
Nisoldipine Inhibits Influenza A Virus Infection by Interfering with Virus Internalization Process

Abstract
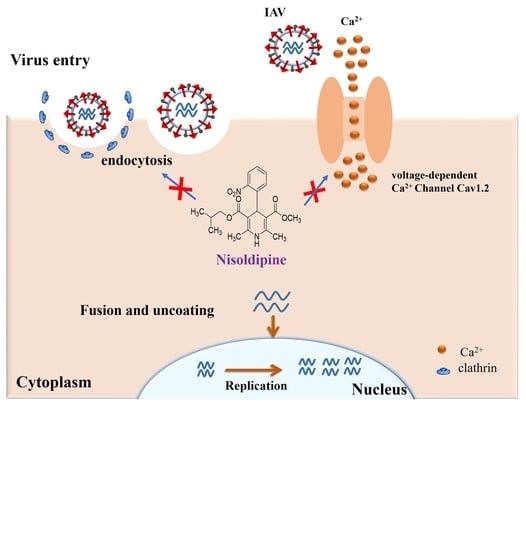
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Cells, Viruses and Plasmids
2.3. Cytotoxicity Studies
2.4. Antiviral Assay and Microscopy
2.5. Plaque Reduction Assay
2.6. Western Blotting and Quantitative Real-Time PCR
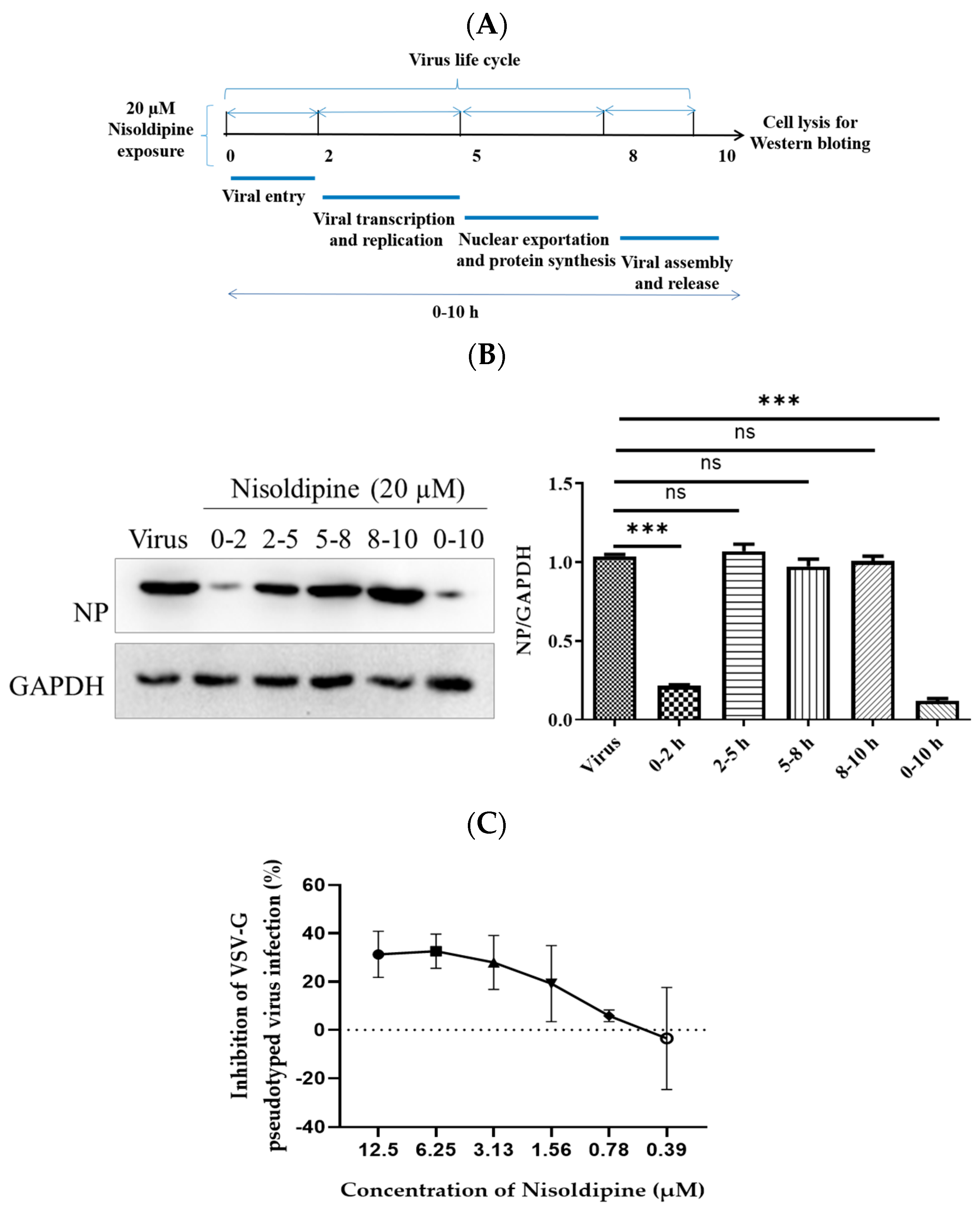
2.7. Time of Addition Study
2.8. Pseudovirus-Based Entry Inhibition Assays
2.9. Mini-Replicon Assay
2.10. IAV Binding, Internalization and Membrane Fusion Assay
2.11. Transferrin Absorption Test
2.12. Transmission Electron Microscopy
2.13. siRNA Knockdown of Cav1.2
2.14. Assessment of Combination Treatment In Vitro
2.15. Fluorescence Confocal Assays
2.16. Statistical Analysis
3. Results
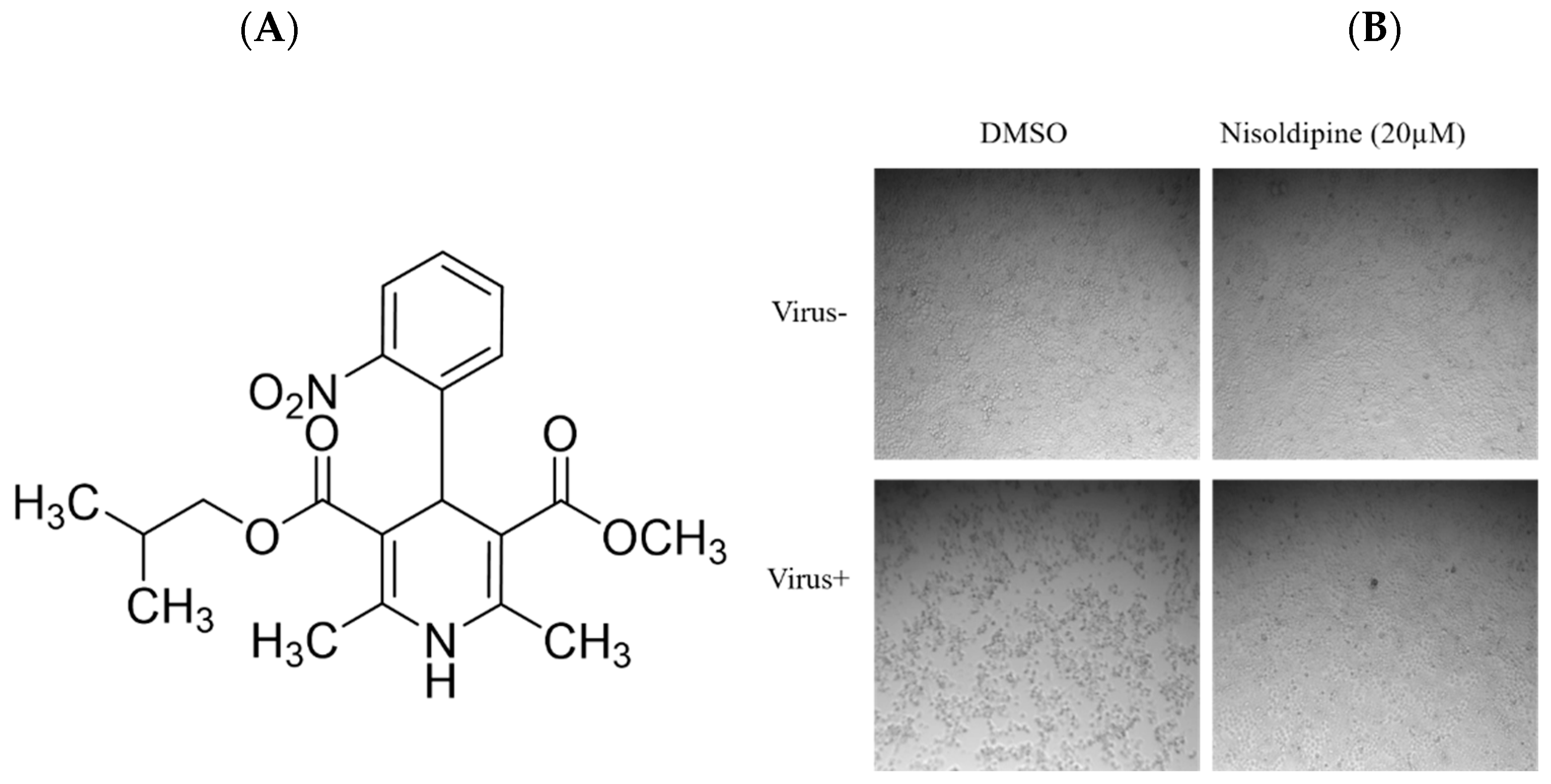
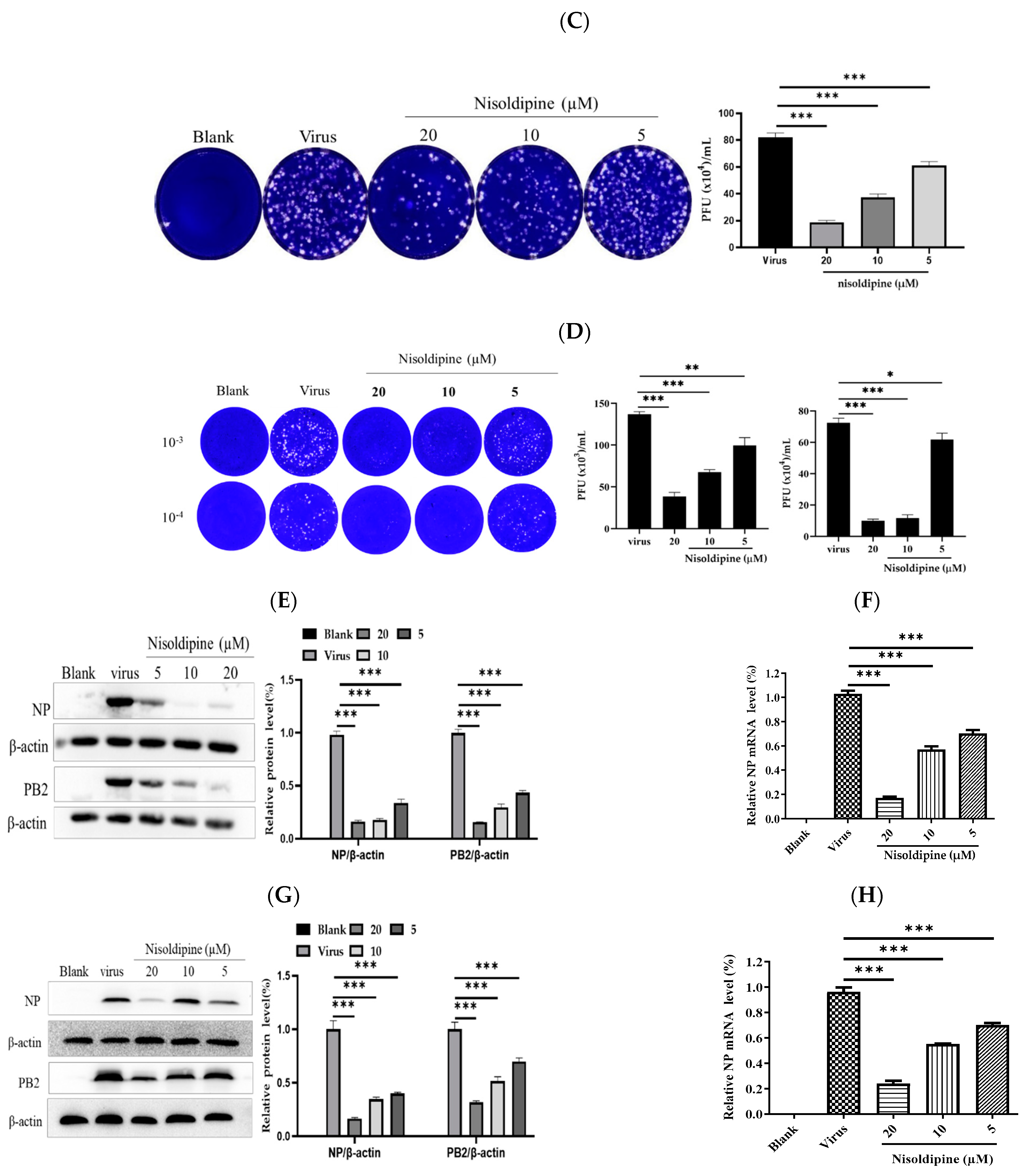
3.1. In Vitro Anti-Influenza Activity of Nisoldipine
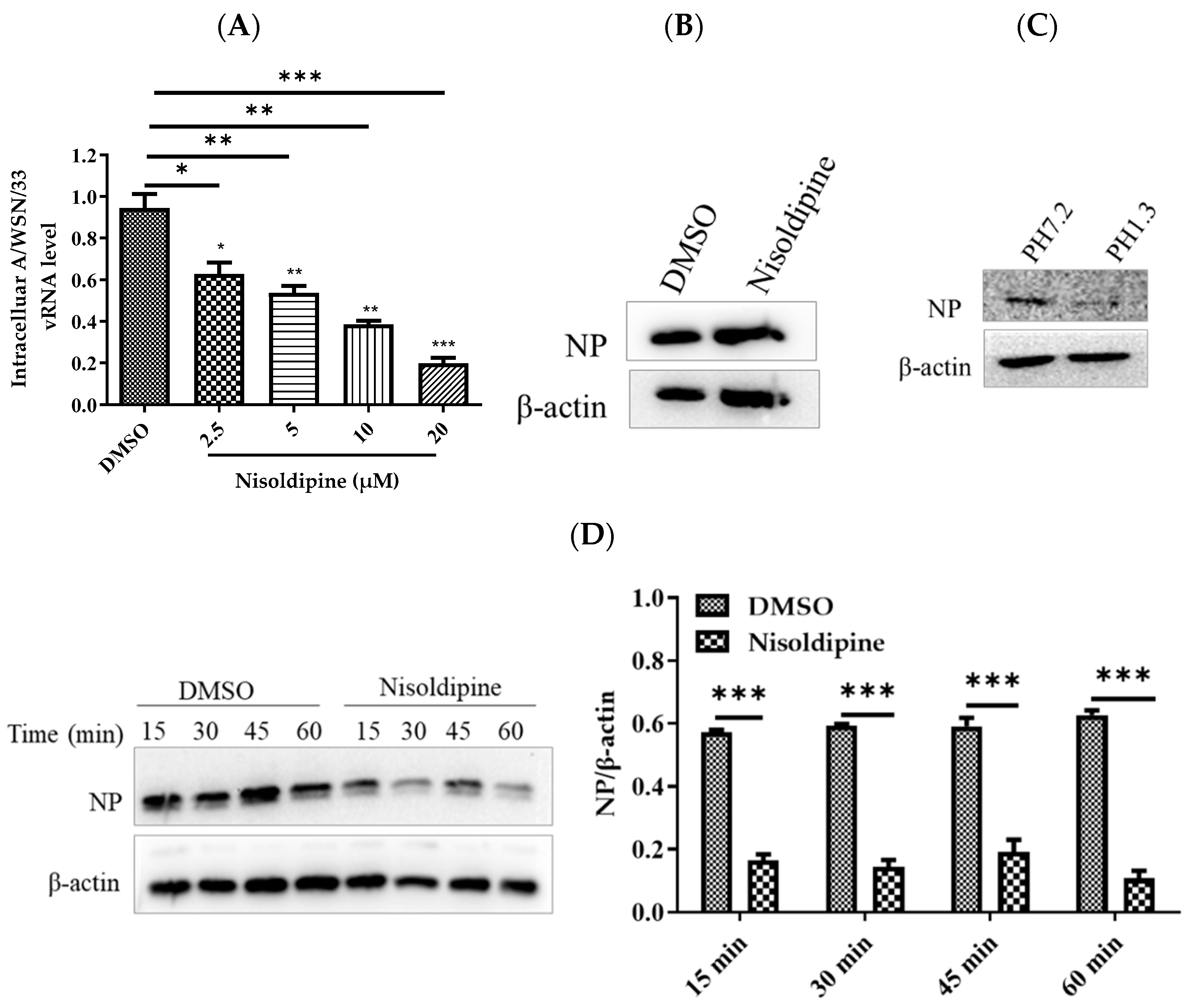
3.2. Nisoldipine Inhibits Viral Entry
3.3. Nisoldipine Exerts Its Antiviral Effect by Interfering with Influenza A Virus Internalization
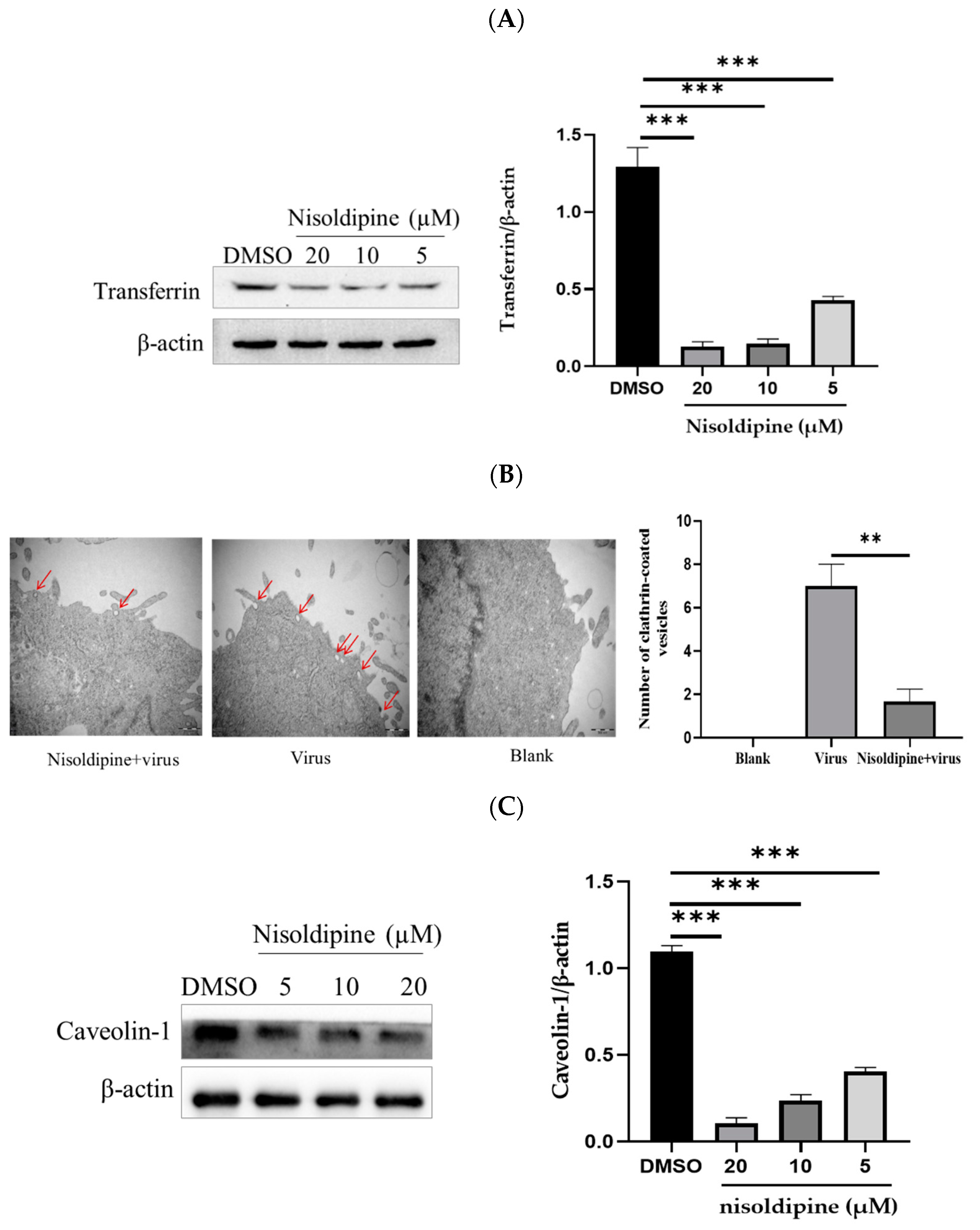
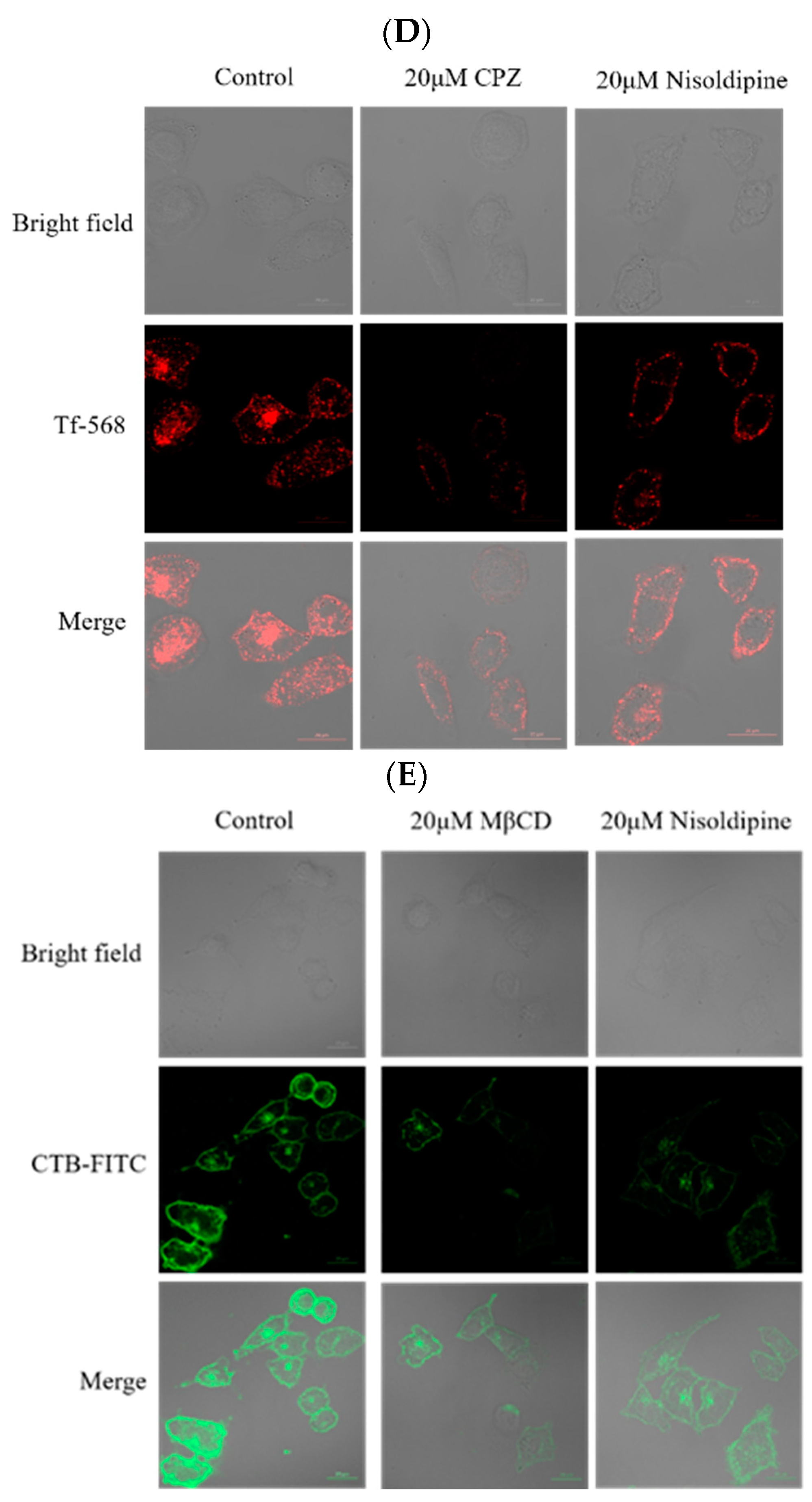
3.4. The Effect of Nisoldipine on Clathrin-Mediated or Clathrin-Independent Endocytic Pathway
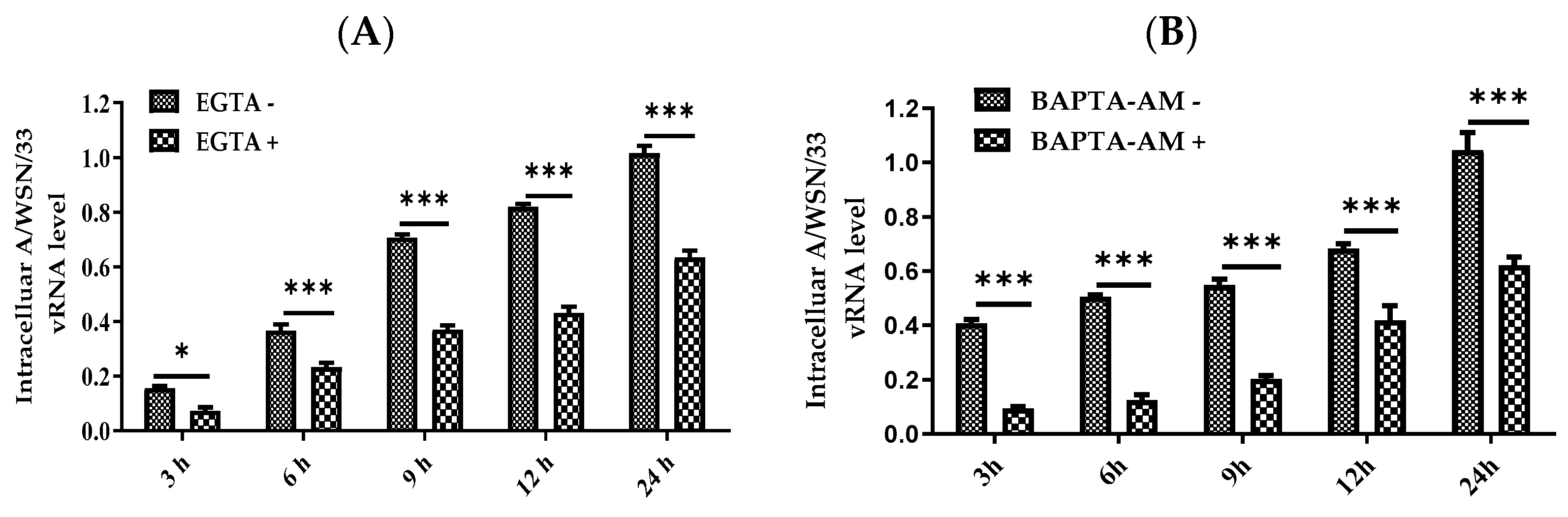
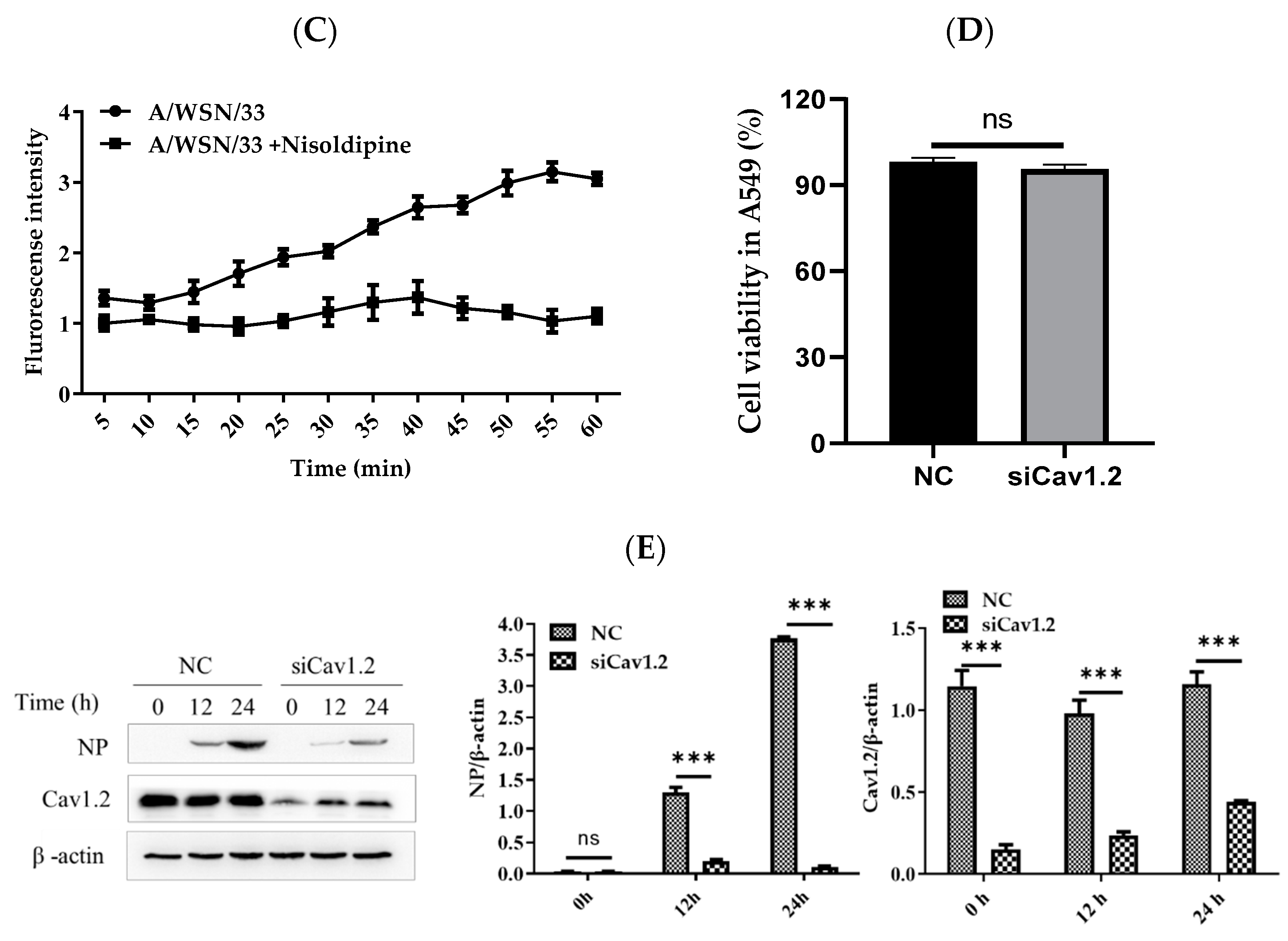
3.5. Nisoldipine Inhibits IAV Infection by Reducing Cellular Ca2+ Uptake
3.6. Effect of siRNA Knockdown of Cav1.2 on the Replication of IAV
3.7. In Vitro Synergistic Anti-Influenza Activity of Nisoldipine with NAI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guidry, J.P.D.; Coman, I.A.; Vraga, E.K.; O’Donnell, N.H.; Sreepada, N. (S)pin the flu vaccine: Recipes for concern. Vaccine 2020, 38, 5498–5506. [Google Scholar] [CrossRef] [PubMed]
- Lambert, L.C.; Fauci, A.S. Influenza vaccines for the future. N. Engl. J. Med. 2010, 363, 2036–2044. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Cichero, E. Fight Against H1N1 Influenza A Virus: Recent Insights Towards the Development of Druggable Compounds. Curr. Med. Chem. 2016, 23, 1802–1817. [Google Scholar] [CrossRef]
- Yang, J.; Huang, Y.; Liu, S. Investigational antiviral therapies for the treatment of influenza. Expert Opin. Investig. Drugs 2019, 28, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Hurt, A.C.; Ho, H.T.; Barr, I. Resistance to anti-influenza drugs: Adamantanes and neuraminidase inhibitors. Expert Rev. Anti-Infect. 2006, 4, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, N.; Koseki, N.; Kaiho, M.; Ariga, T.; Kikuta, H.; Oba, K.; Togashi, T.; Morita, K.; Inagawa, A.; Okamura, A.; et al. Clinical effectiveness of four neuraminidase inhibitors (oseltamivir, zanamivir, laninamivir, and peramivir) for children with influenza A and B in the 2014–2015 to 2016–2017 influenza seasons in Japan. J. Infect. Chemother. 2018, 24, 449–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omoto, S.; Speranzini, V.; Hashimoto, T.; Noshi, T.; Yamaguchi, H.; Kawai, M.; Kawaguchi, K.; Uehara, T.; Shishido, T.; Naito, A.; et al. Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Sci. Rep. 2018, 8, 9633. [Google Scholar] [CrossRef]
- Imai, M.; Yamashita, M.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; Kiso, M.; Murakami, J.; Yasuhara, A.; Takada, K.; Ito, M.; Nakajima, N.; et al. Influenza A variants with reduced susceptibility to baloxavir isolated from Japanese patients are fit and transmit through respiratory droplets. Nat. Microbiol. 2020, 5, 27–33. [Google Scholar] [CrossRef]
- Yang, J.R.; Lin, Y.C.; Huang, Y.P.; Su, C.H.; Lo, J.; Ho, Y.L.; Yao, C.Y.; Hsu, L.C.; Wu, H.S.; Liu, M.T. Reassortment and mutations associated with emergence and spread of oseltamivir-resistant seasonal influenza A/H1N1 viruses in 2005–2009. PLoS ONE 2011, 6, e18177. [Google Scholar] [CrossRef] [Green Version]
- Finberg, R.W.; Lanno, R.; Anderson, D.; Fleischhackl, R.; van Duijnhoven, W.; Kauffman, R.S.; Kosoglou, T.; Vingerhoets, J.; Leopold, L. Phase 2b Study of Pimodivir (JNJ-63623872) as Monotherapy or in Combination With Oseltamivir for Treatment of Acute Uncomplicated Seasonal Influenza A: TOPAZ Trial. J. Infect. Dis. 2019, 219, 1026–1034. [Google Scholar] [CrossRef]
- Han, J.; Perez, J.T.; Chen, C.; Li, Y.; Benitez, A.; Kandasamy, M.; Lee, Y.; Andrade, J.; tenOever, B.; Manicassamy, B. Genome-wide CRISPR/Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication. Cell Rep. 2018, 23, 596–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Wakker, S.I.; Fischer, M.J.E.; Oosting, R.S. New drug-strategies to tackle viral-host interactions for the treatment of influenza virus infections. Eur. J. Pharm. 2017, 809, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Sharma, S.; Kumar, R.; Tripathi, B.N.; Barua, S.; Ly, H.; Rouse, B.T. Host-Directed Antiviral Therapy. Clin. Microbiol. Rev. 2020, 33, e00168-19. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Zhang, Q.; Wu, W.; Chen, L.; Gu, S.; Ye, Y.; Zhong, Y.; Huang, Q.; Liu, S. Inducible Guanylate-Binding Protein 7 Facilitates Influenza A Virus Replication by Suppressing Innate Immunity via NF-kappaB and JAK-STAT Signaling Pathways. J. Virol. 2021, 95, e02038-20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liang, T.; Gu, S.; Ye, Y.; Liu, S. SNW1 interacts with IKKγ to positively regulate antiviral innate immune responses against influenza A virus infection. Microbes Infect. 2020, 22, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Estacio, R.O.; Jeffers, B.W.; Hiatt, W.R.; Biggerstaff, S.L.; Gifford, N.; Schrier, R.W. The effect of nisoldipine as compared with enalapril on cardiovascular outcomes in patients with non-insulin-dependent diabetes and hypertension. N. Engl. J. Med. 1998, 338, 645–652. [Google Scholar] [CrossRef]
- PLoSker, G.L.; Faulds, D. Nisoldipine coat-core. A review of its pharmacology and therapeutic efficacy in hypertension. Drugs 1996, 52, 232–253. [Google Scholar] [CrossRef]
- Fujioka, Y.; Tsuda, M.; Nanbo, A.; Hattori, T.; Sasaki, J.; Sasaki, T.; Miyazaki, T.; Ohba, Y. A Ca2+-dependent signalling circuit regulates influenza A virus internalization and infection. Nat. Commun. 2013, 4, 2763. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, Y.; Nishide, S.; Ose, T.; Suzuki, T.; Kato, I.; Fukuhara, H.; Fujioka, M.; Horiuchi, K.; Satoh, A.O.; Nepal, P.; et al. A Sialylated Voltage-Dependent Ca2+ Channel Binds Hemagglutinin and Mediates Influenza A Virus Entry into Mammalian Cells. Cell Host Microbe 2018, 23, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Pohlmann, S. Cell Entry of Influenza A Viruses: Sweet Talk between HA and Cav1.2. Cell Host Microbe 2018, 23, 697–699. [Google Scholar] [CrossRef]
- Wu, W.; Li, R.; Li, X.; He, J.; Jiang, S.; Liu, S.; Yang, J. Quercetin as an Antiviral Agent Inhibits Influenza A Virus (IAV) Entry. Viruses 2015, 8, 1999–4915. [Google Scholar] [CrossRef] [PubMed]
- Sang, H.; Huang, Y.; Tian, Y.; Liu, M.; Chen, L.; Li, L.; Liu, S.; Yang, J. Multiple modes of action of myricetin in influenza A virus infection. Phytother. Res. 2021, 35, 2797–2806. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Jeong, M.S.; Mun, S.H.; Cho, J.; Seo, M.D.; Kim, H.; Lee, J.; Song, J.H.; Ko, H.J. Antiviral Activity of Chrysin against Influenza Virus Replication via Inhibition of Autophagy. Viruses 2021, 13, 1350. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhao, G.; Zhang, X.; Liu, Z.; Yu, H.; Zheng, B.J.; Zhou, Y.; Jiang, S. Development of a safe and convenient neutralization assay for rapid screening of influenza HA-specific neutralizing monoclonal antibodies. Biochem. Biophys. Res. Commun. 2010, 397, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chu, H.; Zhang, K.; Ye, J.; Singh, K.; Kao, R.Y.; Chow, B.K.; Zhou, J.; Zheng, B.J. A novel small-molecule compound disrupts influenza A virus PB2 cap-binding and inhibits viral replication. J. Antimicrob. Chemother. 2016, 71, 2489–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef]
- Liu, T.; Liu, M.; Chen, F.; Chen, F.; Tian, Y.; Huang, Q.; Liu, S.; Yang, J. A Small-Molecule Compound has Anti-influenza A Virus Activity by Acting as a “PB2 Inhibitor”. Mol. Pharmcol. 2018, 15, 4110–4120. [Google Scholar] [CrossRef]
- Liu, S.; Li, R.; Zhang, R.; Chan, C.C.; Xi, B.; Zhu, Z.; Yang, J.; Poon, V.K.; Zhou, J.; Chen, M.; et al. CL-385319 inhibits H5N1 avian influenza A virus infection by blocking viral entry. Eur. J. Pharm. 2011, 660, 460–467. [Google Scholar] [CrossRef]
- Fujioka, Y.; Tsuda, M.; Hattori, T.; Sasaki, J.; Sasaki, T.; Miyazaki, T.; Ohba, Y. The Ras-PI3K Signaling Pathway Is Involved in Clathrin-Independent Endocytosis and the Internalization of Influenza Viruses. PLoS ONE 2011, 6, e16324. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Ohnishi, S. Uncoating of influenza virus in endosomes. J. Virol. 1984, 51, 497–504. [Google Scholar] [CrossRef]
- Rust, M.J.; Lakadamyali, M.; Zhang, F.; Zhuang, X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 2004, 11, 567–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieczkarski, S.B.; Whittaker, G.R. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 2002, 76, 10455–10464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes-Correia, I.; Eulálio, A.; Nir, S.; Pedroso de Lima, M.C. Caveolae as an additional route for influenza virus endocytosis in MDCK cells. Cell. Mol. Biol. Lett. 2004, 9, 47–60. [Google Scholar] [PubMed]
- Lampejo, T. Influenza and antiviral resistance: An overview. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Lackenby, A.; Besselaar, T.G.; Daniels, R.S.; Fry, A.; Gregory, V.; Gubareva, L.V.; Huang, W.; Hurt, A.C.; Leang, S.K.; Lee, R.T.C.; et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors and status of novel antivirals, 2016-2017. Antivir. Res. 2018, 157, 38–46. [Google Scholar] [CrossRef]
- Nugent, K.M.; Shanley, J.D. Verapamil inhibits influenza A virus replication. Arch. Virol. 1984, 81, 163–170. [Google Scholar] [CrossRef]
- Wang, M.Z.; Tai, C.Y.; Mendel, D.B. Mechanism by which mutations at his274 alter sensitivity of influenza a virus n1 neuraminidase to oseltamivir carboxylate and zanamivir. Antimicrob. Agents Chemother. 2002, 46, 3809–3816. [Google Scholar] [CrossRef] [Green Version]
- Sun, E.Z.; Liu, A.A.; Zhang, Z.L.; Liu, S.L.; Tian, Z.Q.; Pang, D.W. Real-Time Dissection of Distinct Dynamin-Dependent Endocytic Routes of Influenza A Virus by Quantum Dot-Based Single-Virus Tracking. ACS Nano 2017, 11, 4395–4406. [Google Scholar] [CrossRef]
- Sieczkarski, S.B.; Brown, H.A.; Whittaker, G.R. Role of protein kinase C betaII in influenza virus entry via late endosomes. J. Virol. 2003, 77, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Warfield, K.L.; Ruthel, G.; Bavari, S.; Aman, M.J.; Hope, T.J. Ebola virus uses clathrin-mediated endocytosis as an entry pathway. Virology 2010, 401, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Persaud, M.; Martinez-Lopez, A.; Buffone, C.; Porcelli, S.A.; Diaz-Griffero, F. Infection by Zika viruses requires the transmembrane protein AXL, endocytosis and low pH. Virology 2018, 518, 301–312. [Google Scholar] [CrossRef]
- Malin, J.J.; Suárez, I.; Priesner, V.; Fätkenheuer, G.; Rybniker, J. Remdesivir against COVID-19 and Other Viral Diseases. Clin. Microbiol. Rev. 2020, 34, e00162-20. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Boelaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases? Lancet Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.E.; Couette, B.; Marger, L.; Bourinet, E.; Striessnig, J.; Nargeot, J. Voltage-dependent calcium channels and cardiac pacemaker activity: From ionic currents to genes. Prog. Biophys. Mol. Biol. 2006, 90, 38–63. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, L.K.; Li, S.F.; Zhang, S.F.; Wan, W.W.; Zhang, Y.L.; Xin, Q.L.; Dai, K.; Hu, Y.Y.; Wang, Z.B.; et al. Calcium channel blockers reduce severe fever with thrombocytopenia syndrome virus (SFTSV) related fatality. Cell Res. 2019, 29, 739–753. [Google Scholar] [CrossRef]
- Bao, M.N.; Zhang, L.J.; Tang, B.; Fu, D.D.; Li, J.; Du, L.; Hou, Y.N.; Zhang, Z.L.; Tang, H.W.; Pang, D.W. Influenza A Viruses Enter Host Cells via Extracellular Ca2+ Influx-Involved Clathrin-Mediated Endocytosis. ACS Appl. Bio Mater. 2021, 4, 2044–2051. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Inhibition of A/Puerto Rico/8/34 Infection | ||
|---|---|---|---|
| IC50 (μM) a | CC50 (μM) b | SI (CC50/ IC50) c | |
| Nitrendipine | 7.26 ± 1.89 | 171.05 ± 14.15 | 23.56 |
| Nimodipine | 12.83 ± 1.72 | >200 | >15.58 |
| Cilnidipine | 14.26 ± 2.66 | >200 | >14.02 |
| Nisoldipine | 4.74 ± 0.76 | >200 | >42.19 |
| Zanamivir | 1.26 ± 0.30 nM | >200 nM | >158.73 |
| IAV Strains | Inhibition Activity of Nisoldipine | |
|---|---|---|
| IC50 (μM) | CC50 (μM) | |
| A/Puerto Rico/8/34 | 4.74 ± 0.76 | >200 |
| A/FM-1/1/47 | 6.96 ± 0.78 | |
| A/Aichi/2/68 | 5.68 ± 0.61 | |
| A/WSN/33 | 4.47 ± 0.25 | |
| A/PR/8/34 with NA-H274Y a | 5.94 ± 0.55 | |
| H5N1 Pseudovirus | IC50 (µM) |
|---|---|
| A/Qinghai/59/2005 | 5.84 ± 0.64 |
| A/Xinjiang/1/2006 | 3.87 ± 0.51 |
| A/Hong Kong/156/1997 | 4.47 ± 0.47 |
| A/Anhui/1/2005 | 2.59 ± 0.46 |
| Combination Ratio (IC50) a | IC50 Equivalent b | ||
|---|---|---|---|
| Nisoldipine: Zanamivir | Nisoldipine | Zanamivir | c FICI |
| 7:1 | 0.61 | 0.09 | 0.70 |
| 3:1 | 0.49 | 0.17 | 0.66 |
| 1:1 | 0.27 | 0.27 | 0.54 |
| 1:3 | 0.12 | 0.37 | 0.49 d |
| 1:7 | 0.75 | 0.44 | 1.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Li, Y.; Chen, Z.; Chen, L.; Liang, J.; Zhang, C.; Zhang, Z.; Yang, J. Nisoldipine Inhibits Influenza A Virus Infection by Interfering with Virus Internalization Process. Viruses 2022, 14, 2738. https://doi.org/10.3390/v14122738
Huang Y, Li Y, Chen Z, Chen L, Liang J, Zhang C, Zhang Z, Yang J. Nisoldipine Inhibits Influenza A Virus Infection by Interfering with Virus Internalization Process. Viruses. 2022; 14(12):2738. https://doi.org/10.3390/v14122738
Chicago/Turabian StyleHuang, Yingna, Yinyan Li, Zhixuan Chen, Liurong Chen, Jinlong Liang, Chunyu Zhang, Zhengyin Zhang, and Jie Yang. 2022. "Nisoldipine Inhibits Influenza A Virus Infection by Interfering with Virus Internalization Process" Viruses 14, no. 12: 2738. https://doi.org/10.3390/v14122738