
Coinfection of Cotton Plants with Watermelon Mosaic Virus and a Novel Polerovirus in China


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and RNA Extraction
2.2. Small RNA Library Construction, Sequencing, and Data Processing
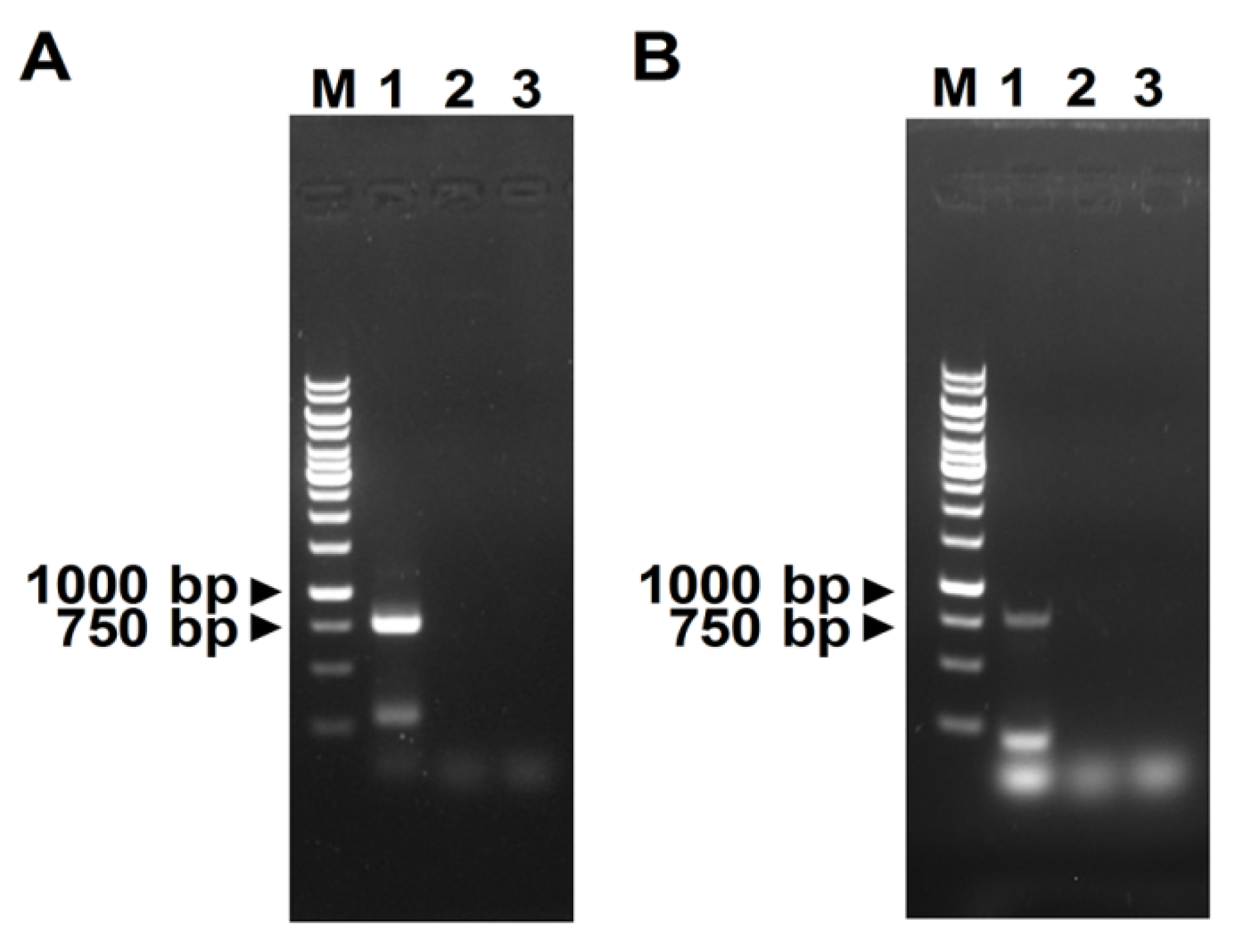
2.3. RT-PCR Validation, Full-Length Genome Amplification, and Sequencing
2.4. Viral Genome Characterization
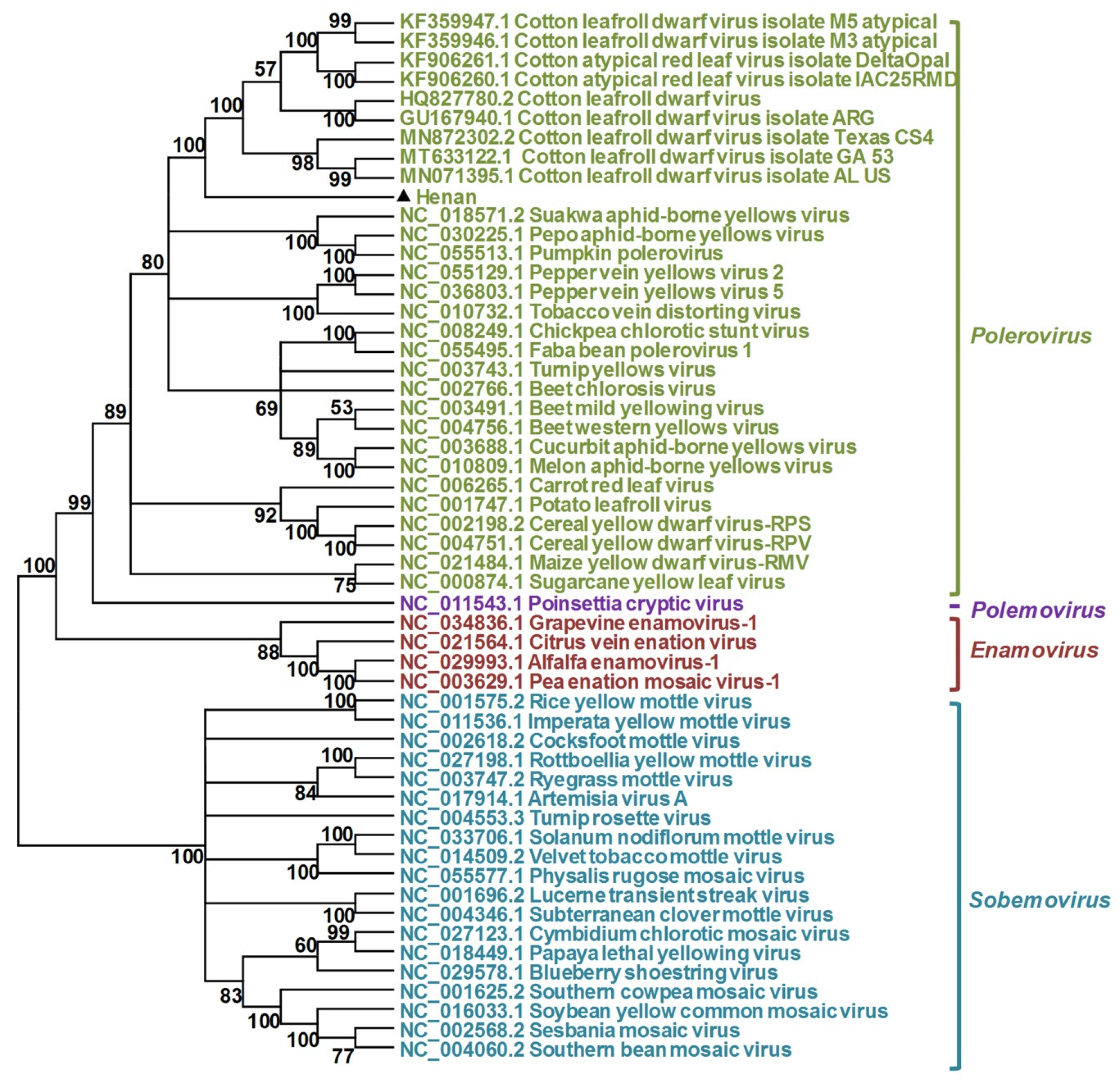
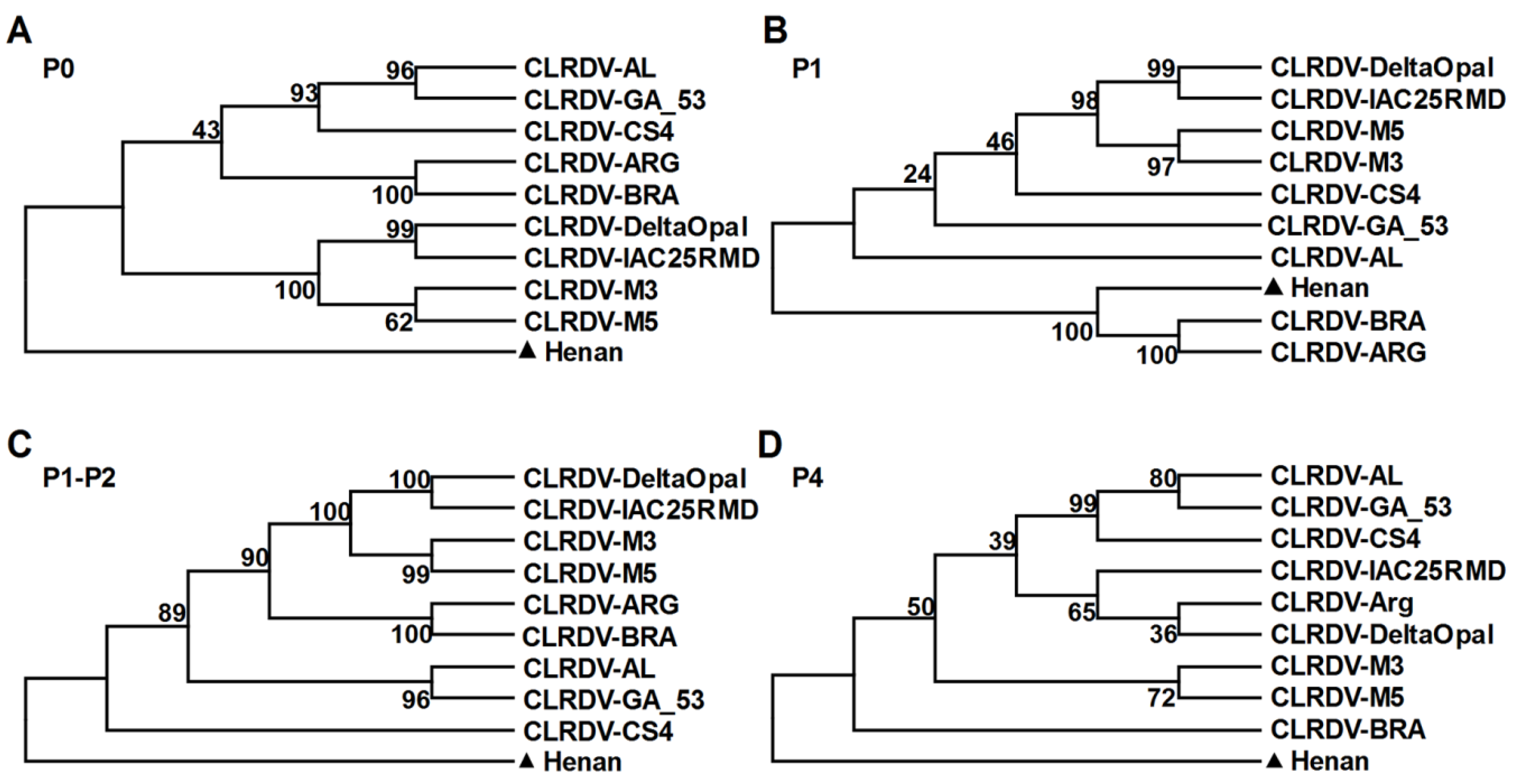
2.5. Phylogenetic Analyses
2.6. Quantitative RT-PCR of Viral RNA
3. Results
3.1. Identification of Two RNA Viruses in Cotton Plants Using Next-Generation Sequencing
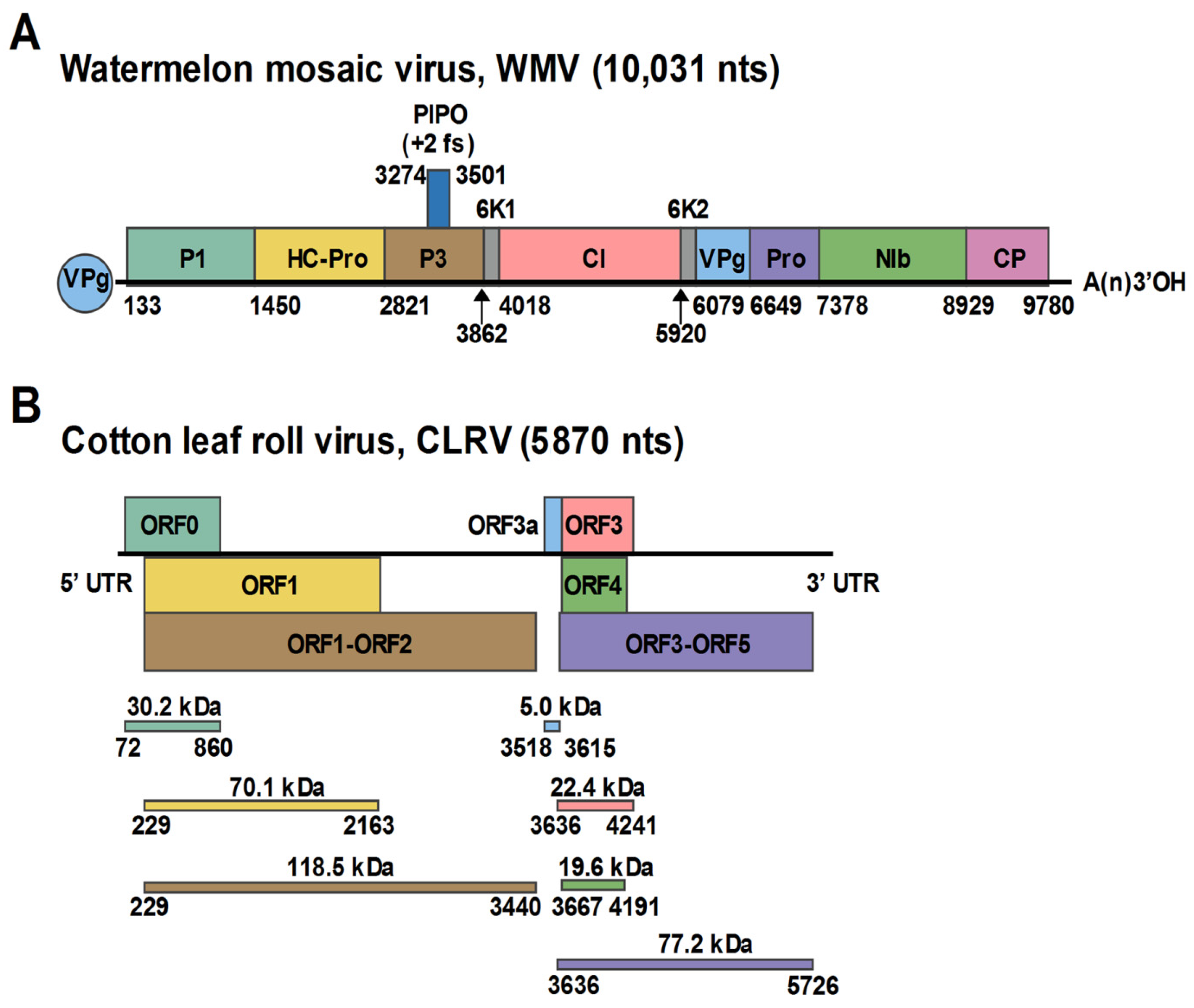
3.2. Characterization of the WMV Isolate
3.3. Characterization of the Polerovirus
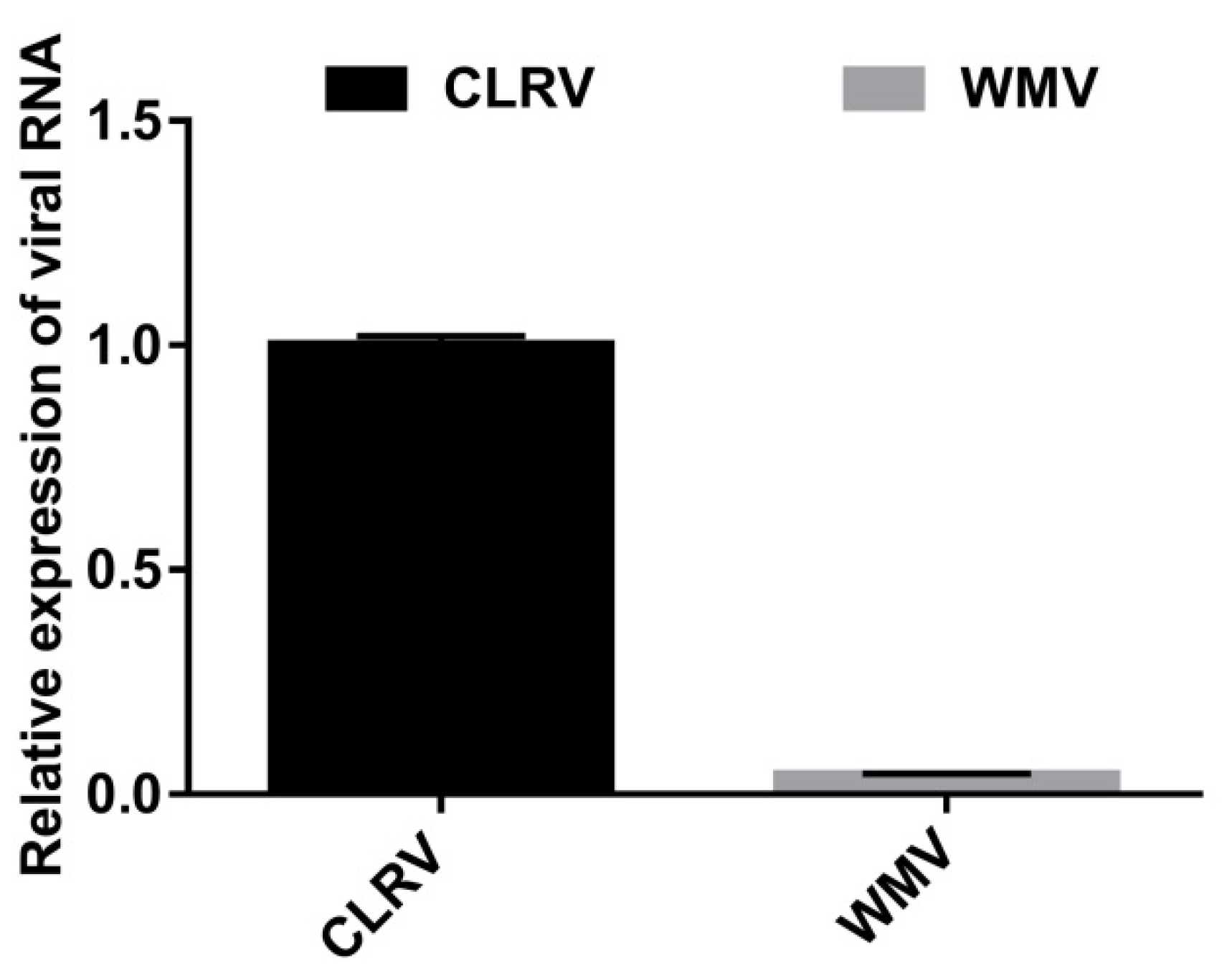
3.4. Relative Abundance of WMV and CLRV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nicaise, V. Crop immunity against viruses: Outcomes and future challenges. Front. Plant Sci. 2014, 5, 660. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Luo, Y.; Lu, R.; Lau, N.; Lai, E.C.; Li, W.X.; Ding, S.W. Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 1606–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Jiang, G.; Wu, J.; Liu, Y.; Qian, Y.; Zhou, X. Characterization of a novel polerovirus infecting maize in China. Viruses 2016, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Xuan, Z.; Xie, J.; Yu, H.; Zhang, S.; Li, R.; Cao, M. Mulberry (Morus alba) is a new natural host of citrus leaf blotch virus in China. Plant Dis. 2021, 105, 716. [Google Scholar] [CrossRef]
- Ma, Y.; Navarro, B.; Zhang, Z.; Lu, M.; Zhou, X.; Chi, S.; Di Serio, F.; Li, S. Identification and molecular characterization of a novel monopartite geminivirus associated with mulberry mosaic dwarf disease. J. Gen. Virol. 2015, 96, 2421–2434. [Google Scholar] [CrossRef]
- Fu, S.; Zhang, T.; He, M.; Sun, B.; Zhou, X.; Wu, J. Molecular characterization of a novel wheat-infecting virus of the family Betaflexiviridae. Arch. Virol. 2021, 166, 2875–2879. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Wang, A. Research advances in potyviruses: From the laboratory bench to the field. Annu. Rev. Phytopathol. 2021, 59, 1–29. [Google Scholar] [CrossRef]
- Revers, F.; Antonio Garcia, J. Molecular biology of potyviruses. Adv. Virus Res. 2015, 92, 101–199. [Google Scholar]
- Redinbaugh, M.G.; Stewart, L.R. Maize lethal necrosis: An emerging, synergistic viral disease. Annu. Rev. Virol. 2018, 5, 301–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, C.; Wang, C.; Qian, Y.; Li, Z.; Hong, J.; Zhou, X. Further characterization of maize chlorotic mottle virus and its synergistic interaction with sugarcane mosaic virus in maize. Sci. Rep. 2017, 7, 39960. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; Garcia, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Firth, A.E.; Miller, W.A.; Scheidecker, D.; Brault, V.; Reinbold, C.; Rakotondrafara, A.M.; Chung, B.Y.; Ziegler-Graff, V. Discovery of a small non-aug-initiated ORF in poleroviruses and luteoviruses that is required for long-distance movement. PLoS Pathog. 2015, 11, e1004868. [Google Scholar] [CrossRef] [Green Version]
- Sattar, M.N.; Kvarnheden, A.; Saeed, M.; Briddon, R.W. Cotton leaf curl disease—An emerging threat to cotton production worldwide. J. Gen. Virol. 2013, 94, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Agrofoglio, Y.C.; Delfosse, V.C.; Casse, M.F.; Hopp, H.E.; Kresic, I.B.; Distefano, A.J. Identification of a new cotton disease caused by an atypical cotton leafroll dwarf virus in Argentina. Phytopathology 2017, 107, 369–376. [Google Scholar] [CrossRef]
- Correa, R.L.; Silva, T.F.; Simoes-Araujo, J.L.; Barroso, P.A.; Vidal, M.S.; Vaslin, M.F. Molecular characterization of a virus from the family Luteoviridae associated with cotton blue disease. Arch. Virol. 2005, 150, 1357–1367. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Guo, W.; Xie, Y.; Xie, Q.; Fan, L.; Zhou, X. Characterization of small interfering RNAs derived from the geminivirus/betasatellite complex using deep sequencing. PLoS ONE 2011, 6, e16928. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Tabassum, A.; Roberts, P.M.; Bag, S. Genome sequence of cotton leafroll dwarf virus infecting cotton in Georgia, USA. Microbiol. Resour. Announc. 2020, 9, e00812-20. [Google Scholar] [CrossRef]
- Ali, A.; Mokhtari, S. Complete genome sequence of cotton leafroll dwarf virus isolated from cotton in Texas, USA. Microbiol. Resour. Announc. 2020, 9, e01587-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avelar, S.; Ramos-Sobrinho, R.; Conner, K.; Nichols, R.L.; Lawrence, K.; Brown, J.K. Characterization of the complete genome and P0 protein for a previously unreported genotype of cotton leafroll dwarf virus, an introduced polerovirus in the United States. Plant Dis. 2020, 104, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Distefano, A.J.; Bonacic Kresic, I.; Hopp, H.E. The complete genome sequence of a virus associated with cotton blue disease, cotton leafroll dwarf virus, confirms that it is a new member of the genus Polerovirus. Arch. Virol. 2010, 155, 1849–1854. [Google Scholar] [CrossRef]
- Mukherjee, A.K.; Mukherjee, P.K.; Kranthi, S. Genetic similarity between cotton leafroll dwarf virus and chickpea stunt disease associated virus in India. Plant Pathol. J. 2016, 32, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, A.; Bag, S.; Suassuna, N.D.; Conner, K.N.; Chee, P.; Kemerait, R.C.; Roberts, P. Genome analysis of cotton leafroll dwarf virus reveals variability in the silencing suppressor protein, genotypes and genomic recombinants in the USA. PLoS ONE 2021, 16, e0252523. [Google Scholar] [CrossRef]
- Ll, D. Family Luteoviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Andrew, M.Q.K., Elliot, L., Michael, J.A., Carstens, E.B., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2012; pp. 1045–1053. [Google Scholar]
- SYLLER, J. Facilitative and antagonistic interactions between plant viruses in mixed infections. Mol. Plant. Pathol. 2012, 13, 204–216. [Google Scholar] [CrossRef]
- Savenkov, E.I.; Valkonen, J.P. Potyviral helper-component proteinase expressed in transgenic plants enhances titers of Potato leaf roll virus but does not alleviate its phloem limitation. Virology 2001, 283, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Barker, H. Invasion of non-phloem tissue in Nicotiana clevelandii by potato leafroll luteovirus is enhanced in plants also infected with potato Y potyvirus. J. Gen. Virol. 1987, 68, 1223–1227. [Google Scholar] [CrossRef]
- Ryabov, E.V.; Fraser, G.; Mayo, M.A.; Barker, H.; Taliansky, M. Umbravirus gene expression helps potato leafroll virus to invade mesophyll tissues and to be transmitted mechanically between plants. Virology 2001, 286, 363–372. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Nucleotide Identity (%) of Complete Genome | Nucleotide and Amino Acid Identity (%, nt/aa) | ||||||
|---|---|---|---|---|---|---|---|---|
| ORF0/P0 | ORF1/P1 | ORF1-ORF2/P1-P2 | ORF3/P3 | ORF3a/P3a | ORF4/P4 | ORF3-ORF5/P3-P5 | ||
| CLRDV-AL | 90.92 | 85.86/80.00 | 88.08/85.02 | 90.33/90.17 | 93.4/96.02 | 98.55/100 | 94.10/86.78 | 92.29/92.80 |
| CLRDV-ARG | 91.46 | 85.75/80.77 | 88.54/85.18 | 90.83/90.45 | 93.56/95.02 | 99.28/100 | 94.28/87.93 | 92.91/93.66 |
| CLRDV-CS4 | 91.33 | 85.6/78.85 | 88.96/86.43 | 90.92/90.64 | 93.73/96.02 | 98.55/100 | 94.48/87.93 | 92.48/93.66 |
| CLRDV-Delta | 91.55 | 86.00/76.92 | 88.28/84.56 | 90.80/89.70 | 93.73/96.02 | 99.28/100 | 94.67/89.08 | 93.06/94.38 |
| CLRDV-GA53 | 91.45 | 85.37/79.23 | 88.65/85.34 | 91.02/90.26 | 93.40/96.02 | 99.28/100 | 94.1/87.36 | 92.43/93.08 |
| CLRDV-HQ | 91.7 | 86.13/80.77 | 89.00/85.80 | 91.08/90.82 | 94.22/96.02 | 97.82/100 | 95.05/90.23 | 93.20/94.38 |
| CLRDV-IAC | 91.48 | 86.13/77.31 | 88.34/84.71 | 90.77/89.89 | 93.73/96.02 | 99.28/100 | 94.67/89.08 | 92.86/94.38 |
| CLRDV-M5 | 91.74 | 86.26/78.46 | 88.75/85.02 | 91.05/89.79 | 94.06/96.02 | 98.55/100 | 94.67/89.08 | 93.10/93.80 |
| CLRDV-M3 | 91.7 | 86.51/79.23 | 88.75/85.02 | 91.01/89.61 | 92.73/96.52 | 98.55/100 | 94.28/87.93 | 93.15/94.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Du, M.; Li, S.; Zhou, X. Coinfection of Cotton Plants with Watermelon Mosaic Virus and a Novel Polerovirus in China. Viruses 2021, 13, 2210. https://doi.org/10.3390/v13112210
Yang X, Du M, Li S, Zhou X. Coinfection of Cotton Plants with Watermelon Mosaic Virus and a Novel Polerovirus in China. Viruses. 2021; 13(11):2210. https://doi.org/10.3390/v13112210
Chicago/Turabian StyleYang, Xiuling, Min Du, Shupeng Li, and Xueping Zhou. 2021. "Coinfection of Cotton Plants with Watermelon Mosaic Virus and a Novel Polerovirus in China" Viruses 13, no. 11: 2210. https://doi.org/10.3390/v13112210