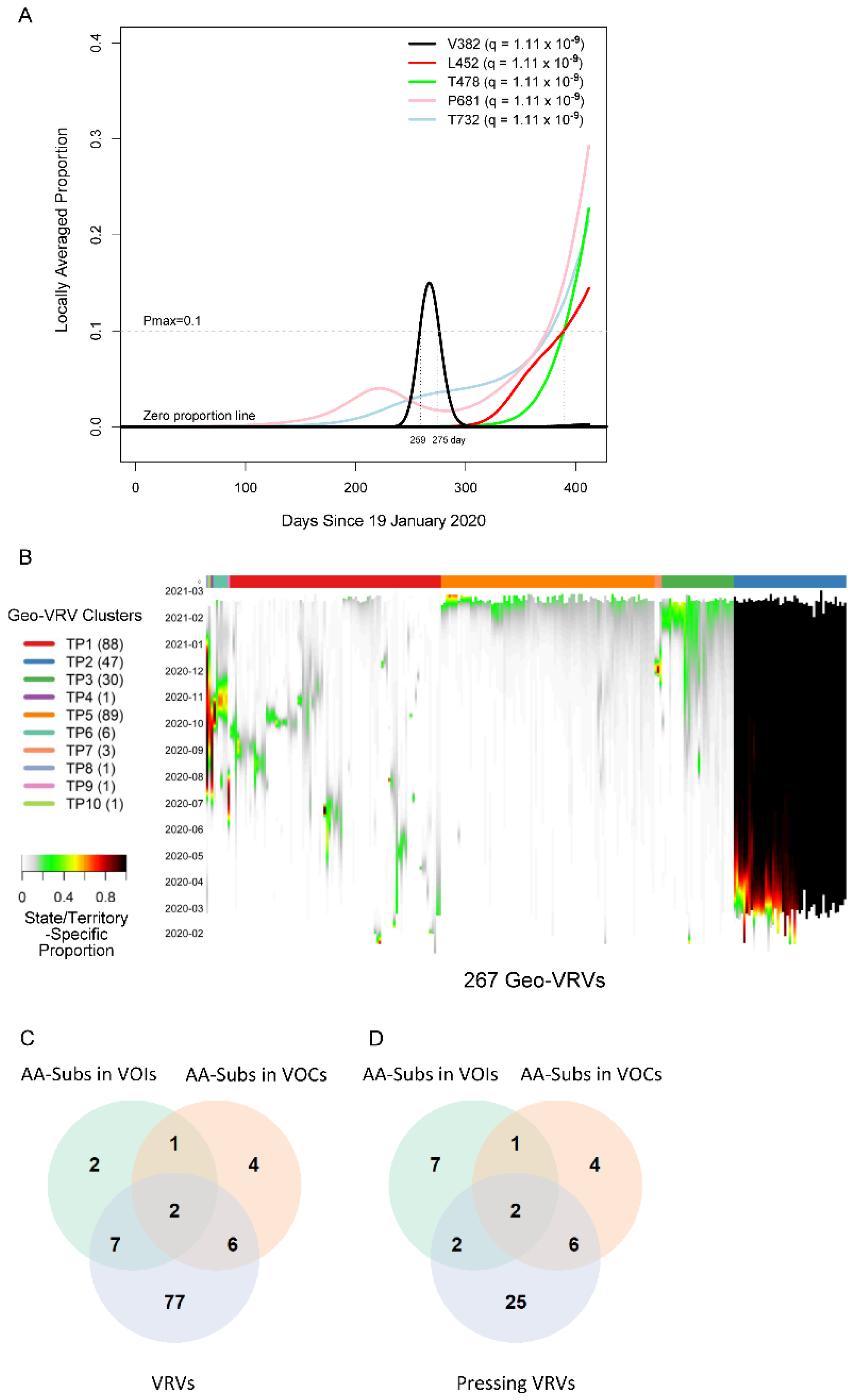
2.1. Ab Initio Discovery of VRVs
2.2. Comparison with AA Positions Where Substitutions Have Been Identified within US-Circulating VOIs and VOCs
2.3. Timely Detection of Emerging VRVs
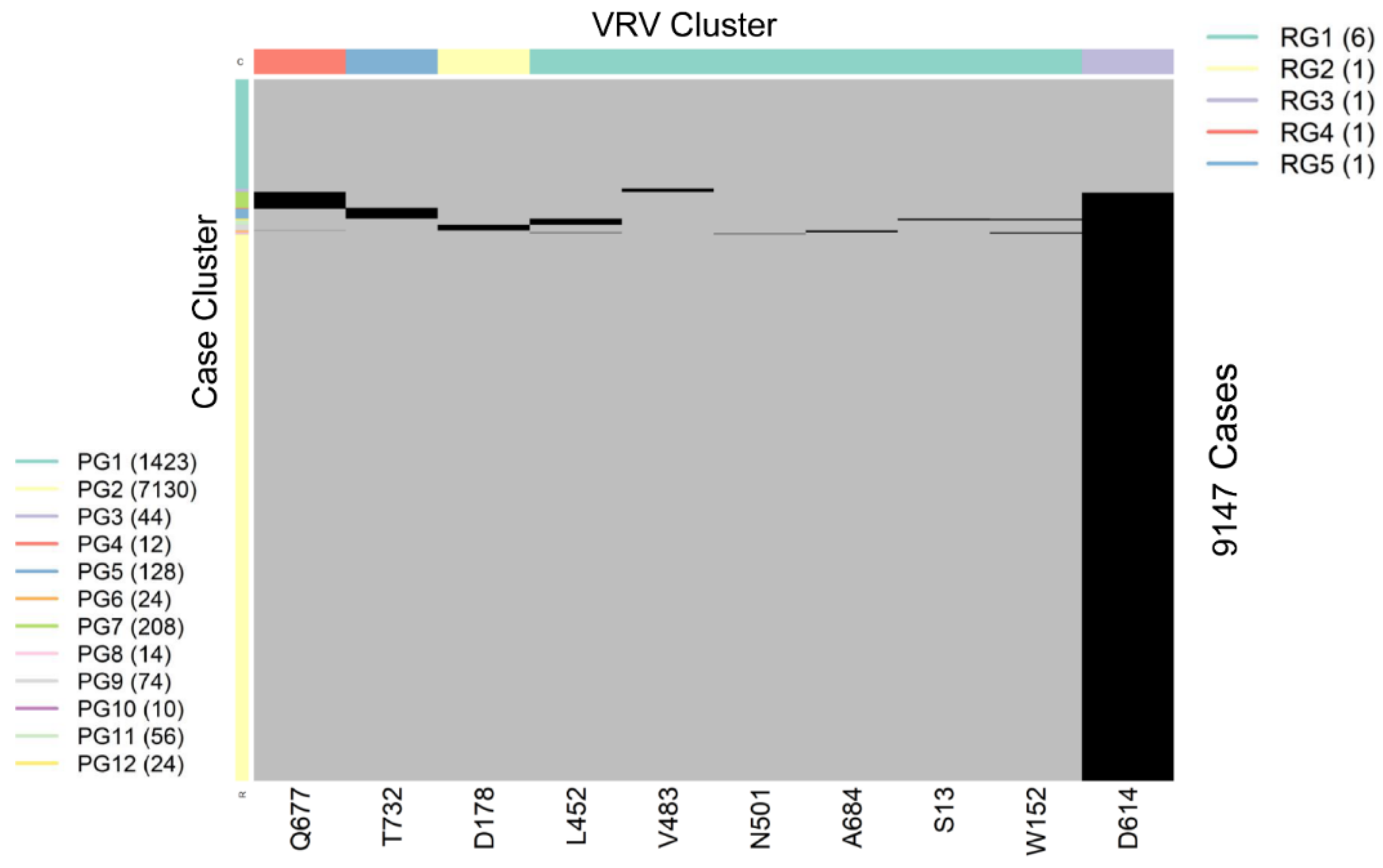
2.4. VRV-Haplotypes
2.5. Naming VRV-Haplotypes via PANGO Lineages
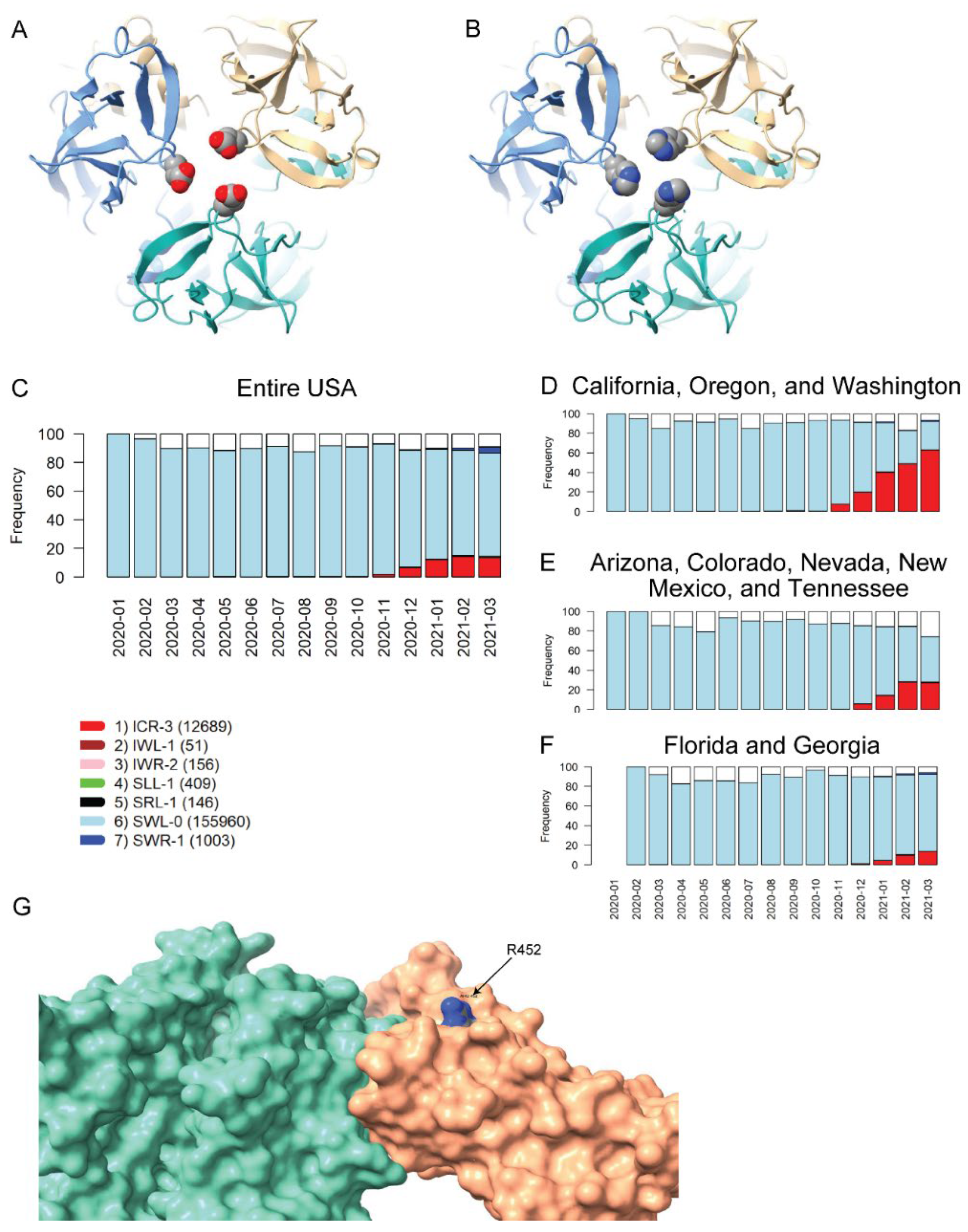
2.6. Impact of VRV Haplotypes on Viral Structure
3. Materials and Methods
3.1. Spike AA Sequences
3.2. Sequence Alignment and Transformation to VRV Indicators
3.3. Statistical Learning Strategy (SLS)
3.3.1. Modeling VRV Temporal Dynamics
3.3.2. Visual Representation of Temporal Dynamics
3.3.3. Missing Residue Imputation
3.3.4. VRV-Haplotypes
3.3.5. Homology Modeling of Selected Haplotype Mutants
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [Green Version]
- US Centers for Disease Control and Prevention. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variant-info.html (accessed on 4 December 2021).
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-Gonzalez, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell 2021, 184, 3426–3437.e8. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Voloch, C.M.; da Silva Francisco, R., Jr.; de Almeida, L.G.P.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; Guimaraes, A.P.C.; Mariani, D.; da Costa, R.M.; Ferreira, O.C., Jr.; et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. J. Virol. 2021, 95, e00119-21. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, A.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Collier, D.A.; De Marco, A.; Ferreira, I.; Meng, B.; Datir, R.P.; Walls, A.C.; Kemp, S.A.; Bassi, J.; Pinto, D.; Silacci-Fregni, C.; et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 2021, 593, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Supasa, P.; Zhou, D.; Dejnirattisai, W.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Nutalai, R.; Tuekprakhon, A.; et al. Reduced neutralization of SARS-CoV-2 B.1.1.7 variant by convalescent and vaccine sera. Cell 2021, 184, 2201–2211.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants, B.1.351 and B.1.1.7. bioRxiv 2021, 428137. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; St Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383. [Google Scholar] [CrossRef]
- Madhi, S.A.; Baillie, V.; Cutland, C.L.; Voysey, M.; Koen, A.L.; Fairlie, L.; Padayachee, S.D.; Dheda, K.; Barnabas, S.L.; Bhorat, Q.E.; et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. N. Engl. J. Med. 2021, 384, 1885–1898. [Google Scholar] [CrossRef]
- Rubin, R. COVID-19 vaccines vs variants-determining how much immunity is enough. JAMA 2021, 325, 1241–1243. [Google Scholar] [CrossRef]
- Altmann, D.M.; Boyton, R.J.; Beale, R. Immunity to SARS-CoV-2 variants of concern. Science 2021, 371, 1103–1104. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.A.; Read, A.F. Monitor for COVID-19 vaccine resistance evolution during clinical trials. PLoS Biol. 2020, 18, e3001000. [Google Scholar] [CrossRef]
- Maison, D.P.; Ching, L.L.; Shikuma, C.M.; Nerurkar, V.R. Genetic characteristics and phylogeny of 969-bp S gene sequence of SARS-CoV-2 from Hawaii reveals the worldwide emerging P681H mutation. Hawaii J. Health Soc. Welf. 2021, 80, 52–61. [Google Scholar] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Rouchka, E.C.; Chariker, J.H.; Chung, D. Variant analysis of 1040 SARS-CoV-2 genomes. PLoS ONE 2020, 15, e0241535. [Google Scholar]
- Bindayna, K.M.; Crinion, S. Variant analysis of SARS-CoV-2 genomes in the Middle East. Microb. Pathog. 2021, 153, 104741. [Google Scholar] [CrossRef] [PubMed]
- Studdert, D.M.; Hall, M.A.; Mello, M.M. Partitioning the curve-interstate travel restrictions during the Covid-19 pandemic. N. Engl. J. Med. 2020, 383, e83. [Google Scholar] [CrossRef]
- Arora, P.; Pohlmann, S.; Hoffmann, M. Mutation D614G increases SARS-CoV-2 transmission. Signal. Transduct. Target. Ther. 2021, 6, 101. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Z.; Jackson, C.B.; Mou, H.H.; Ojha, A.; Peng, H.Y.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.D.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N.; et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 2021, 372, 525–530. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Q.; Wei, P.; Chen, Z.; Aviszus, K.; Yang, J.; Downing, W.; Jiang, C.; Liang, B.; Reynoso, L.; et al. The basis of a more contagious 501Y.V1 variant of SARS-CoV-2. Cell Res. 2021, 31, 720–722. [Google Scholar] [CrossRef]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, 373, 648–654. [Google Scholar] [CrossRef]
- Shu, Y.L.; McCauley, J. GISAID: Global initiative on sharing all influenza data-from vision to reality. Eurosurveillance 2017, 22, 2–4. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, Z.; Chen, Z.; Huang, X.; Xu, M.; He, T.; Zhang, Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020, 92, 667–674. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [Green Version]
- Hastie, T.J. Generalized Additive Models; Chapman and Hall: New York, NY, USA, 1990. [Google Scholar]
- Storey, J.D.; Taylor, J.E.; Siegmund, D. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: A unified approach. J. R. Stat. Soc. 2004, 66, 187–205. [Google Scholar] [CrossRef]
- Wood, S.N.; Pya, N.; Safken, B. Smoothing parameter and model selection for general smooth models. J. Am. Stat. Assoc. 2016, 111, 1548–1563. [Google Scholar] [CrossRef]
- Murtagh, F.; Legendre, P. Ward’s hierarchical agglomerative clustering method: Which algorithms implement Ward’s criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- Scheet, P.; Stephens, M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Neerukonda, S.N.; Vassel, R.; Lusvarghi, S.; Wang, R.; Echegaray, F.; Bentley, L.; Eakin, A.E.; Erlandson, L.J.; Katzelnick, L.C.; Weiss, C.D.; et al. SARS-CoV-2 Delta Variant Displays Moderate Resistance to Neutralizing Antibodies and Spike Protein Properties of Higher Soluble ACE2 Sensitivity, Enhanced Cleavage and Fusogenic Activity. Viruses 2021, 13, 2485. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. bioRxiv 2021, 456173. [Google Scholar] [CrossRef]
- Rolland, M.; Gilbert, P.B. Sieve analysis to understand how SARS-CoV-2 diversity can impact vaccine protection. PLoS Pathog. 2021, 17, e1009406. [Google Scholar] [CrossRef]
- Follmann, D.; Fintzi, J.; Fay, M.P.; Janes, H.E.; Baden, L.R.; El Sahly, H.M.; Fleming, T.R.; Mehrotra, D.V.; Carpp, L.N.; Juraska, M.; et al. A deferred-vaccination design to assess durability of COVID-19 vaccine effect after the placebo group is vaccinated. Ann. Intern. Med. 2021, 174, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Addetia, A.; Crawford, K.H.D.; Dingens, A.; Zhu, H.; Roychoudhury, P.; Huang, M.L.; Jerome, K.R.; Bloom, J.D.; Greninger, A.L. Neutralizing antibodies correlate with protection from SARS-CoV-2 in humans during a fishery vessel outbreak with a high attack rate. J. Clin. Microbiol. 2020, 58, 11. [Google Scholar] [CrossRef]
- Letizia, A.G.; Ge, Y.; Vangeti, S.; Goforth, C.; Weir, D.L.; Kuzmina, N.A.; Balinsky, C.A.; Chen, H.W.; Ewing, D.; Soares-Schanoski, A.; et al. SARS-CoV-2 seropositivity and subsequent infection risk in healthy young adults: A prospective cohort study. Lancet Respir. Med. 2021, 9, 712–720. [Google Scholar] [CrossRef]
- Earle, K.A.; Ambrosino, D.M.; Fiore-Gartland, A.; Goldblatt, D.; Gilbert, P.B.; Siber, G.R.; Dull, P.; Plotkin, S.A. Evidence for antibody as a protective correlate for COVID-19 vaccines. Vaccine 2021, 39, 4423–4428. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Phillips, D.J.; White, T.; Sayal, H.; Aley, P.K.; Bibi, S.; Dold, C.; Fuskova, M.; Gilbert, S.C.; Hirsch, I.; et al. Correlates of protection against symptomatic and asymptomatic SARS-CoV-2 infection. Nat. Med. 2021, 27, 2032–2040. [Google Scholar] [CrossRef]
- Gilbert, P.B.; Montefiori, D.C.; McDermott, A.B.; Fong, Y.; Benkeser, D.; Deng, W.; Zhou, H.; Houchens, C.R.; Martins, K.; Jayashankar, L.; et al. Immune correlates analysis of the mRNA-1273 COVID-19 vaccine efficacy clinical trial. Science 2021, eab3435. [Google Scholar] [CrossRef]
- McMahan, K.; Yu, J.; Mercado, N.B.; Loos, C.; Tostanoski, L.H.; Chandrashekar, A.; Liu, J.; Peter, L.; Atyeo, C.; Zhu, A.; et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature 2021, 590, 630–634. [Google Scholar] [CrossRef]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell 2021, 184, 2348.e6–2364.e6. [Google Scholar] [CrossRef]
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N.; et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| L5 | S13 | V70 | T95 | W152 | D253 | L452 | S477 | E484 | N501 | A570 | D614 | Q677 | P681 | A701 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reporting Day * | 301 | 301 | 301 | 301 | 301 | 87 | 301 | 331 | 87 | 362 | 362 | 362 | 362 | 362 | 362 |
| Earliest SLS Detection Day Across All States | 11 | 159 | 329 | 149 | 381 | 98 | 381 | 405 | 371 | 206 | 404 | 10 | 20 | 11 | 176 |
| Alabama | 63–186 | - | - | - | - | - | - | - | 404 | - | - | 63 | 253 | - | - |
| Alaska | - | - | - | - | - | - | - | - | - | - | - | 56 | 305–323 | 383 | - |
| Arizona | - | - | - | - | - | - | - | - | - | - | - | 26 | 357–400 | 353 | - |
| Arkansas | - | - | - | - | - | - | - | - | - | - | - | 56 | 329 | - | - |
| California | - | 398 | - | - | 402 | - | 390 | - | - | - | - | 45 | - | 374 | - |
| Colorado | 286 | - | - | - | - | - | - | - | - | - | - | 45 | 286 | 370 | - |
| Connecticut | - | - | - | - | - | - | - | - | - | 314 | - | 43 | 191 | 378 | - |
| DC | - | - | - | - | - | - | - | - | 391 | - | - | 47 | - | 344 | - |
| Delaware | - | - | - | - | - | - | - | - | - | - | - | 52 | 384 | 288 | - |
| Florida | - | - | - | - | - | - | - | - | - | - | - | 33 | 368 | 389 | - |
| Georgia | - | - | - | - | - | - | - | - | - | - | - | 41 | 345 | 407 | - |
| Hawaii | 175–190 | - | - | - | - | - | - | - | - | - | - | 46 | 374 | 174–376 | - |
| Idaho | - | - | - | - | - | - | - | - | - | - | - | 53 | - | - | - |
| Illinois | - | - | - | - | - | - | - | - | - | - | - | 24 | 366 | 380 | - |
| Indiana | - | - | - | - | - | - | - | - | - | - | - | 48 | 370 | 383 | - |
| Iowa | - | - | - | - | - | - | - | - | - | - | - | 48 | 273–388 | - | - |
| Kansas | - | - | - | - | - | 260–291 | - | - | - | - | - | 47 | - | 385 | - |
| Kentucky | - | - | - | - | - | - | - | - | - | - | - | 59 | 389–397 | - | - |
| Louisiana | - | - | - | - | - | - | - | - | - | - | - | 50 | 307 | 368 | - |
| Maine | - | - | - | - | - | - | - | - | 404 | 371 | - | 51 | - | - | - |
| Maryland | - | - | - | - | 407 | - | 394 | - | 390 | - | - | 45 | - | 230 | - |
| Massachusetts | - | - | - | - | - | - | - | - | - | 206 | - | 10 | 346 | 298 | - |
| Michigan | - | - | - | 149–177 | - | - | - | - | - | 264–273 | - | 50 | 361 | - | - |
| Minnesota | 186–294 | - | - | - | - | - | - | - | - | - | - | 46 | 297 | 387 | - |
| Mississippi | 144–215 | - | - | - | - | - | - | - | - | - | - | 42 | 353 | 392 | - |
| Missouri | - | - | - | - | - | - | - | - | - | - | - | 47 | - | 364–384 | - |
| Montana | - | - | - | - | - | - | - | - | - | - | - | 68 | - | - | - |
| Nebraska | - | - | - | - | - | - | - | - | - | - | - | 46 | - | 387 | - |
| Nevada | - | 392 | - | - | 396 | - | 393 | - | - | - | - | 37 | 391 | 394 | - |
| New Hampshire | - | - | - | - | - | - | - | - | - | 374 | - | 41 | 349–390 | 364 | - |
| New Jersey | - | - | - | - | - | - | 402 | - | - | - | - | 44 | - | 276 | - |
| New Mexico | - | - | - | - | - | - | - | - | - | - | - | 50 | 291 | 387 | 233–252 |
| New York | 11–13 | - | - | - | - | - | 386 | - | 414 | - | - | 11 | - | 11 | - |
| North Carolina | - | - | - | - | - | - | - | - | - | - | - | 44 | - | 382 | - |
| North Dakota | - | - | - | - | - | - | - | - | - | - | - | 57 | 328–363 | - | - |
| Ohio | - | - | - | - | - | - | 405 | - | - | - | - | 20 | 20 | 391 | - |
| Oklahoma | - | - | - | - | - | - | - | - | - | - | - | 54 | 306 | - | - |
| Oregon | - | 397 | - | - | - | - | 398 | - | - | - | - | 45 | - | - | - |
| Pennsylvania | - | - | - | - | - | - | - | - | - | - | - | 44 | 394 | 317 | - |
| Puerto Rico | - | - | - | - | - | 185–252 | - | - | - | - | - | 49 | - | 347 | - |
| Rhode Island | - | - | - | - | - | - | - | 405 | 371–384 | 356 | - | 40 | 358–398 | 379 | - |
| South Carolina | - | - | - | - | - | - | - | - | - | - | - | 46 | 405 | 368–389 | - |
| Tennessee | 50–141 | - | - | - | - | - | - | - | - | - | - | 50 | 318 | - | - |
| Texas | - | - | - | - | - | - | - | - | - | - | - | 23 | 360 | 378 | - |
| Utah | - | 159–173 | - | - | - | 98–190 | - | - | - | - | - | 44 | - | 358 | - |
| Virginia | - | - | 329–331 | - | - | - | - | - | 397 | - | - | 47 | 384 | 359 | 176–185 |
| Washington | - | 406 | - | - | 411 | - | 403 | - | - | 410 | - | 50 | 373 | - | - |
| Wisconsin | - | - | - | - | - | - | 409 | - | - | 265–271 | - | 12 | 299–407 | 411 | - |
| Wyoming | - | 381 | - | - | 381 | - | 381 | - | - | - | - | 51 | - | 392 | - |
| Other States | - | - | - | - | - | - | - | - | 393 | 404 | 404 | 34 | - | 368 | 375–381 |
| ID | VRV-Haplotype | Freq | L + | Haplotypic Polymorphism-Number of Substitutions (Frequency) |
|---|---|---|---|---|
| Washington | ||||
| W1 | S13-W152-L452-V483-N501-D614-A684 | 104 | 4 | ICRVNGA-4(20)/IWRVNGA-3(4)/SCRVNGA-3(5)/SLLVNGA-2(5)/SRLVNGA-2(4)/SWLVTGA-2(4)/SWLVYDA-1(1)/SWLVYGA-2(5)/SWQVNGA-2(2)/SWRVNGA-2(54) |
| W2 | D614-Q677-T732 | 12 | 3 | GHS-3(11)/XXX-3(1) |
| W3 | D614-T732 | 128 | 2 | GA-2(126)/GI-2(2) |
| W4 | D614-Q677 | 208 | 2 | DH-1(9)/GH-2(110)/GP-2(89) |
| W5 | D178-D614 | 74 | 2 | GG-2(70)/NG-2(4) |
| W6 | D614 | 7130 | 1 | G-1(7125)/N-1(5) |
| New York | ||||
| N1 | L5-L54-E132-Y453-T478-E484-D614-P681-T732 | 172 | 9 | LLEYKEGHA-4(168)/LLEYKEGHT-3(4) |
| N2 | L5-L54-E132-Y453-T478-E484-D614-T732 | 651 | 8 | FLEYREGT-3(4)/FLEYTEDT-1(11)/FLEYTEGA-3(3)/FLEYTEGS-3(1)/FLEYTEGT-2(266)/FLEYTKGA-4(1)/FLEYTKGT-3(44)/LLEYKEGT-2(3)/ LLEYTAGT-2(1)/LLEYTEGA-2(51)/LLEYTEGI-2(2)/LLEYTEGS-2(24)/LLEYTKGS-3(2)/LLEYTKGT-2(171)/LLEYTQGT-2(8)/LLQYTEGT-2(59) |
| N3 | D80-F157-L452-D614-P681-T859-D950 | 132 | 7 | DFLGHID-3(108)/DFLGPID-2(18)/DFLGPNH-3(4)/DSLGPNH-4(2) |
| N4 | D80-F157-L452-D614-T859-D950 | 637 | 6 | DFQGND-3(4)/DFRGID-3(15)/DFRGNH-4(1)/DFRGTD-2(120)/DFRNTD-2(2)/DSRGNH-5(3)/DSRGTD-3(2)/GFRGND-4(1)/GFRGNH-5(1)/GSLGNH-5(9)/GSRGND-5(10)/GSRGNH-6(455)/GSRGNY-6(1)/GSRGTD-4(13) |
| N5 | S494-D614-P681-T716 | 514 | 4 | PGHI-4(367)/PGHT-3(55)/PGPT-2(52)/SGHI-3(19)/SGHT-2(8)/SGPI-2(13) |
| N6 | D614-P681 | 1161 | 2 | GH-2(1124)/GL-2(4)/GR-2(32)/GS-2(1) |
| N7 | D614 | 10822 | 1 | D-0(1)/G-1(10821) |
| Hap-Load | Freq | Unknown | A.2.4 | B.1 | B.1.1 | B.1.1.1 | B.1.1.171 | B.1.1.222 | B.1.1.29 | B.1.1.304 | B.1.1.317 | B.1.152 | B.1.165 | B.1.166 | B.1.2 | B.1.215 | B.1.234 | B.1.256 | B.1.324 | B.1.350 | B.1.354 | B.1.360 | B.1.399 | B.1.94 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (1) D80-F157-L452-D614-T859-D950 | ||||||||||||||||||||||||
| DSRGNH-5 | 63 | 58 | 5 | |||||||||||||||||||||
| GSLGNH-5 | 9 | 9 | ||||||||||||||||||||||
| GSRGND-5 | 21 | 19 | 1 | |||||||||||||||||||||
| GSRGNH-6 | 539 | 522 | 1 | 2 | 3 | 5 | ||||||||||||||||||
| (2) D80-S155-F157-L452-T859-D950 | ||||||||||||||||||||||||
| DRSRNH-5 | 39 | 39 | ||||||||||||||||||||||
| GRSRND-5 | 3 | 3 | ||||||||||||||||||||||
| GRSRNH-6 | 30 | 30 | ||||||||||||||||||||||
| GSSRNH-5 | 509 | 492 | 1 | 2 | 3 | 5 | ||||||||||||||||||
| (3) G142-E180-D614-Q677-S940 | ||||||||||||||||||||||||
| SEGHF-4 | 3 | 1 | 1 | 1 | ||||||||||||||||||||
| SVGHF-5 | 353 | 353 | ||||||||||||||||||||||
| SVGHS-4 | 273 | 2 | 1 | 262 | 8 | |||||||||||||||||||
| (4) S155-F157-L452-T859-D950 | ||||||||||||||||||||||||
| RSRND-4 | 3 | 3 | ||||||||||||||||||||||
| RSRNH-5 | 69 | 69 | ||||||||||||||||||||||
| SSRNH-4 | 533 | 511 | 1 | 7 | 3 | 5 | ||||||||||||||||||
| (5) S13-W152-L452-D614 | ||||||||||||||||||||||||
| ICLG-3 | 43 | 1 | 36 | 3 | ||||||||||||||||||||
| ICRG-4 | 795 | 51 | 557 | 1 | 4 | 10 | 14 | 10 | 34 | 2 | 72 | |||||||||||||
| IWRG-3 | 120 | 1 | 77 | 7 | 2 | 28 | ||||||||||||||||||
| SCRG-3 | 30 | 4 | 16 | 4 | ||||||||||||||||||||
| (6) S494-D614-P681-T716 | ||||||||||||||||||||||||
| PGHI-4 | 521 | 467 | 1 | 1 | 1 | 20 | 3 | |||||||||||||||||
| PGHT-3 | 194 | 100 | 8 | 3 | 31 | 2 | 3 | 29 | 1 | |||||||||||||||
| RGHI-4 | 3 | 3 | ||||||||||||||||||||||
| SGHI-3 | 38 | 19 | 3 | 1 | 4 | |||||||||||||||||||
| (7) T478-D614-P681-T732 | ||||||||||||||||||||||||
| KGHA-4 | 2132 | 11 | 17 | 2 | 14 | 2029 | 18 | 1 | 12 | 2 | ||||||||||||||
| KGHS-4 | 6 | |||||||||||||||||||||||
| KGHT-3 | 159 | 4 | 57 | 3 | 67 | 8 | 1 | |||||||||||||||||
| KGPA-3 | 5 | 1 | 3 | 1 | ||||||||||||||||||||
| TGHA-3 | 85 | 13 | 63 | 2 | 1 | 2 | 2 | |||||||||||||||||
| (8) F157-L452-D614-T859 | ||||||||||||||||||||||||
| FQGN-3 | 22 | 22 | ||||||||||||||||||||||
| FRGI-3 | 15 | 14 | 1 | |||||||||||||||||||||
| FRGN-3 | 5 | 5 | ||||||||||||||||||||||
| SLGN-3 | 11 | 10 | 1 | |||||||||||||||||||||
| SRGN-4 | 625 | 601 | 1 | 7 | 3 | 6 | ||||||||||||||||||
| SRGT-3 | 37 | 33 | ||||||||||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.P.; Lybrand, T.P.; Gilbert, P.B.; Hawn, T.R.; Schiffer, J.T.; Stamatatos, L.; Payne, T.H.; Carpp, L.N.; Geraghty, D.E.; Jerome, K.R. Tracking SARS-CoV-2 Spike Protein Mutations in the United States (January 2020—March 2021) Using a Statistical Learning Strategy. Viruses 2022, 14, 9. https://doi.org/10.3390/v14010009
Zhao LP, Lybrand TP, Gilbert PB, Hawn TR, Schiffer JT, Stamatatos L, Payne TH, Carpp LN, Geraghty DE, Jerome KR. Tracking SARS-CoV-2 Spike Protein Mutations in the United States (January 2020—March 2021) Using a Statistical Learning Strategy. Viruses. 2022; 14(1):9. https://doi.org/10.3390/v14010009
Chicago/Turabian StyleZhao, Lue Ping, Terry P. Lybrand, Peter B. Gilbert, Thomas R. Hawn, Joshua T. Schiffer, Leonidas Stamatatos, Thomas H. Payne, Lindsay N. Carpp, Daniel E. Geraghty, and Keith R. Jerome. 2022. "Tracking SARS-CoV-2 Spike Protein Mutations in the United States (January 2020—March 2021) Using a Statistical Learning Strategy" Viruses 14, no. 1: 9. https://doi.org/10.3390/v14010009