D. involucrata is an important Chinese tree species with a long history as a fine wood and ornamental tree, and possesses significant ecological value. Unfortunately, factors such as poor adaptability, strict habitat requirements, long seed dormancy, seed abortion, and human interference have led to a sharp decline in the natural population of
D. involucrata, making it an endangered species [
1]. Although many scholars have researched methods to save this species, including seed dormancy release, cutting seedlings, and organ regeneration plants, asexual reproduction of
D. involucrata remains difficult [
2]. Somatic embryogenesis has proven advantageous in large-scale plant seedling production, and can preserve plant genetic resources as well as save endangered species from extinction [
41]. In our previous study, we established a somatic embryogenesis and plant regeneration protocol for
D. involucrata. In the present study, we have investigated the key step in this process, namely, the molecular mechanism of EC induction, laying a scientific foundation for future research into the molecular mechanism of somatic embryogenesis in
D. involucrata. This work contributes to a comprehensive understanding of the mechanism of somatic embryogenesis, which is important for genetic transformation and the preservation of germplasm resources.
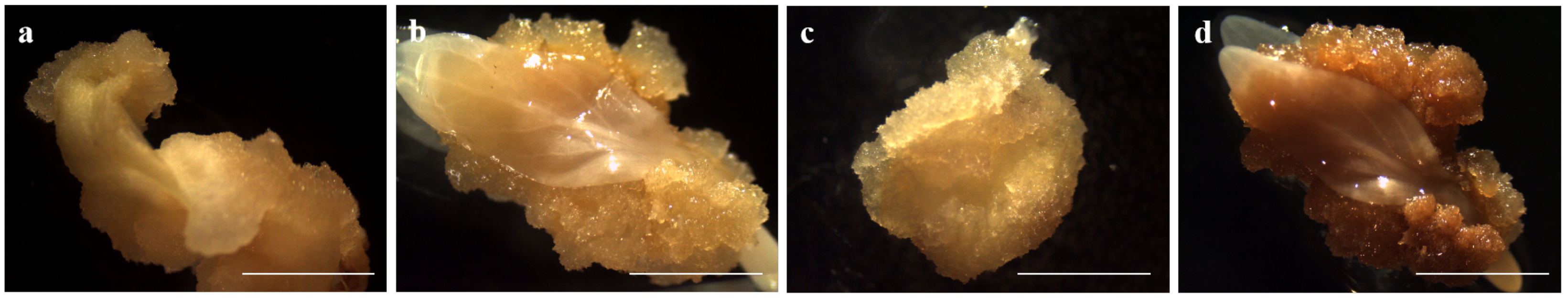
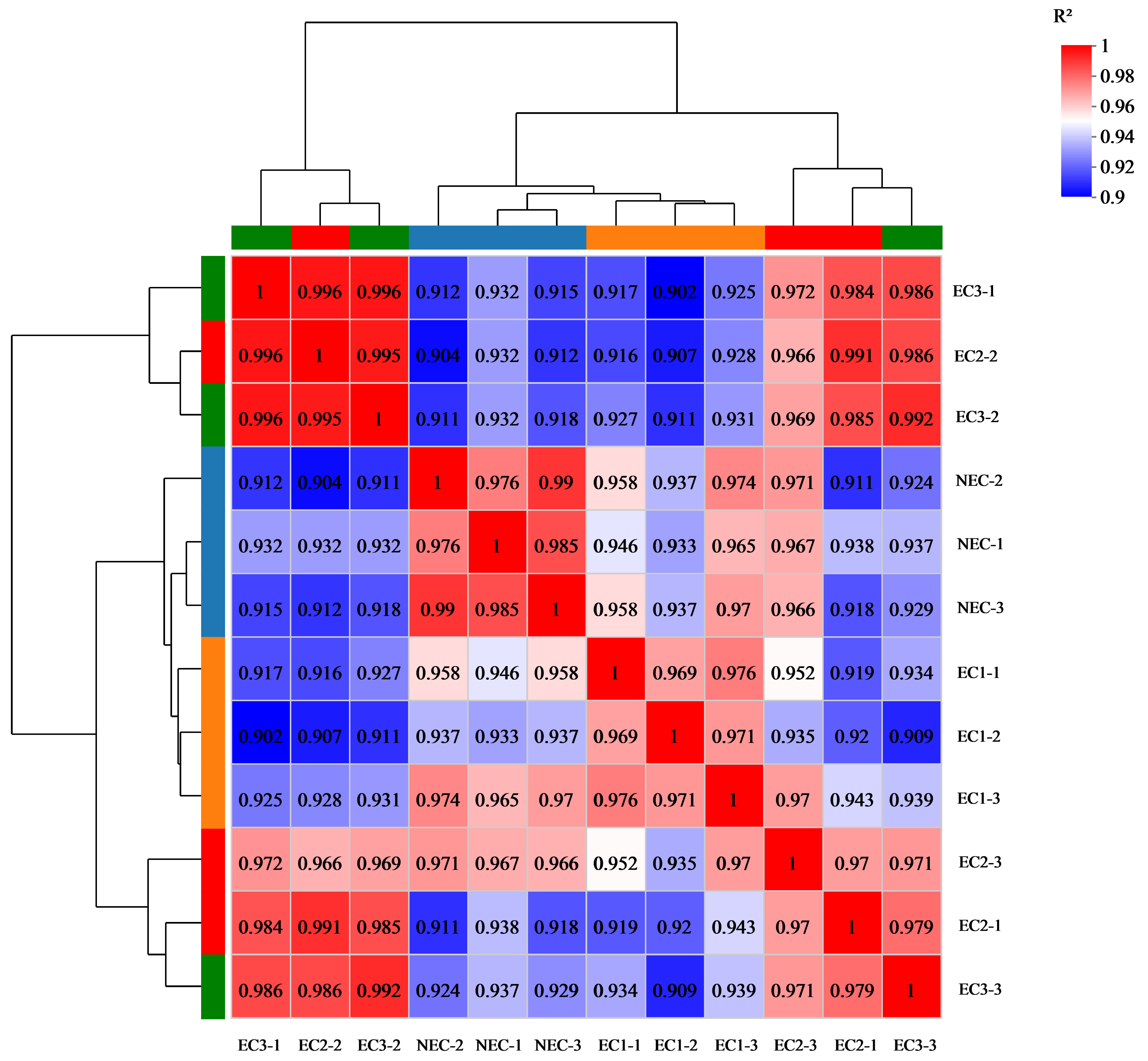
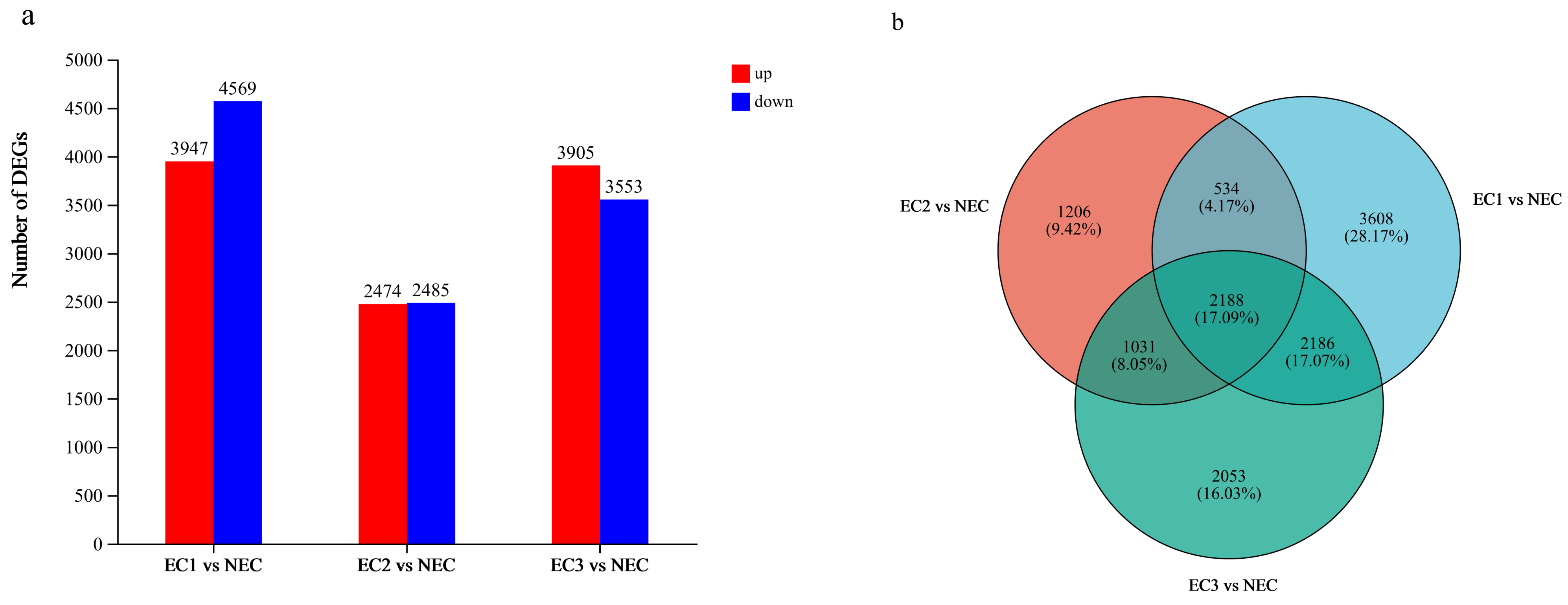
During somatic embryogenesis, ECs and NECs exhibit distinct developmental rates. For the first time, we used RNA-seq to compare global transcriptome reprogramming, revealing the regulatory mechanisms underlying EC formation. We identified a total of 12,806 DEGs which may contribute to callus differentiation. To further investigate these DEGs, we classified them using GO and KEGG analysis (
Figure 4). GO term cluster analysis showed that the most abundant category was biological processes (1037 DEGs), followed by molecular functions (1138 DEGs) and cellular components (1192 DEGs). KEGG functional classification revealed that the DEGs were significantly enriched in nineteen pathways, mainly metabolism, genetic information processing, and environmental information processing, suggesting their involvement in the induction of EC. Additionally, this study is the first to report transcriptome data for the embryogenic callus of
D. involucrata. We assembled a total of 131,109 unigenes, of which 52,420 unigenes were annotated in at least one of the databases, providing sufficient information for further gene function analysis.
4.1. The Role of Growth Hormone, Cytokinin, and Stress-Related Genes
The low homologous matching rate between the transcriptome sequencing data of
D. involucrata EC and NEC can be attributed to the lack of genomic information. Although we screened 46 genes related to plant hormones and stress by comparing the sequencing data of
D. involucrata with key genes and functions reported in other model plants, this approach has limitations, as important genes possibly related to the EC of
D. involucrata remained unannotated and unexplored. This issue has been reported in other species-related studies as well [
42].
Plant somatic cells can undergo dedifferentiation and redifferentiation, resulting in the formation of EC with cell pluripotency, ultimately leading to the development of embryos in somatic embryogenesis. Hormonal treatments and stress are commonly used to induce this process [
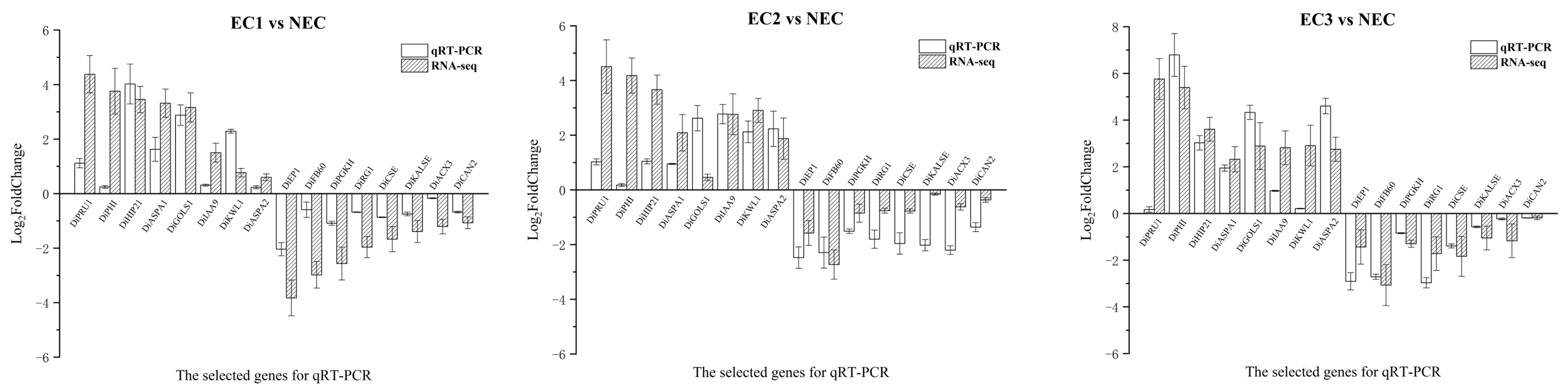
43]. The genome-wide expression patterns during EC induction may differ among various plant species. In this study, we identified genes related to plant hormones and stress that exhibited consistent differential expression patterns by comparing three EC treatments with NEC treatment. Among these, the DEGs from families such as
Aux,
IAA,
ARF,
GH3,
AHP,
ARR, and
CYCD were associated with the auxin and cytokinin signaling pathways. This result is consistent with the expression patterns of genes related to EC formation in
Arabidopsis thaliana (L.) Heynh. (
A. thaliana),
Carica papaya L. (
C. papaya), and other plant species [
44,
45]. The addition of exogenous auxin during the induction of embryogenic callus in plants can significantly affect the synthesis and metabolism of endogenous auxin. During EC induction of
Neolamarckia cadamba (Roxb.) Bosser (
N. cadamba), Li et al. [
46] found
Aux/IAA (
auxin/indole-3-acetic acid) genes,
GH3 genes, and
SAUR (
small auxin-up RNA) genes related to auxin through sequencing at different stages of EC induction. These genes were dynamically expressed, with significant upregulation and downregulation during early and later stages of callus differentiation. Our study identified auxin-related genes with a common expression trend in EC compared to NEC under different treatments, with most of these family genes being upregulated in EC. This finding suggests that auxin-responsive genes may be involved in the regulatory network of cell differentiation and regeneration during the growth of
D. involucrata EC. Although there have been few reports on the mechanism of cytokinin action, most studies have suggested its coordination with auxin in the process of EC and somatic embryogenesis, promoting cell growth, division, differentiation, and other pathways [
18].
This study identified multiple genes that are related to cell response to adverse stress during the process of EC induction. For instance, the
ABI gene, which plays a vital role in the ABA signaling pathway, might be associated with environmental stimuli such as maturation, aging, or stress [
34]. Additionally, the
BRI family gene
DiBRI1 and
BSK family gene
DiBSK1.1 might respond to plant water stress [
35]. Furthermore,
TGA family genes might play a significant role in the response to heavy metal stress [
37]. Lastly,
XTH family genes have been reported to play a critical role in drought stress in
Glycine max (L.) Merr. (
G. max) [
47]. However, the expression of these genes related to cell response to adverse stress is inconsistent during the induction of EC, indicating that the regulation mechanism of stress-related genes in the process of EC differentiation is complex and that their specific gene function requires further research and analysis.
4.2. Molecular Regulation of Somatic Embryo-Related Genes in Healing Tissues
Plant cell regeneration research suggests that certain genes may be specifically expressed during the transformation of somatic or vegetative cells into EC, providing them with meristem ability when cultured in vitro [
48]. To identify common somatic embryogenesis-related genes, we compared transcript abundance from three sets of EC treatments. Although certain genes showed inconsistent expression trends, we found 80 genes with consistent expression trends and obvious expression levels, including genes in the
BBM family,
WUS family,
GRF family (
growth-regulating factor),
SERK family,
WOX family, etc. These gene families are known to promote plant regeneration and development during somatic embryogenesis [
39].
BBM gene, a member of the
AP2/ERF transcription factor family, was first identified in somatic embryo cells of
Brassica napus L. (
B. napus), and has been found to be helpful in somatic embryo induction [
49]. The participation of the
BBM gene in the EC differentiation of
D. involucrata was detected in the present study, where
DiBBM2 transcript abundance was significantly higher in the three EC treatment groups than in the NEC treatment group. This suggests that the
BBM gene may promote cell dedifferentiation and regeneration ability during the process of embryogenic callus differentiation in
D. involucrata.
The
WOX family is a plant-specific family of transcription factors mostly involved in important biological processes such as growth, development, and responses to abiotic stress [
50].
WUS was the first gene identified in the
WOX family, and studies have shown that it has a similar effect to
BBM.
WUS mainly plays a vital role in cellular processes such as the maintenance of shoot apical meristem and the totipotency of plant cells [
51]. There are many successful reports of these two genes being used in plant genetic transformation, including
Zea mays L. (
Z. mays),
Oryza sativa L. (
O. sativa),
Sorghum bicolor (L.) Moench (
S. bicolor), and other plants. Their application in meristem induction and maintenance, plant regeneration, and genetic transformation technology systems has been successful [
48].
In this study, multiple genes of multiple
WOX families were detected in
D. involucrata callus. However, members of the same gene family showed different expression patterns. For example, the downregulated expression of the
WOX11 gene and the upregulated expression of the
WOX1,
WOX5, and
WOX8 genes suggest that the regulation of the auxin signaling network during callus differentiation is extremely complex. The negative feedback of
WOX11 may affect the upregulated expression of
WIND (
wound-induced dedifferentiation) to further promote cell reprogramming and organ regeneration. This phenomenon has been reported in
A. thaliana, where
WOX13 expression is rapidly induced after injury, mainly through the activity of the
AP2/ERF transcription factor
WIND1. Additionally,
WOX13 negative feedback can directly upregulate
WIND2 and
WIND3 [
13]. The overexpression of
WOX5 shows its close relationship with callus induction. For instance, Zhao et al. [
52] found that in
Triticum aestivum L. (
T. aestivum) the
TaWOX5 gene was mainly expressed in roots and callus induced by auxin and cytokinin, suggesting that
TaWOX5 may be related to root formation and hormone regulation of somatic embryogenesis, which is similar to the results of this study.
GRFs are a plant-specific family of transcription factors that often form a complex with the transcription cofactor
GIF (
GRF-Interacting Factor) [
53]. Our study identified multiple
GRF genes that were significantly and dynamically expressed in EC. This finding is consistent with a previous study by Luo et al. [
54] which demonstrated that the
GRF-GIF complex regulates the transition between stem cells and their rapidly dividing daughter cells, thereby promoting cell proliferation and endowing proliferating cells with meristem potential during organogenesis. Our results support the notion that
GRF family genes are closely involved in the differentiation and proliferation of
D. involucrata callus. Previous studies have shown that using
GRFs or
GRF-GIF complexes from different species can improve plant transformation and regeneration efficiency by promoting cell proliferation and differentiation. For instance, the
GRF4-GIF1 complex has been shown to significantly enhance the transformation efficiency of
T. aestivum,
O. sativa, and
Citrus reticulata Blanco. (
C. reticulata) [
55,
56].
SERK was initially discovered during somatic cell development of
Daucus carota L. (
D. carota) [
57]. Subsequently, studies in plants such as
A. thaliana,
O. sativa,
T. aestivum, and
Z. mays have demonstrated that the upregulation of
SERK is indicative of explants possessing somatic embryogenesis ability, which further enhances genetic transformation efficiency [
18,
48]. In this study, the
SERK gene was screened and found to be significantly upregulated in the EC of
D. involucrata, suggesting that somatic embryo-related genes are vital for the induction of embryogenic callus. This highlights the potential of EC for somatic embryogenesis, emphasizing the importance of EC as a prerequisite for somatic embryogenesis.
In addition, our study revealed a significant upregulation of a large number of
SAUR family genes in EC treated with auxin and cytokinin.
SAUR is a type of auxin early response gene that is closely linked to plant growth and development [
58], as confirmed by our results. Moreover, ZANIN et al. [
59] discovered that
CaSAUR12 and
CaSAUR18 of
Coffea arabica L. (
C. arabica) are highly expressed in all stages of somatic embryonic development, suggesting that these genes may be involved in auxin-induced cell growth and expansion. Thus, we infer that
SAUR genes are closely related to somatic embryogenesis of
D. involucrata.
Understanding the mechanism of EC is critical in the study of plant somatic embryogenesis as well as to plant improvement and reproduction [
11]. In this study, we investigated the EC of
D. involucrata and found that it is closely related to somatic embryogenesis. These findings can advance our knowledge of the initial stages of somatic embryogenesis in this tree species, including the dynamic changes of endogenous hormone levels and potential key regulatory genes. Somatic embryogenesis can be promoted by regulating these key elements, leading to faster plant regeneration through improved culture conditions and ultimately the improving efficiency of plant genetic transformation [
60].















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}