
Downregulation of Cell Surface Major Histocompatibility Complex Class I Expression Is Mediated by the Left-End Transcription Unit of Fowl Adenovirus 9

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Plasmids
2.3. Antibodies
2.4. Quantitative RT-PCR
2.5. Western Blotting
2.6. Flow Cytometry
2.7. Internalization and Restoration Assays
2.8. Immunofluorescence Microscopy
3. Results
3.1. FAdV-4, FAdV-8a, and FAdV-9 Downregulate Cell Surface MHC-I
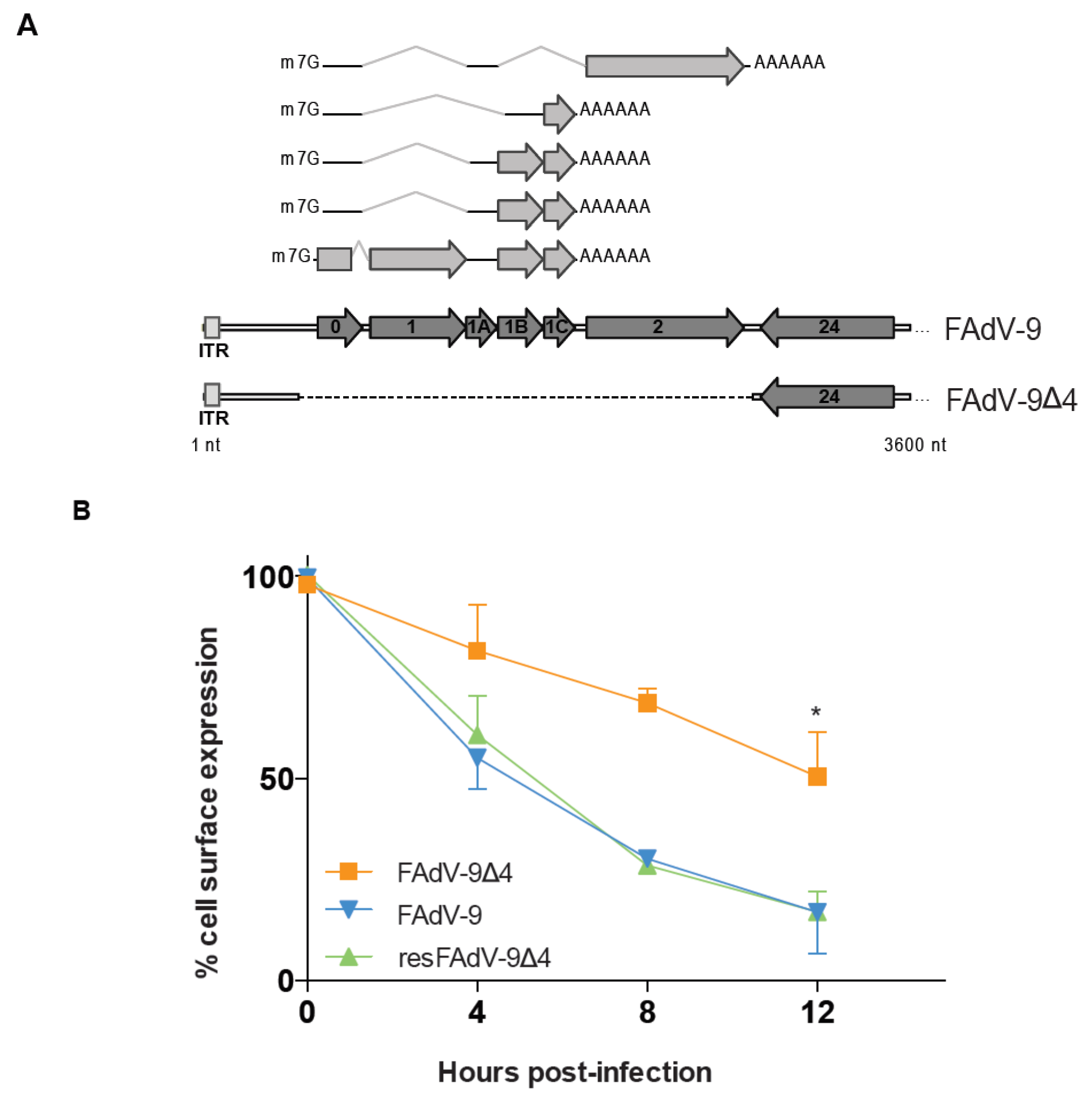
3.2. FAdV-9-Mediated Downregulation of Cell Surface MHC-I in CH-SAH Cells Depends in Part on the FE1 Transcription Unit
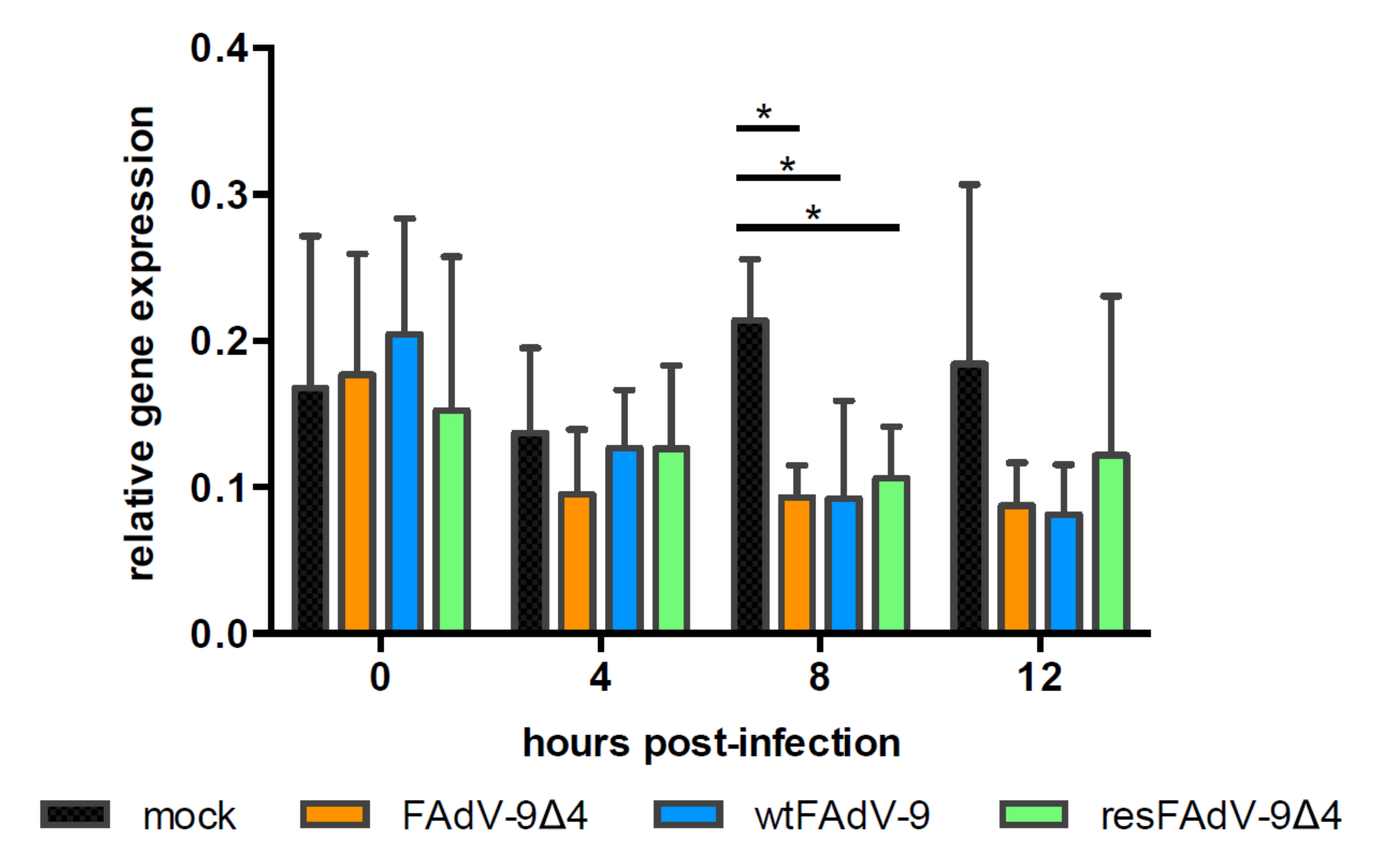
3.3. FAdV Induced Downregulation of MHC-I Transcription Is FE1 Unit Independent
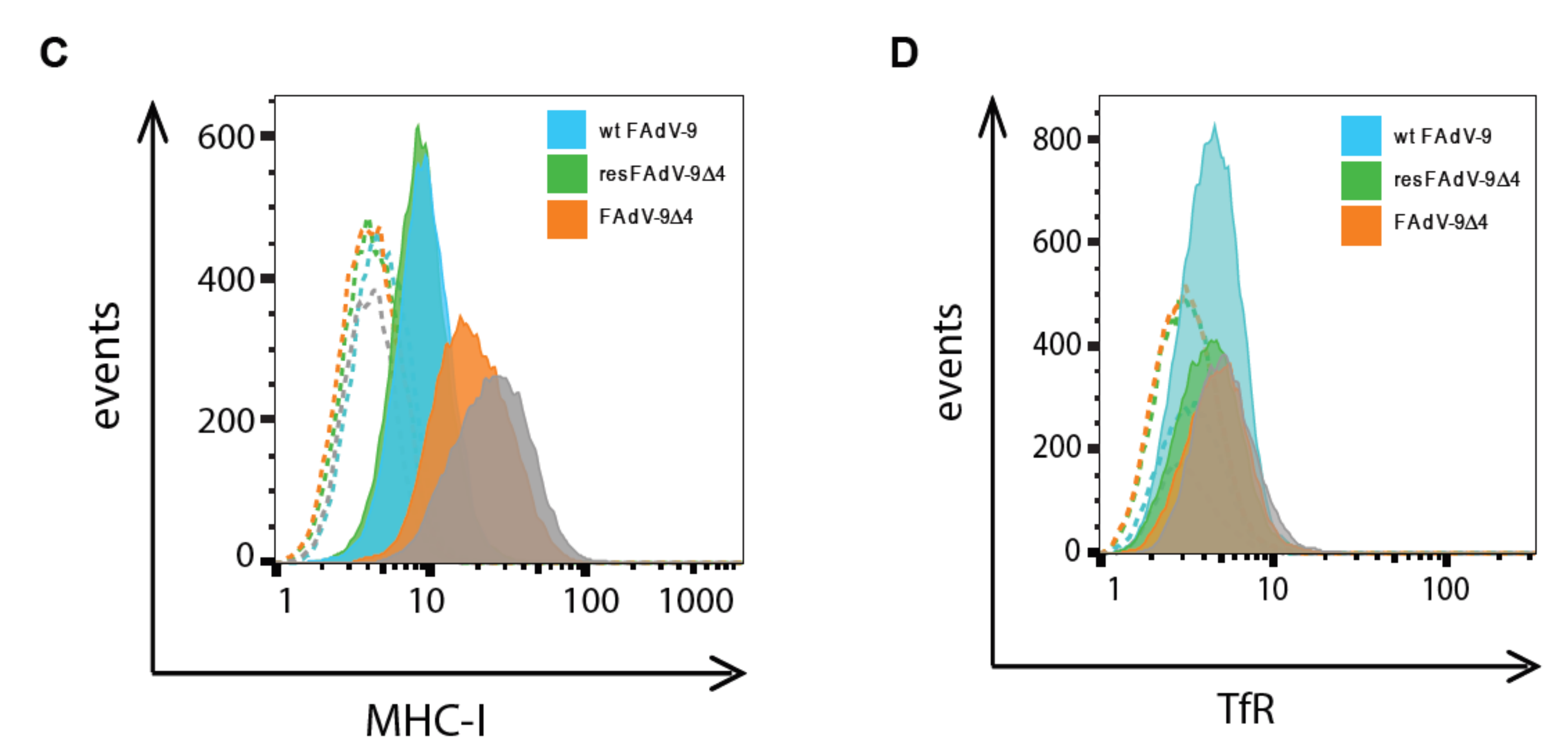
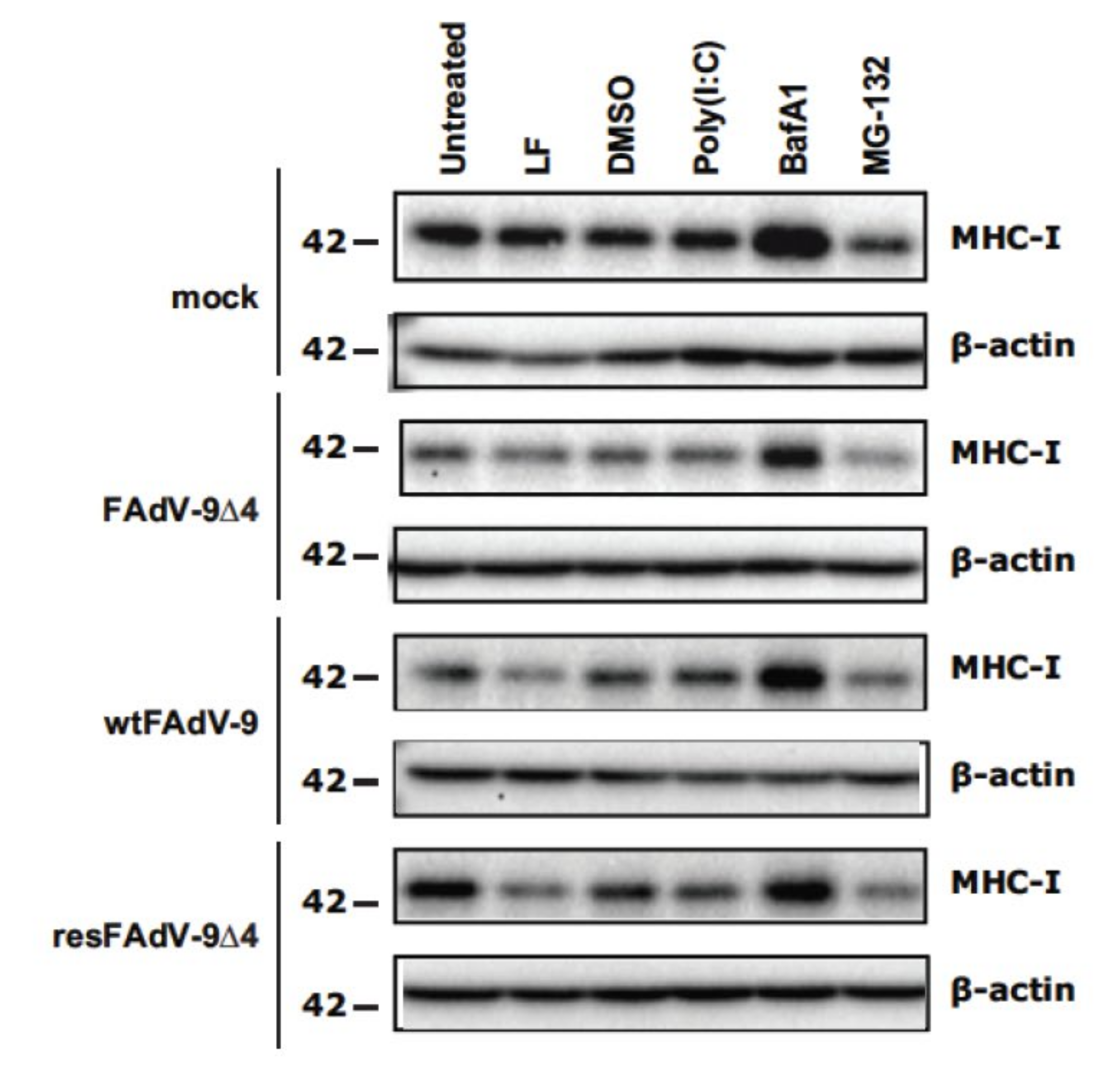
3.4. FAdV Induced Downregulation of Total MHC-I Levels Is FE1 Unit Independent
3.5. The Presence of the FE1 Transcription Unit of FAdV-9 Does Not Affect the Rate of MHC-I Internalization, but Reduces the Rate of MHC-I Cell Surface Restoration
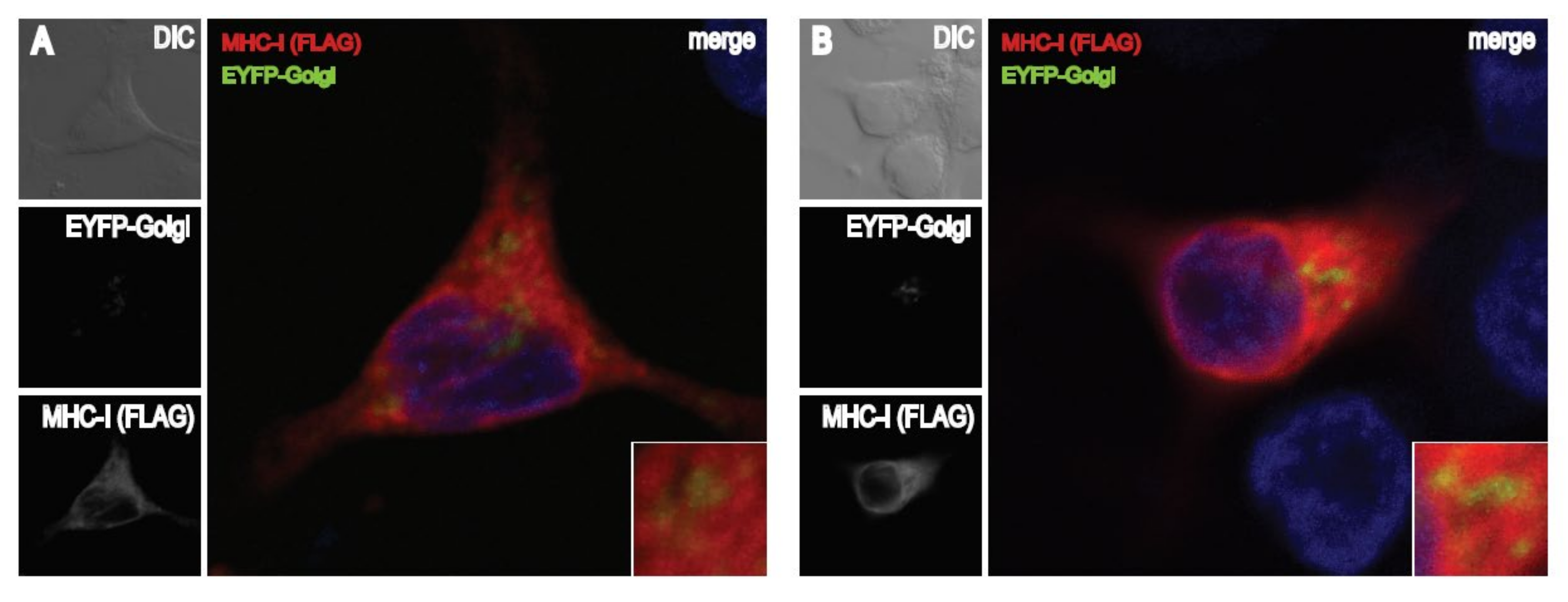
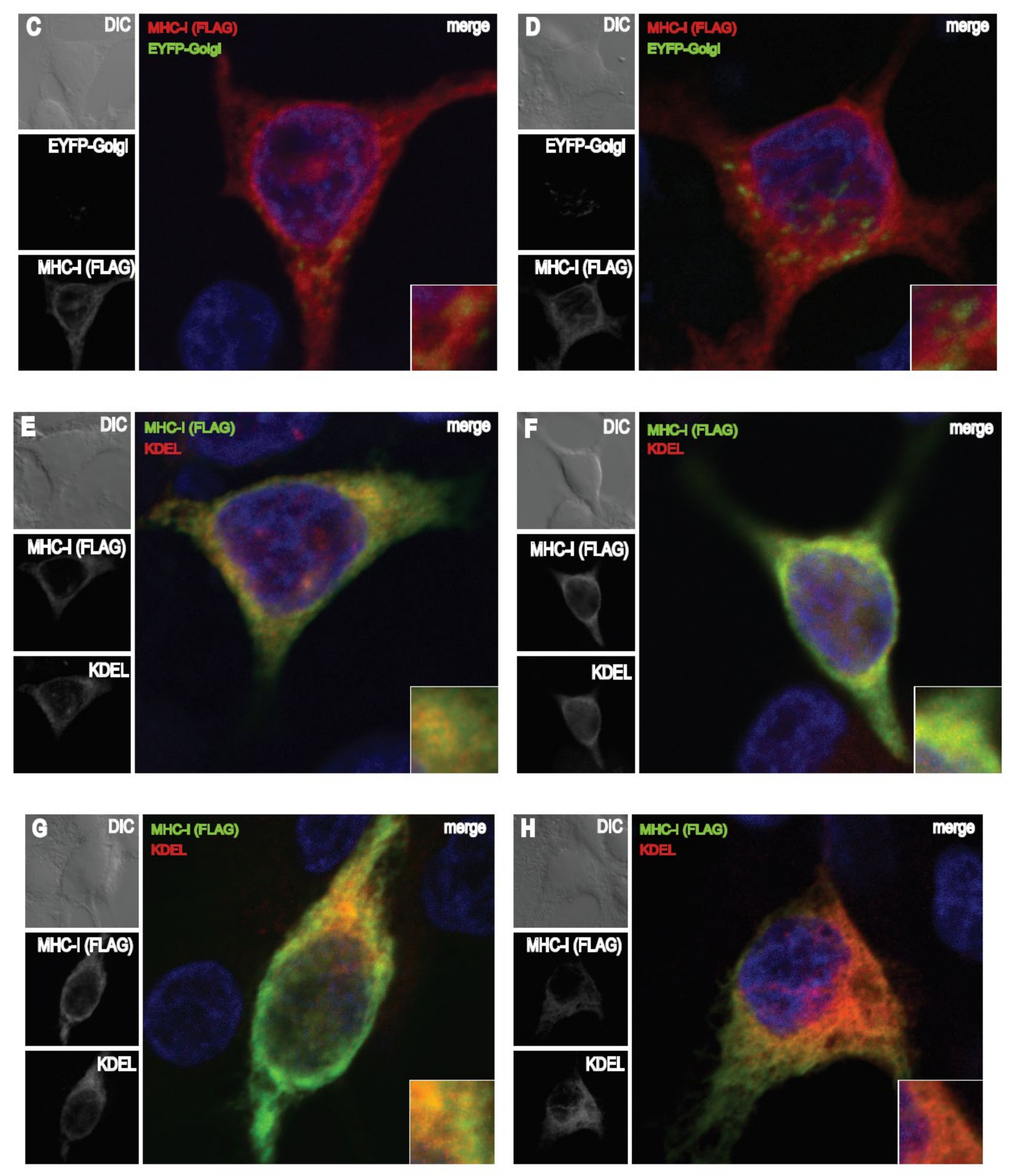
3.6. The FE1 Transcription Unit of FAdV-9 during Infection Enhances Accumulation of MHC-I in the Endoplasmic Reticulum
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alcami, A.; Koszinowski, U.H. Viral mechanisms of immune evasion. Immunol. Today 2000, 21, 447–455. [Google Scholar] [CrossRef]
- Chiocca, S.; Kurzbauer, R.; Schaffner, G.; Baker, A.; Mautner, V.; Cotten, M. The complete DNA sequence and genomic organization of the avian adenovirus CELO. J. Virol. 1996, 70, 2939–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojkic, D.; Nagy, E. The complete nucleotide sequence of fowl adenovirus type 8. J. Gen. Virol. 2000, 81, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Harrach, B.; Benkő, M. Adenoviruses (Adenoviridae). In Encyclopedia of Virology, 4th ed.; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: Oxford, UK, 2021; pp. 3–16. [Google Scholar] [CrossRef]
- Hess, M. Aviadenovirus Infections. In Diseases of Poultry, 13th ed.; Swayne, D., Glisson, J., McDougald, L., Nolan, L., Suarez, D., Nair, V., Eds.; John Wiley and Sons: Iowa City, IA, USA, 2013; pp. 290–300. [Google Scholar]
- Schachner, A.; Marek, A.; Grafl, B.; Hess, M. Detailed molecular analyses of the hexon loop-1 and fibers of fowl aviadenoviruses reveal new insights into the antigenic relationship and confirm that specific genotypes are involved in field outbreaks of inclusion body hepatitis. Vet. Microbiol. 2016, 186, 13–20. [Google Scholar] [CrossRef]
- Schachner, A.; Matos, M.; Grafl, B.; Hess, M. Fowl adenovirus-induced diseases and strategies for their control—A review on the current global situation. Avian. Pathol. 2018, 47, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Ojkic, D.; Krell, P.J.; Nagy, E. Unique features of fowl adenovirus 9 gene transcription. Virology 2002, 302, 274–285. [Google Scholar] [CrossRef]
- Davison, A.J.; Benko, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895. [Google Scholar] [CrossRef] [PubMed]
- Corredor, J.C.; Nagy, E. A region at the left end of the fowl adenovirus 9 genome that is non-essential in vitro has consequences in vivo. J. Gen. Virol. 2010, 91, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Sharif, S.; Nagy, E. Oral inoculation of chickens with a candidate fowl adenovirus 9 vector. Clin. Vaccine Immunol. 2013, 20, 1189–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Qin, X.; Krell, P.; Lu, R.; Sharif, S.; Nagy, E. Characterization and functional studies of fowl adenovirus 9 dUTPase. Virology 2016, 497, 251–261. [Google Scholar] [CrossRef]
- Deng, L.; Griffin, B.D.; Pei, Y.; Leishman, D.; McBey, B.A.; Sharif, S.; Nagy, E. Fowl Aviadenovirus 9 dUTPase Plays a Role in Regulation of the Host Immune Response. Viral Immunol. 2017, 30, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.H.; Bouvier, M. MHC class I antigen presentation: Learning from viral evasion strategies. Nat. Rev. Immunol. 2009, 9, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.N.; Powis, S.J. Pathogen evasion strategies for the major histocompatibility complex class I assembly pathway. Immunology 2008, 124, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.P.; Fennie, E.H. Adenovirus early region 1A modulation of interferon antiviral activity. J. Virol. 1987, 61, 787–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, D.J.; Ricciardi, R.P. Adenovirus type 12 E1A gene represses accumulation of MHC class I mRNAs at the level of transcription. Virology 1988, 165, 303–305. [Google Scholar] [CrossRef]
- Burgert, H.G.; Kvist, S. An adenovirus type 2 glycoprotein blocks cell surface expression of human histocompatibility class I antigens. Cell 1985, 41, 987–997. [Google Scholar] [CrossRef]
- Cox, J.H.; Bennink, J.R.; Yewdell, J.W. Retention of adenovirus E19 glycoprotein in the endoplasmic reticulum is essential to its ability to block antigen presentation. J. Exp. Med. 1991, 174, 1629–1637. [Google Scholar] [CrossRef] [Green Version]
- Zuniga, M.C. Lessons in detente or know thy host: The immunomodulatory gene products of myxoma virus. J. Biosci. 2003, 28, 273–285. [Google Scholar] [CrossRef]
- Alexander, H.; Huber, P.; Cao, J.; Krell, P.; Nagy, E. Growth characteristics of fowl adenovirus type 8 in a chicken hepatoma cell line. J. Virol. Methods 1998, 74, 9–14. [Google Scholar] [CrossRef]
- Abdul-Careem, M.F.; Hunter, B.D.; Sarson, A.J.; Mayameei, A.; Zhou, H.; Sharif, S. Marek’s disease virus-induced transient paralysis is associated with cytokine gene expression in the nervous system. Viral Immunol. 2006, 19, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Careem, M.F.; Hunter, B.D.; Sarson, A.J.; Parvizi, P.; Haghighi, H.R.; Read, L.; Heidari, M.; Sharif, S. Host responses are induced in feathers of chickens infected with Marek’s disease virus. Virology 2008, 370, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Sugawara, S.; Abo, T.; Kumagai, K. A simple method to eliminate the antigenicity of surface class I MHC molecules from the membrane of viable cells by acid treatment at pH 3. J. Immunol. Methods 1987, 100, 83–90. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Yee, M.B.; Erazo, A.; Abendroth, A.; Kinchington, P.R. Downregulation of class I major histocompatibility complex surface expression by varicella-zoster virus involves open reading frame 66 protein kinase-dependent and -independent mechanisms. J. Virol. 2007, 81, 9034–9049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartee, E.; Mansouri, M.; Hovey Nerenberg, B.T.; Gouveia, K.; Früh, K. Downregulation of Major Histocompatibility Complex Class I by Human Ubiquitin Ligases Related to Viral Immune Evasion Proteins. J. Virol. 2004, 78, 1109–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, E.A.; Hammond, C.; Cresswell, P. Misfolded major histocompatibility complex class I heavy chains are translocated into the cytoplasm and degraded by the proteasome. Proc. Natl. Acad. Sci. USA 1997, 94, 1896–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Washietl, S.; Eisenhaber, F. Reannotation of the CELO genome characterizes a set of previously unassigned open reading frames and points to novel modes of host interaction in avian adenoviruses. BMC Bioinform. 2003, 4, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Goff, F.; Méderlé-Mangeot, I.; Jestin, A.; Langlois, P. Deletion of open reading frames 9, 10 and 11 from the avian adenovirus CELO genome: Effect on biodistribution and humoral responses. J. Gen. Virol. 2005, 86, 2019–2027. [Google Scholar] [CrossRef]
- Corredor, J.C.; Krell, P.J.; Nagy, E. Sequence analysis of the left end of fowl adenovirus genomes. Virus Genes 2006, 33, 95–106. [Google Scholar] [CrossRef]
- Griffin, B.D.; Nagy, E. Coding potential and transcript analysis of fowl adenovirus 4: Insight into upstream ORFs as common sequence features in adenoviral transcripts. J. Gen. Virol. 2011, 92, 1260–1272. [Google Scholar] [CrossRef]
- Ashrafi, G.H.; Tsirimonaki, E.; Marchetti, B.; O’Brien, P.M.; Sibbet, G.J.; Andrew, L.; Campo, M.S. Down-regulation of MHC class I by bovine papillomavirus E5 oncoproteins. Oncogene 2002, 21, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, R.; Wade-Glass, M.; Rabson, M.S.; Yang, Y.C. The E5 transforming gene of bovine papillomavirus encodes a small, hydrophobic polypeptide. Science 1986, 233, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Stoddart, C.A.; Linquist-Stepps, V.; Moreno, M.E.; McCune, J.M. IFN-alpha secretion by type 2 predendritic cells up-regulates MHC class I in the HIV-1-infected thymus. J. Immunol. 2002, 168, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Petti, L.; Nilson, L.A.; DiMaio, D. Activation of the platelet-derived growth factor receptor by the bovine papillomavirus E5 transforming protein. EMBO J. 1991, 10, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B.; Ashrafi, G.H.; Tsirimonaki, E.; O’Brien, P.M.; Campo, M.S. The bovine papillomavirus oncoprotein E5 retains MHC class I molecules in the Golgi apparatus and prevents their transport to the cell surface. Oncogene 2002, 21, 7808–7816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, M.R.; Wonderlich, E.R.; Roeth, J.F.; Leonard, J.A.; Collins, K.L. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog. 2008, 4, e1000131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, A.W.; Howley, P.M.; Ploegh, H.L. A human herpesvirus 7 glycoprotein, U21, diverts major histocompatibility complex class I molecules to lysosomes. J. Virol. 2001, 75, 12347–12358. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, M.E.; Jung, J.U.; Ploegh, H.L. Kaposi’s Sarcoma-Associated Herpesvirus K3 Utilizes the Ubiquitin-Proteasome System in Routing Class I Major Histocompatibility Complexes to Late Endocytic Compartments. J. Virol. 2002, 76, 5522–5531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, Y.; Griffin, B.; de Jong, J.; Krell, P.J.; Nagy, E. Rapid generation of fowl adenovirus 9 vectors. J. Virol. Methods 2015, 223, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Corredor, J.C.; Krell, P.J.; Nagy, E. Fowl adenovirus 9 ORF19, a lipase homolog, is nonessential for virus replication and is suitable for foreign gene expression. Virus Res. 2019, 260, 129–134. [Google Scholar] [CrossRef]
- Halenius, A.; Gerke, C.; Hengel, H. Classical and non-classical MHC I molecule manipulation by human cytomegalovirus: So many targets-but how many arrows in the quiver? Cell. Mol. Immunol. 2015, 12, 139–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehrmann, H.; Cotten, M. Characterization of CELO virus proteins that modulate the pRb/E2F pathway. J. Virol. 1999, 73, 6517–6525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, T.M.; Guix, S.; Katayama, K.; Crawford, S.E.; Estes, M.K. Inhibition of cellular protein secretion by norwalk virus nonstructural protein p22 requires a mimic of an endoplasmic reticulum export signal. PLoS ONE 2010, 5, e13130. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.; Bellamy, A.R.; Taylor, J.A. Immobilization of the early secretory pathway by a virus glycoprotein that binds to microtubules. EMBO J. 2000, 19, 6465–6474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffat, K.; Knox, C.; Howell, G.; Clark, S.J.; Yang, H.; Belsham, G.J.; Ryan, M.; Wileman, T. Inhibition of the secretory pathway by foot-and-mouth disease virus 2BC protein is reproduced by coexpression of 2B with 2C, and the site of inhibition is determined by the subcellular location of 2C. J. Virol. 2007, 81, 1129–1139. [Google Scholar] [CrossRef] [Green Version]
- Cornell, C.T.; Kiosses, W.B.; Harkins, S.; Whitton, J.L. Coxsackievirus B3 Proteins Directionally Complement Each Other To Downregulate Surface Major Histocompatibility Complex Class I. J. Virol. 2007, 81, 6785–6797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousnier, A.; Swieboda, D.; Pinto, A.; Guedán, A.; Rogers, A.V.; Walton, R.; Johnston, S.L.; Solari, R. Human Rhinovirus 16 Causes Golgi Apparatus Fragmentation without Blocking Protein Secretion. J. Virol. 2014, 88, 11671–11685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deitz, S.B.; Dodd, D.A.; Cooper, S.; Parham, P.; Kirkegaard, K. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. USA 2000, 97, 13790–13795. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griffin, B.D.; Corredor, J.C.; Pei, Y.; Nagy, É. Downregulation of Cell Surface Major Histocompatibility Complex Class I Expression Is Mediated by the Left-End Transcription Unit of Fowl Adenovirus 9. Viruses 2021, 13, 2211. https://doi.org/10.3390/v13112211
Griffin BD, Corredor JC, Pei Y, Nagy É. Downregulation of Cell Surface Major Histocompatibility Complex Class I Expression Is Mediated by the Left-End Transcription Unit of Fowl Adenovirus 9. Viruses. 2021; 13(11):2211. https://doi.org/10.3390/v13112211
Chicago/Turabian StyleGriffin, Bryan D., Juan Carlos Corredor, Yanlong Pei, and Éva Nagy. 2021. "Downregulation of Cell Surface Major Histocompatibility Complex Class I Expression Is Mediated by the Left-End Transcription Unit of Fowl Adenovirus 9" Viruses 13, no. 11: 2211. https://doi.org/10.3390/v13112211