
Study on the Origin and Classification of Two Poplar Species on the Qinghai–Tibet Plateau
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Sites Investigation and Material Collection
2.2. Determination of Morphological Traits
2.3. DNA Extraction and Resequencing
2.4. Phylogenetic Relationship Analysis
2.5. Gene Flow and Hybridization Detection
2.6. Population Genomic Analyses in Sliding Windows
2.7. Population Historical Dynamics Analysis
2.8. Positive Selection Analysis
2.9. Prediction of Suitable Distribution Area of Species
2.10. Statistical Analysis of Data
3. Results
3.1. Distribution and Habitat of Two Taxa of Poplar
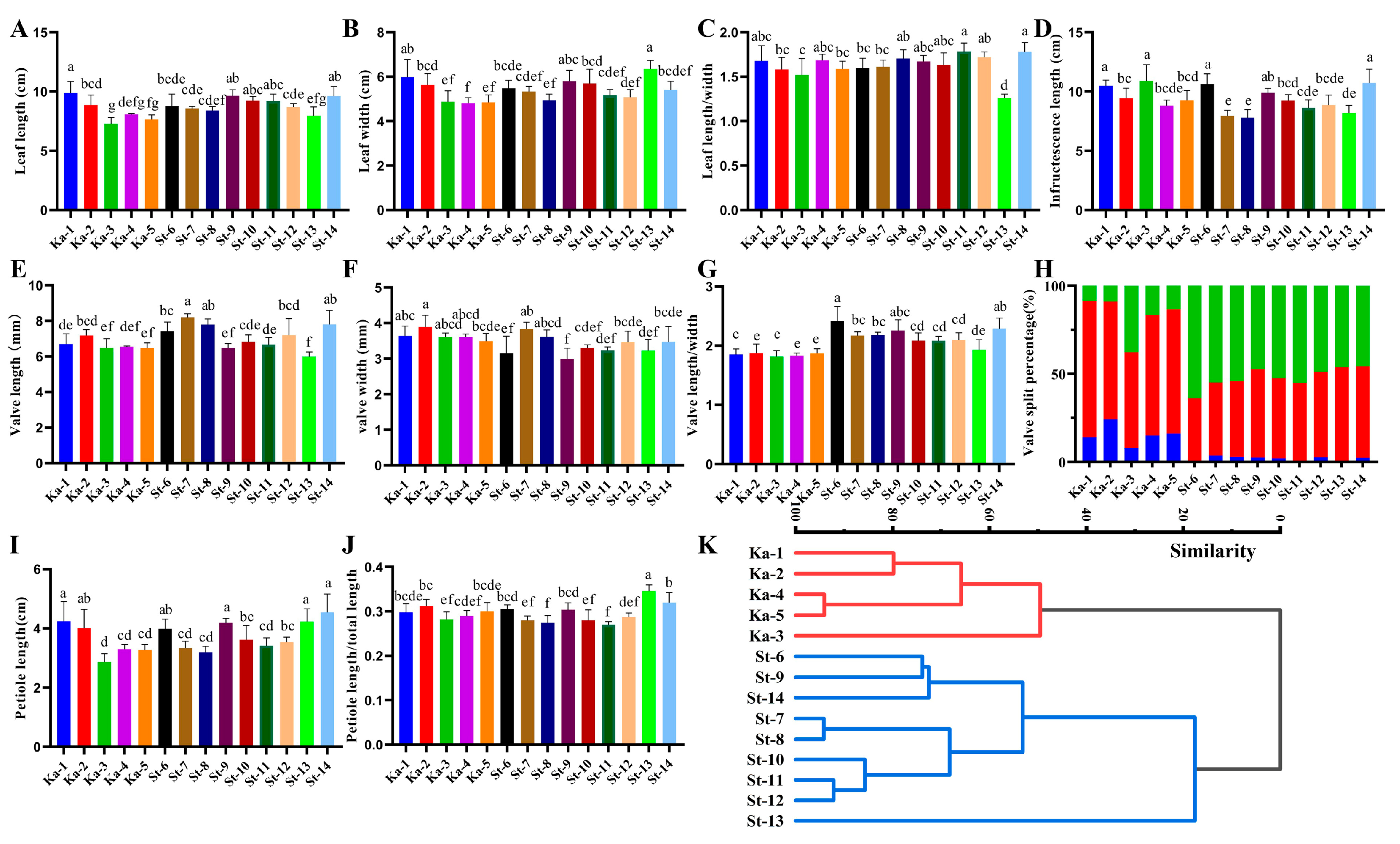
3.2. Morphological Traits of Two Taxa of Poplar
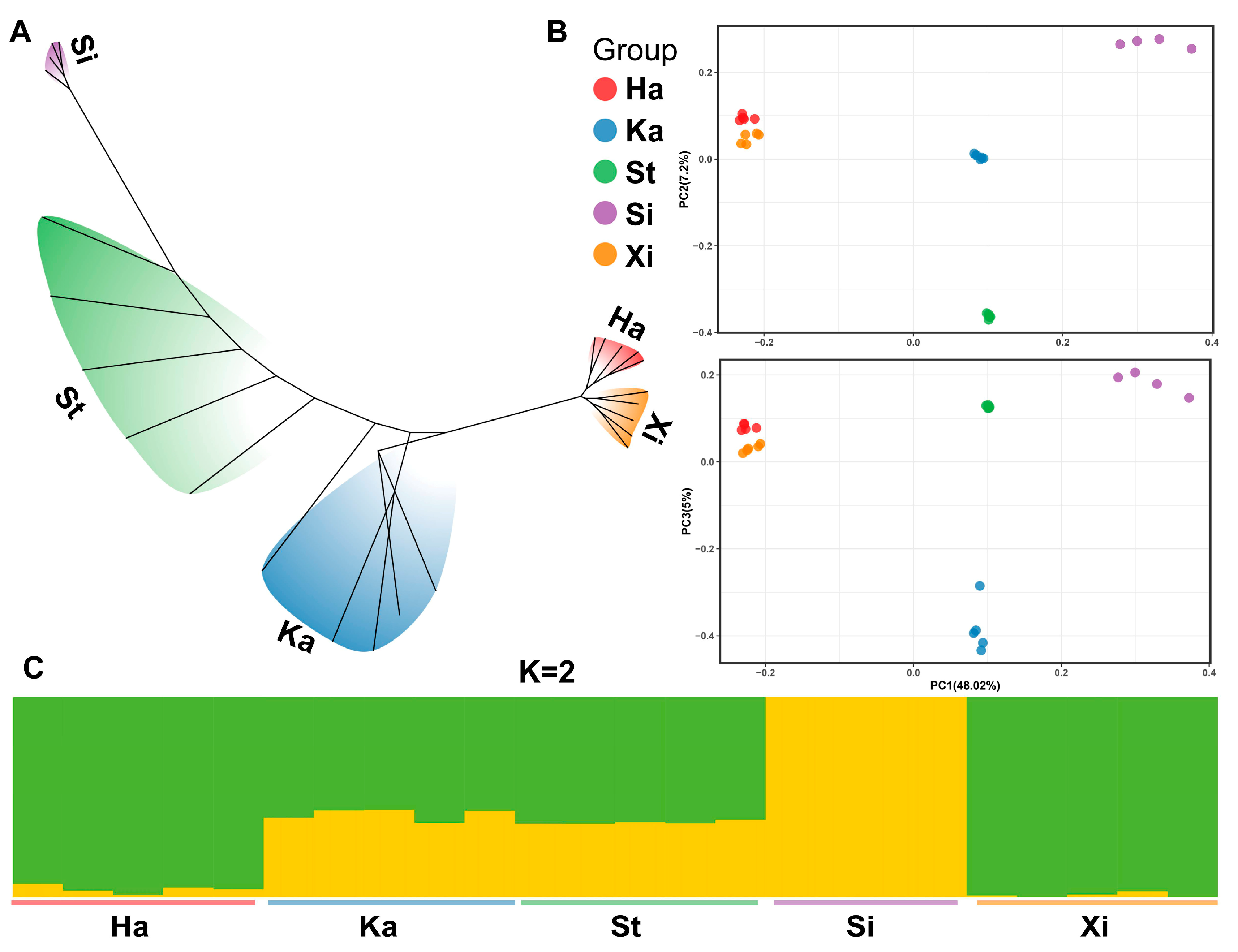
3.3. Phylogenetic Analysis
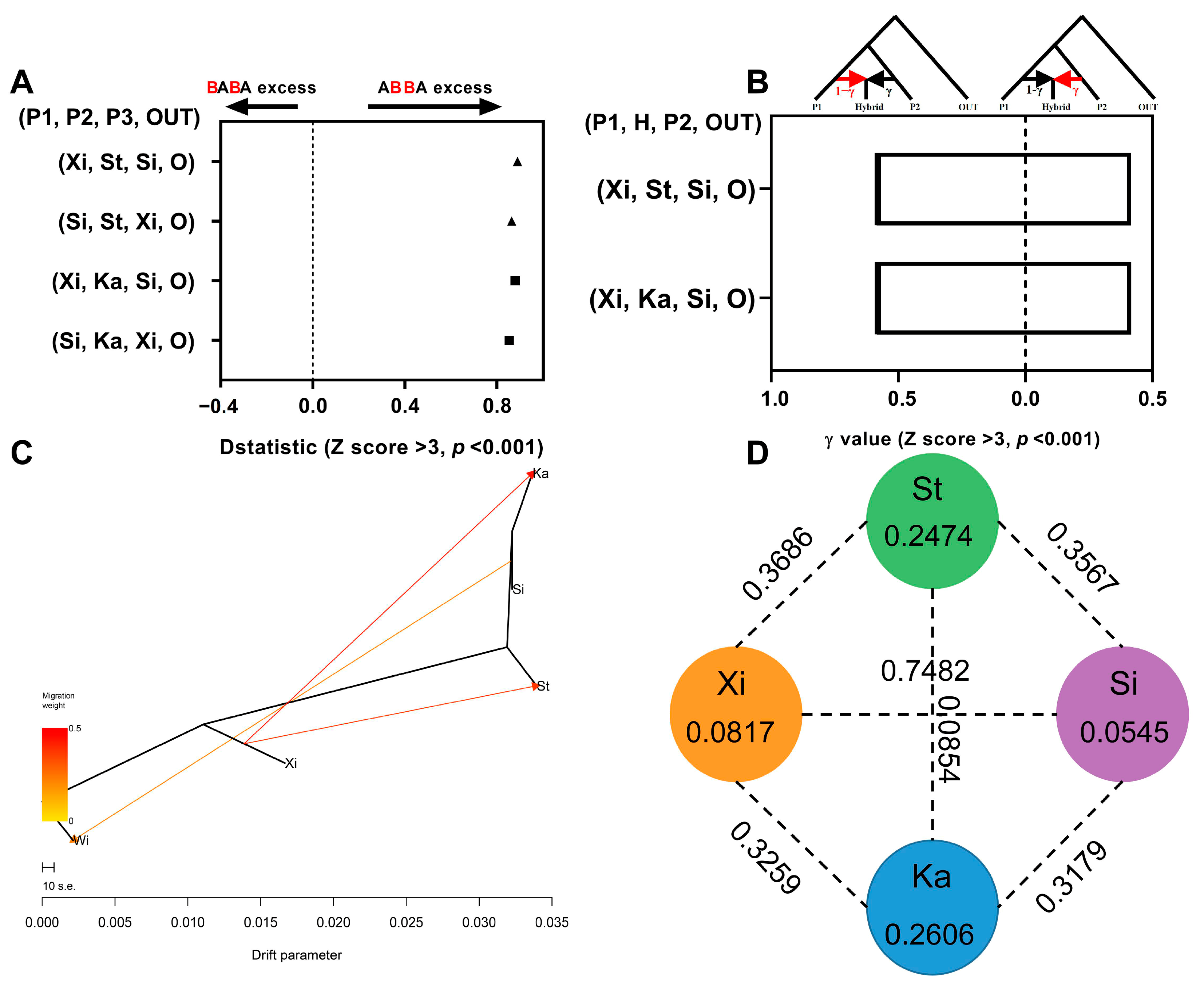
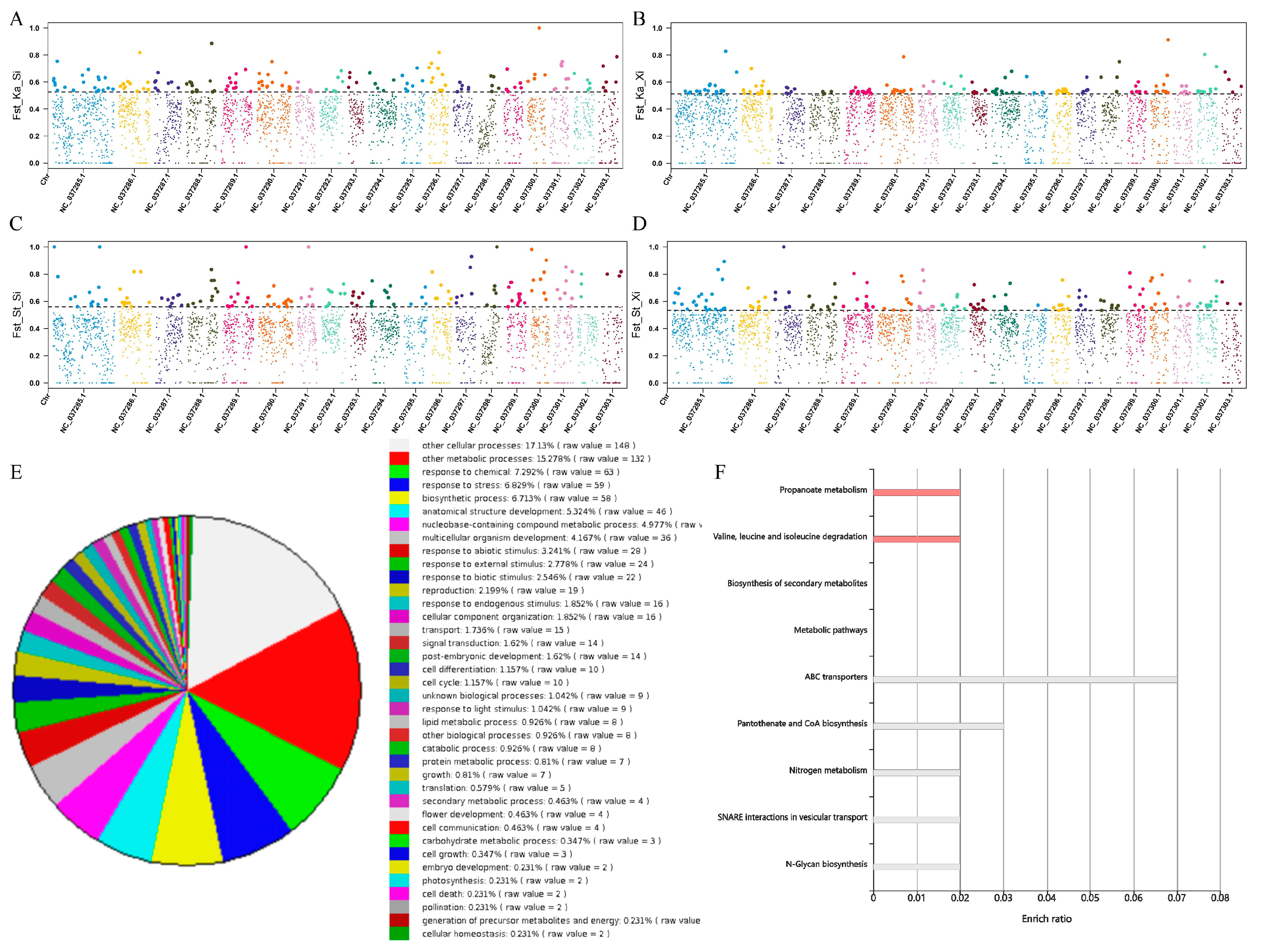
3.4. Gene Flow Detection and Genetic Differentiation Analysis
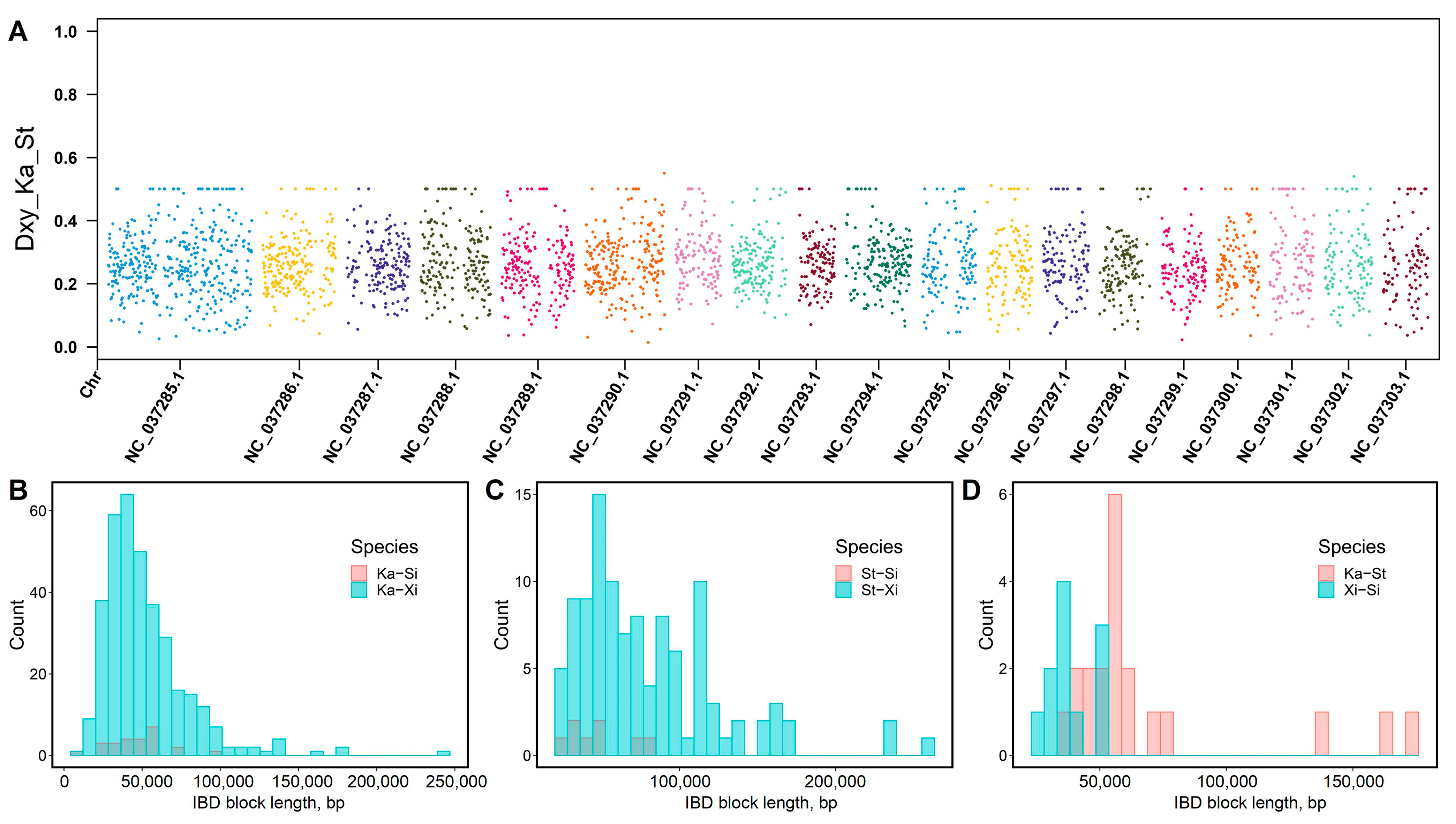
3.5. Homology Relationship Detection
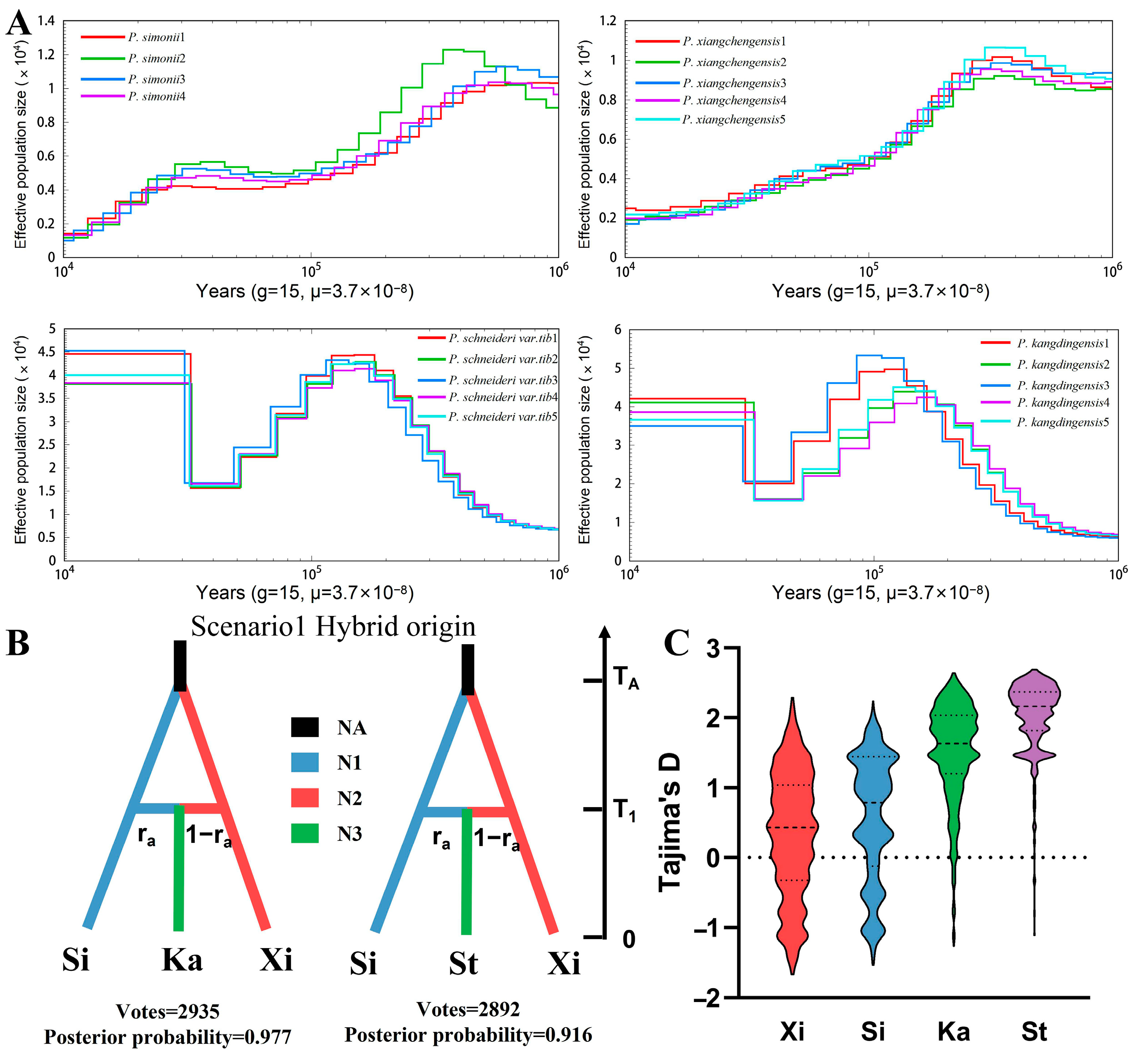
3.6. Reconstruction of Population History
3.7. Positive Selection Analysis
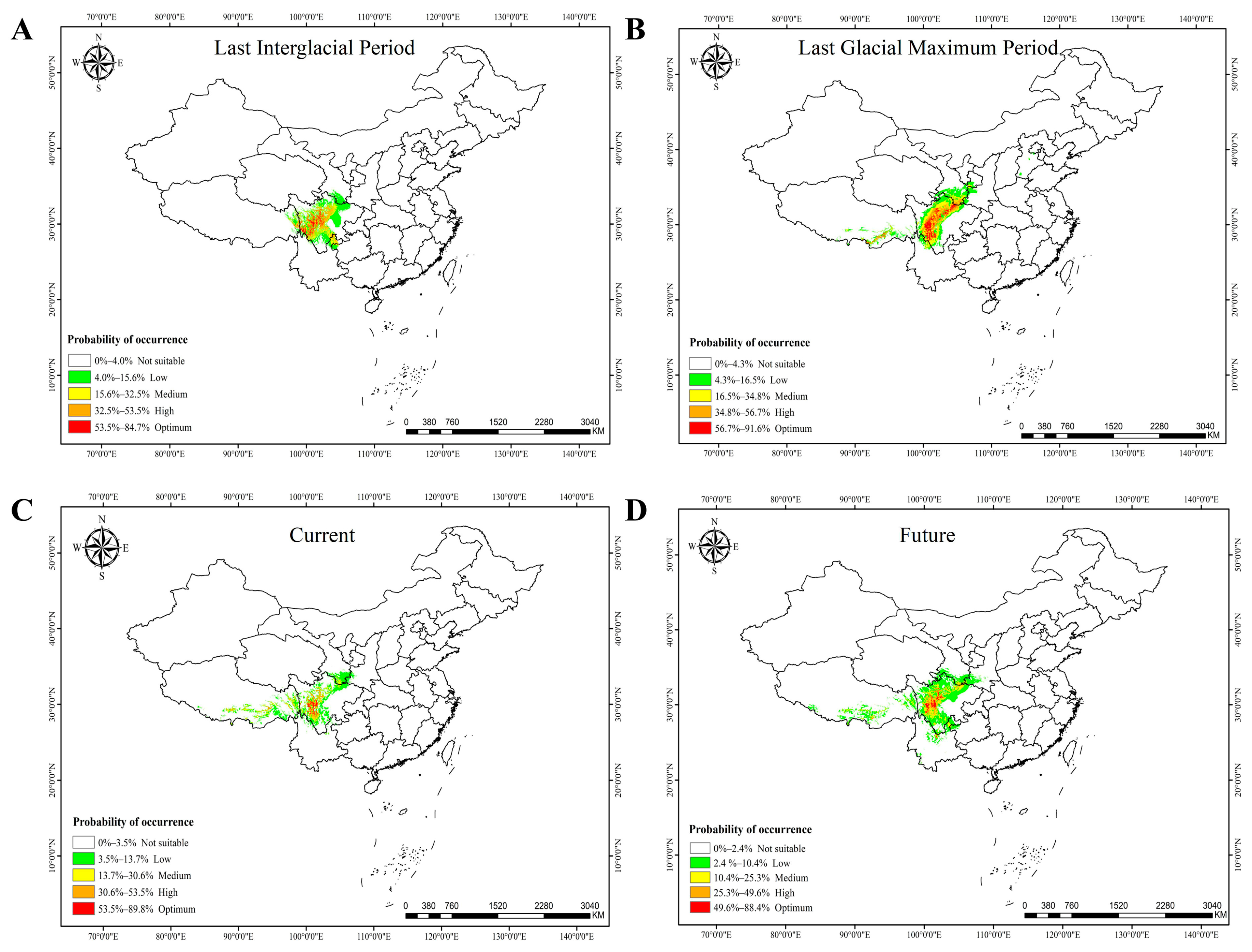
3.8. Prediction of Suitable Distribution Area
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamzeh, M.; Dayanandan, S. Phylogeny of Populus (Salicaceae) based on nucleotide sequences of chloroplast TRNT-TRNF region and nuclear rDNA. Am. J. Bot. 2004, 91, 1398–1408. [Google Scholar] [CrossRef] [PubMed]
- Eckenwalder, J.E. Systematics and evolution of Populus. In Biology of Populus and Its Implications for Management and Conservation. Part I; Stettler, R.F., Bradshaw, H.D., Jr., Heilman, P.E., Hinkley, T.M., Eds.; NRC Research Press; National Research Council of Canada: Ottawa, ON, Canada, 1996; Chapter 1; pp. 7–32. [Google Scholar]
- Dickmann, D.I.; Kuzovkina, J. Poplars and Willows of the world, with emphasis on silviculturally important species. In Poplars and Willows: Trees for Society and the Environment; CABI: Wallingford, UK, 2014; pp. 8–91. [Google Scholar]
- Braatne, J.H.; Hinckley, T.M.; Stettler, R.F. Influence of soil water on the physiological and morphological components of plant water balance in Populus trichocarpa, Populus deltoides and their F1 hybrids. Tree Physiol. 1992, 11, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G. Populus: Arabidopsis for Forestry. Do We Need a Model Tree? Ann. Bot. 2002, 90, 681–689. [Google Scholar] [CrossRef]
- Tuskan, G.A.; DiFazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef]
- Jansson, S.; Douglas, C.J. Populus: A Model System for Plant Biology. Annu. Rev. Plant Biol. 2007, 58, 435–458. [Google Scholar] [CrossRef]
- Fang, Z.F.; Zhao, S.D.; Skvortsov, A.K. Salicaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China; Missouri Botancial Garden: St. Louis, MI, USA, 1994; Volume 4, pp. 139–274. [Google Scholar]
- Lu, Z.-Q.; Tian, B.; Liu, B.-B.; Yang, C.; Liu, J.-Q. Origin of Ostryopsis intermedia (Betulaceae) in the southeast Qinghai-Tibet Plateau through hybrid speciation. J. Syst. Evol. 2014, 52, 250–259. [Google Scholar] [CrossRef]
- Wu, S.; Wang, Y.; Wang, Z.; Shrestha, N.; Liu, J. Species divergence with gene flow and hybrid speciation on the Qinghai–Tibet Plateau. New Phytol. 2022, 234, 392–404. [Google Scholar] [CrossRef]
- Wan, X.Q.; Zhang, F.; Zhong, Y.; Ding, Y.H.; Wang, C.L.; Hu, T.X. Study of genetic relationships and phylogeny of the native Populus in Southwest China based on nucleotide sequences of chloroplast trnT–trnF and nuclear DNA. Plant Syst. Evol. 2013, 299, 57–65. [Google Scholar] [CrossRef]
- Liu, J. Geographical types of Populus of Sect. Tacamahaca in western Sichuan plateau. J. Sichuan For. Sci. Technol. 1997, 18, 36–39. [Google Scholar]
- Zong, D.; Zhou, A.; Zhang, Y.; Zou, X.; Li, D.; Duan, A.; He, C. Characterization of the complete chloroplast genomes of five Populus species from the western Sichuan plateau, southwest China: Comparative and phylogenetic analyses. PeerJ 2019, 7, e6386. [Google Scholar] [CrossRef]
- Wang, C.; Tung, S.L. New taxa of poplar (I). Bull. Bot. Res. 1979, 1, 17–29. [Google Scholar]
- Chen, K.; Peng, Y.; Wang, Y.; Korpelainen, H.; Li, C. Genetic relationships among poplar species in section Taca-mahaca (Populus L.) from western Sichuan, China. Plant Sci. 2007, 172, 196–203. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Malinsky, M.; Matschiner, M.; Svardal, H. Dsuite-Fast D-statistics and related admixture evidence from VCF files. Mol. Ecol. Resour. 2021, 21, 584–595. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534, Erratum in Mol. Biol. Evol. 2020, 37, 2461. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.-Y. A Draft Sequence of the Neandertal Genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef]
- Martin, S.H.; Davey, J.W.; Jiggins, C.D. Evaluating the Use of ABBA–BABA Statistics to Locate Introgressed Loci. Mol. Biol. Evol. 2014, 32, 244–257. [Google Scholar] [CrossRef]
- Zhang, B.-W.; Xu, L.-L.; Li, N.; Yan, P.-C.; Jiang, X.-H.; Woeste, K.E.; Lin, K.; Renner, S.S.; Zhang, D.-Y.; Bai, W.-N. Phylogenomics Reveals an Ancient Hybrid Origin of the Persian Walnut. Mol. Biol. Evol. 2019, 36, 2451–2461. [Google Scholar] [CrossRef]
- Blischak, P.D.; Chifman, J.; Wolfe, A.D.; Kubatko, L.S. HyDe: A Python Package for Genome-Scale Hybridization Detection. Syst. Biol. 2018, 67, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.; Pritchard, J. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef] [PubMed]
- Korunes, K.L.; Samuk, K. Pixy: Unbiased estimation of nucleotide diversity and divergence in the presence of missing data. Mol. Ecol. Resour. 2021, 21, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Browning, B.L.; Browning, S.R. Improving the accuracy and efficiency of Identity-by-Descent detection in population data. Genetics 2013, 194, 459–471. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, L.; Sang, Y.; Lai, Q.; Zhang, X.; Jia, C.; Long, Z.; Wu, J.; Ma, T.; Mao, K. Demographic History and Natural Selection Shape Patterns of Deleterious Mutation Load and Barriers to Introgression across Populus Genome. Mol. Biol. Evol. 2022, 39, msac008. [Google Scholar] [CrossRef] [PubMed]
- Ingvarsson, P.K. Multilocus Patterns of Nucleotide Polymorphism and the Demographic History of Populus tremula. Genetics 2008, 180, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Collin, F.D.; Durif, G.; Raynal, L.; Lombaert, E.; Gautier, M.; Vitalis, R.; Marin, J.M.; Estoup, A. Extending approximate Bayesian computation with supervised machine learning to infer demographic history from genetic polymorphisms using DIYABC Random Forest. Mol. Ecol. Resour. 2021, 21, 2598–2613. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L. Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies By Use of Localized Haplotype Clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef]
- Radosavljevic, A.; Anderson, R.P. Making better Maxent models of species distributions: Complexity, overfitting and evaluation. J. Biogeogr. 2014, 41, 629–643. [Google Scholar] [CrossRef]
- Hamilton, A.J. Species diversity or biodiversity? J. Environ. Manag. 2005, 75, 89–92. [Google Scholar] [CrossRef]
- Cervera, M.T.; Storme, V.; Soto, A.; Ivens, B.; Van Montagu, M.; Rajora, O.; Boerjan, W. Intraspecific and interspecific genetic and phylogenetic relationships in the genus Populus based on AFLP markers. Theor. Appl. Genet. 2005, 111, 1440–1456. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, J.; Li, E.; Xu, S.; Zhan, Z.; Zhang, X.; Yang, Z.; Guo, F.; Liu, K.; Liu, D.; et al. Phylogenomics and Biogeography of Populus Based on Comprehensive Sampling Reveal Deep-Level Relationships and Multiple Intercontinental Dispersals. Front. Plant Sci. 2022, 13, 813177. [Google Scholar] [CrossRef]
- Yu, J.; Niu, Y.; You, Y.; Cox, C.J.; Barrett, R.L.; Trias-Blasi, A.; Guo, J.; Wen, J.; Lu, L.; Chen, Z. Integrated phylogenomic analyses unveil reticulate evolution in Parthenocissus (Vitaceae), highlighting speciation dynamics in the Himalayan-Hengduan Mountains. New Phytol. 2023, 238, 888–903. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-P.; Cao, L.; Lin, X.-R.; Ding, Y.-M.; Liang, Y.; Zhang, D.-Y.; Pang, E.-L.; Renner, S.S.; Bai, W.-N. Dead-End Hybridization in Walnut Trees Revealed by Large-Scale Genomic Sequence Data. Mol. Biol. Evol. 2022, 39, msab308. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Mao, X.; Wang, J.; Zhang, L.; Jiang, Y.; Geng, Y.; Ma, T.; Cai, L.; Huang, S.; Hollingsworth, P.; et al. Pervasive hybridization during evolutionary radiation of Rhododendron subgenus Hymenanthes in mountains of southwest China. Natl. Sci. Rev. 2022, 9, nwac276. [Google Scholar] [CrossRef]
- Arnold, M.L. Natural Hybridization and Evolution; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Abbott, R.; Albach, D.; Ansell, S.; Arntzen, J.W.; Baird, S.J.; Bierne, N.; Boughman, J.; Brelsford, A.; Buerkle, C.A.; Buggs, R. Hybridization and speciation. J. Evol. Biol. 2013, 26, 229–246. [Google Scholar] [CrossRef]
- Payseur, B.A.; Rieseberg, L.H. A genomic perspective on hybridization and speciation. Mol. Ecol. 2016, 25, 2337–2360. [Google Scholar] [CrossRef]
- Ellstrand, N.C.; Whitkus, R.; Rieseberg, L.H. Distribution of spontaneous plant hybrids. Proc. Natl. Acad. Sci. USA 1996, 93, 5090–5093. [Google Scholar] [CrossRef]
- Todesco, M.; Pascual, M.A.; Owens, G.L.; Ostevik, K.L.; Moyers, B.T.; Hübner, S.; Heredia, S.M.; Hahn, M.A.; Caseys, C.; Bock, D.G.; et al. Hybridization and extinction. Evol. Appl. 2016, 9, 892–908. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Y.; He, H.; He, G.; Deng, X.W. From hybrid genomes to heterotic trait output: Challenges and opportunities. Curr. Opin. Plant Biol. 2022, 66, 102193. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.-N.; Yan, P.-C.; Zhang, B.-W.; Woeste, K.E.; Lin, K.; Zhang, D.-Y. Demographically idiosyncratic responses to climate change and rapid Pleistocene diversification of the walnut genus Juglans (Juglandaceae) revealed by whole-genome sequences. New Phytol. 2018, 217, 1726–1736. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, T.; Wang, Z.; Lu, Z.; Li, Y.; Fu, C.; Chen, X.; Zhao, M.; Olson, M.S.; Liu, J. Genomic effects of population collapse in a critically endangered ironwood tree Ostrya rehderiana. Nat. Commun. 2018, 9, 5449. [Google Scholar] [CrossRef] [PubMed]
- Salojärvi, J.; Smolander, O.P.; Nieminen, K.; Rajaraman, S.; Safronov, O.; Safdari, P.; Lamminmäki, A.; Immanen, J.; Lan, T.; Tanskanen, J. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nat. Genet. 2017, 49, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zheng, P.; Dong, S.; Zhan, X.; Wu, Q.; Guo, X.; Hu, Y.; He, W.; Zhang, S.; Fan, W. Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat. Genet. 2013, 45, 67–71. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.-J.; Mi, J.-X.; Huang, J.-L.; He, F.; Chen, L.-H.; Wan, X.-Q. Study on the Origin and Classification of Two Poplar Species on the Qinghai–Tibet Plateau. Forests 2023, 14, 1003. https://doi.org/10.3390/f14051003
Shi Y-J, Mi J-X, Huang J-L, He F, Chen L-H, Wan X-Q. Study on the Origin and Classification of Two Poplar Species on the Qinghai–Tibet Plateau. Forests. 2023; 14(5):1003. https://doi.org/10.3390/f14051003
Chicago/Turabian StyleShi, Yu-Jie, Jia-Xuan Mi, Jin-Liang Huang, Fang He, Liang-Hua Chen, and Xue-Qin Wan. 2023. "Study on the Origin and Classification of Two Poplar Species on the Qinghai–Tibet Plateau" Forests 14, no. 5: 1003. https://doi.org/10.3390/f14051003