
Identification and Characterization of Salt-Responsive MicroRNAs in Taxodium hybrid ‘Zhongshanshan 405’ by High-Throughput Sequencing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Small RNA Library Construction and High-Throughput Sequencing
2.3. Data Filtering and Mapping Reads
2.4. Identification of Known MicroRNAs and Novel MicroRNAs
2.5. Analyzing sRNA Expression
2.6. Target Prediction
2.7. Validation of miRNAs’ Expression by Real-Time Quantitative PCR (QRT-PCR)
3. Results
3.1. Deep-Sequencing of sRNAs
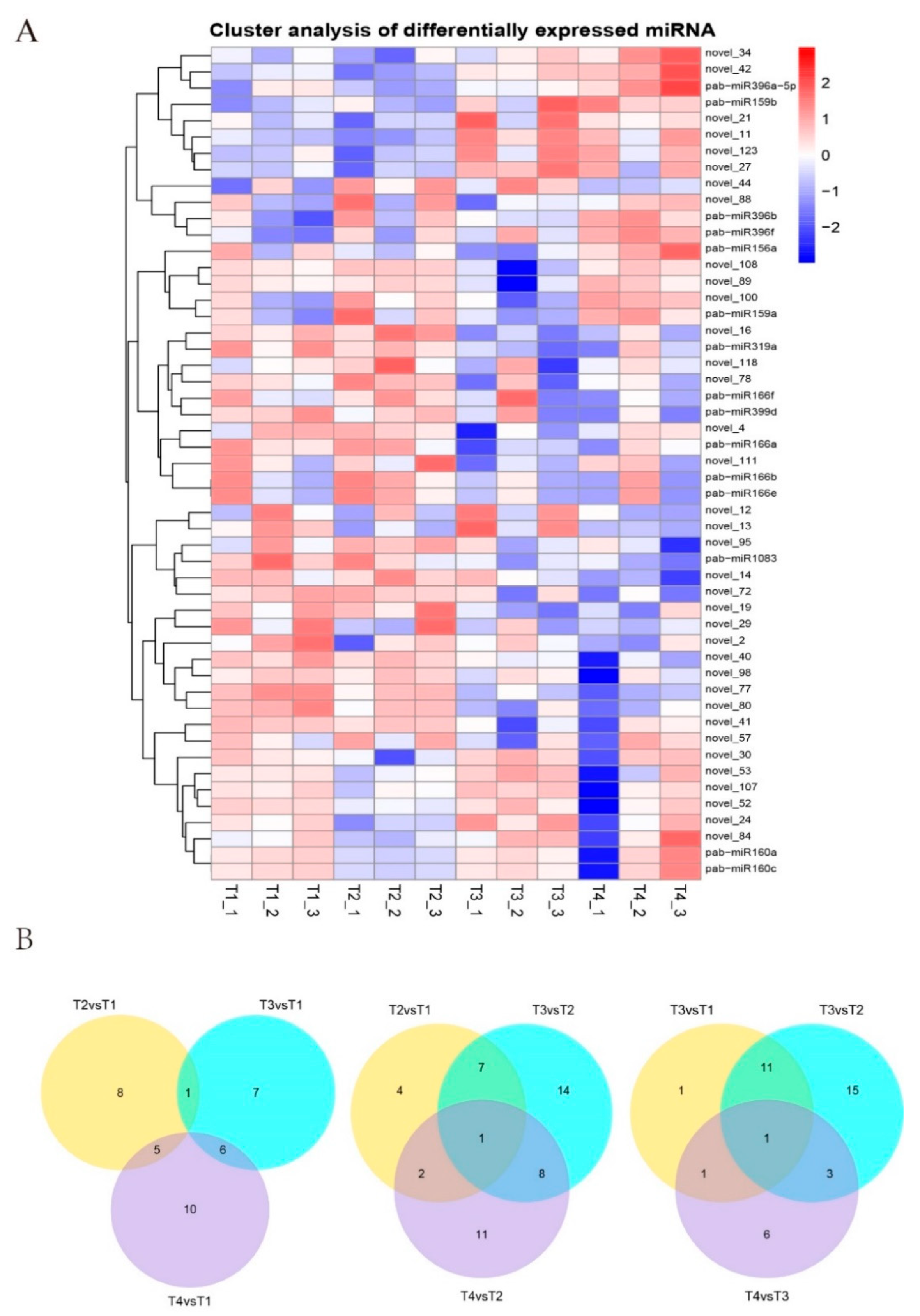
3.2. Identification of Known miRNAs and Novel miRNAs
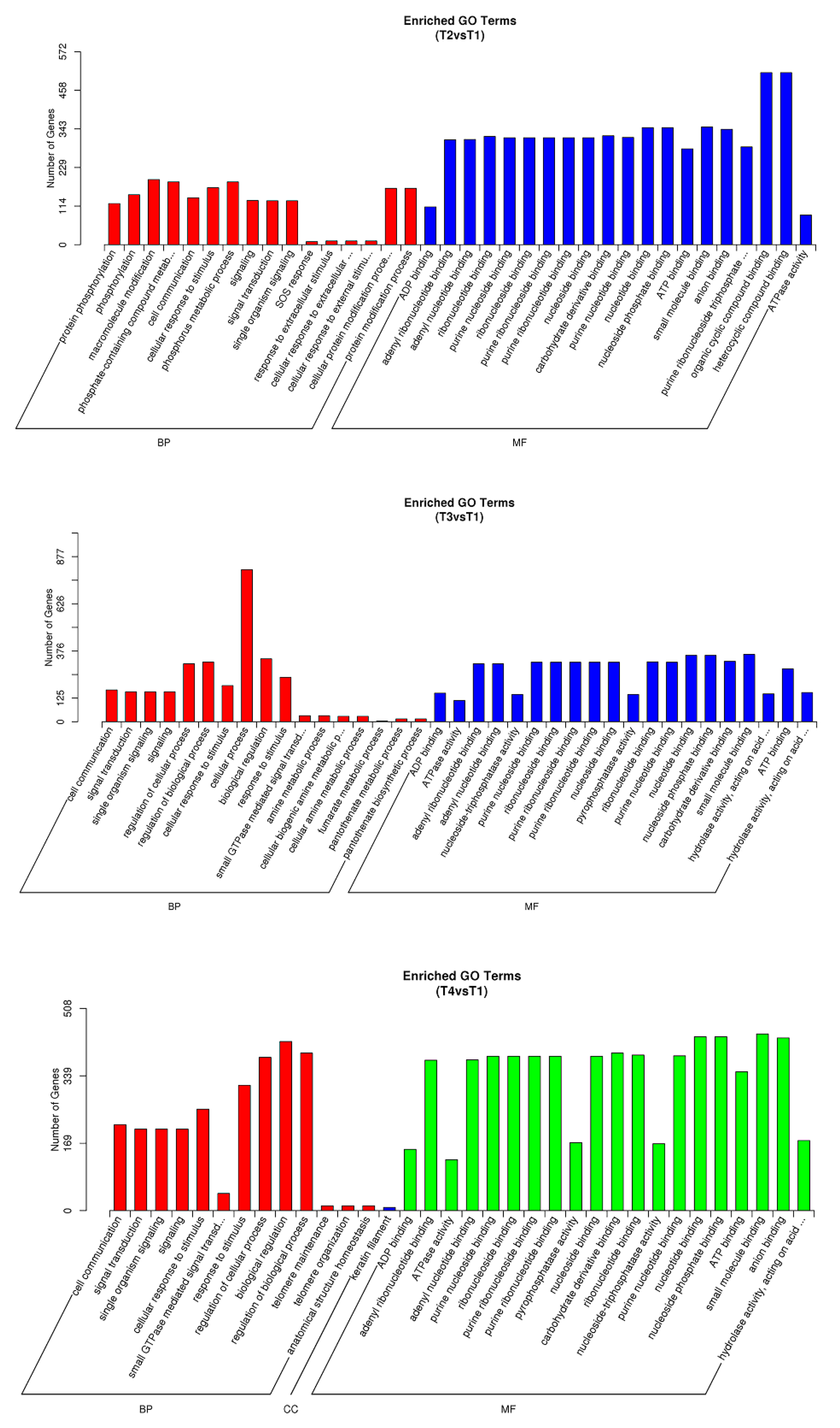
3.3. Target Prediction for Known and Novel miRNAs
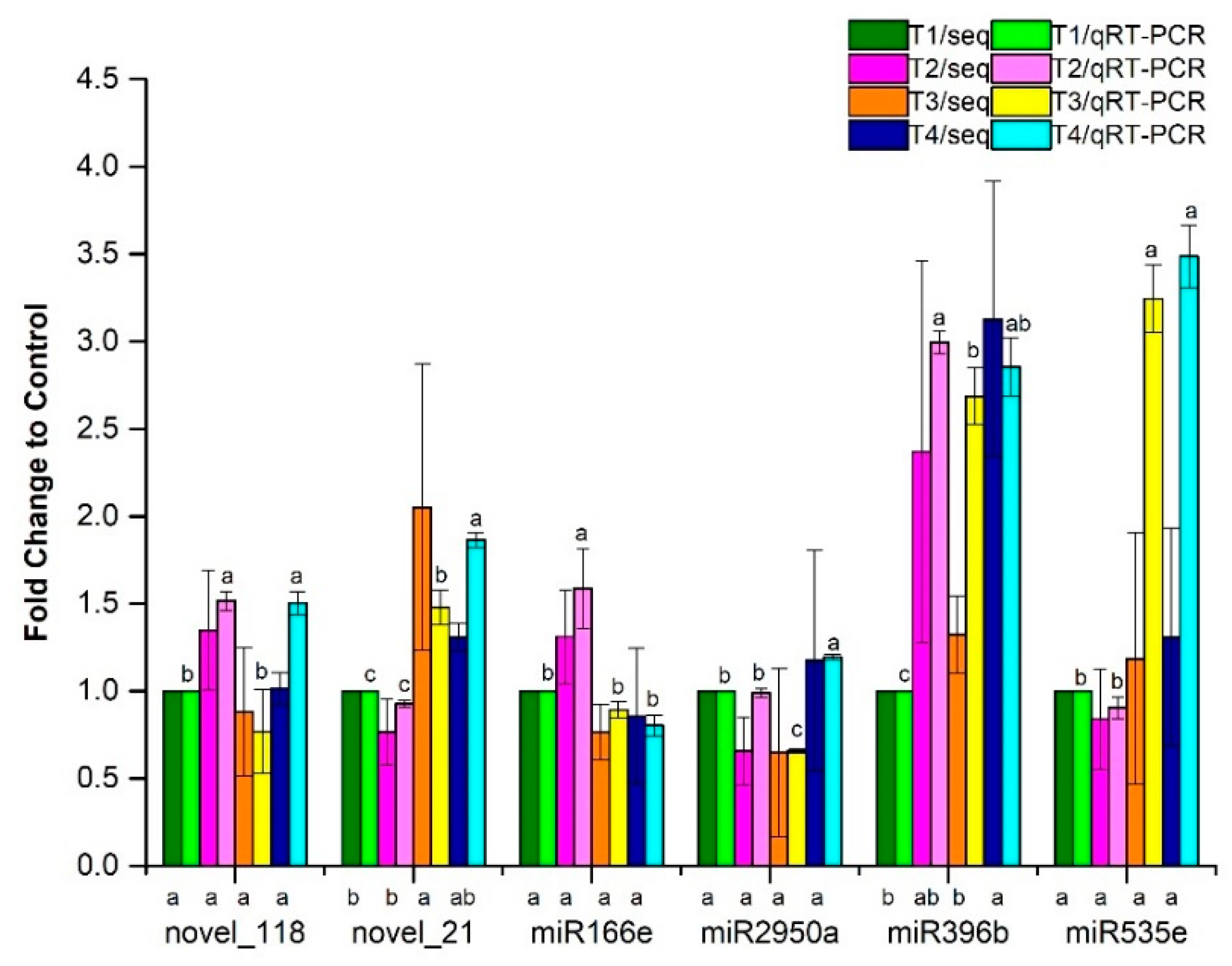
3.4. QRT-PCR Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Paul, D.; Lade, H. Plant-growth-promoting rhizobacteria to improve crop growth in saline soils: A review. Agron. Sustain. Dev. 2014, 34, 737–752. [Google Scholar] [CrossRef]
- Akula, R.; Ravishankar, G.A. Influence of abiotic stress signals on secondary metabolites in plants. Plant Signal. Behav. 2011, 6, 1720–1731. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Creech, D.L.; Krauss, K.W.; Yunlong, Y.; Kulhavy, D.L. Can we improve the salinity tolerance of genotypes of taxodium by using varietal and hybrid crosses? HortScience 2010, 45, 1773–1778. [Google Scholar] [CrossRef] [Green Version]
- Creech, D.; Zhou, L.; Yunlong, Y.; Eguiluz-Piedra, T. Can Taxodium be improved. Arnoldia 2011, 69, 11–20. [Google Scholar]
- Qi, B.; Yang, Y.; Yin, Y.; Xu, M.; Li, H. De novo sequencing, assembly, and analysis of the Taxodium ‘Zhongshansa’ roots and shoots transcriptome in response to short-term waterlogging. BMC Plant Biol. 2014, 14, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gu, C.; Xuan, L.; Hua, J.; Shi, Q.; Fan, W.; Yin, Y.; Yu, F. Identification of suitable reference genes in Taxodium ‘Zhongshanshan’under abiotic stresses. Trees 2017, 31, 1519–1530. [Google Scholar] [CrossRef]
- Xuan, L.; Hua, J.; Zhang, F.; Wang, Z.; Pei, X.; Yang, Y.; Yin, Y.; Creech, D.L. Identification and functional analysis of Thadh1 and Thadh4 genes involved in tolerance to waterlogging stress in Taxodium hybrid ‘Zhongshanshan 406’. Genes 2021, 12, 225. [Google Scholar] [CrossRef]
- Yu, C.; Xu, S.; Yin, Y. Transcriptome analysis of the Taxodium ‘Zhongshanshan 405’ roots in response to salinity stress. Plant Physiol. Biochem. 2016, 100, 156–165. [Google Scholar] [CrossRef]
- Pattanayak, D.; Solanke, A.U.; Kumar, P.A. Plant RNA interference pathways: Diversity in function, similarity in action. Plant Mol. Biol. Report. 2013, 31, 493–506. [Google Scholar] [CrossRef]
- Sunkar, R.; Li, Y.-F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef]
- Saini, A.; Li, Y.; Jagadeeswaran, G.; Sunkar, R. Role of microRNAs in plant adaptation to environmental stresses. In Micrornas in Plant Development and Stress Responses; Springer: Berlin/Heidelberg, Germany, 2012; pp. 219–232. [Google Scholar]
- Gupta, O.P.; Sharma, P. Potential role of small RNAs during stress in plants. In Molecular Approaches in Plant Abiotic Stress; CRC Press: Boca Raton, FL, USA, 2013; pp. 67–89. [Google Scholar]
- Alzahrani, S.M.; Alaraidh, I.A.; Khan, M.A.; Migdadi, H.M.; Alghamdi, S.S.; Alsahli, A.A. Identification and characterization of salt-responsive microRNAs in Vicia faba by high-throughput sequencing. Genes 2019, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhang, L.; Wang, H.; Liu, Z.; Zhang, Z.; Zheng, Y. Differential expression of miRNAs in response to salt stress in maize roots. Ann. Bot. 2009, 103, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shriram, V.; Kumar, V.; Devarumath, R.M.; Khare, T.S.; Wani, S.H. MicroRNAs as potential targets for abiotic stress tolerance in plants. Front. Plant Sci. 2016, 7, 817. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Xu, L.; Wang, Y.; Yu, R.; Zhu, X.; Luo, X.; Gong, Y.; Wang, R.; Limera, C.; Zhang, K. Identification of novel and salt-responsive miRNAs to explore miRNA-mediated regulatory network of salt stress response in radish (Raphanus sativus L.). BMC Genom. 2015, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. Mirdeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. Mirevo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-J.; Ma, Y.-K.; Chen, T.; Wang, M.; Wang, X.-J. Psrobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Lee, E.J.; Jiang, J.; Anasuya, S.; Yang, L.Q.; Elton, T.S.; Chen, C. Real-time PCR quantification of precursor and mature microRNA. Methods 2008, 44, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Jiang, B.; Zhao, B.; Mao, X.; Lu, J.; Jin, B.; Wang, L. Liquid profiling in plants: Identification and analysis of extracellular metabolites and miRNAs in pollination drops of Ginkgo biloba. Tree Physiol. 2020, 40, 1420–1436. [Google Scholar] [CrossRef]
- Xie, F.; Stewart Jr, C.N.; Taki, F.A.; He, Q.; Liu, H.; Zhang, B. High-throughput deep sequencing shows that micro RNA s play important roles in switchgrass responses to drought and salinity stress. Plant Biotechnol. J. 2014, 12, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Han, X.; Li, Y.; Wang, J.; Wang, D.; Wang, S.; Fu, X.; Ye, W. Comparative analysis of cotton small RNAs and their target genes in response to salt stress. Genes 2017, 8, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Khare, T.; Shriram, V.; Wani, S.H. Plant small RNAs: The essential epigenetic regulators of gene expression for salt-stress responses and tolerance. Plant Cell Rep. 2018, 37, 61–75. [Google Scholar] [CrossRef]
- Lu, W.; Li, J.; Liu, F.; Gu, J.; Guo, C.; Xu, L.; Zhang, H.; Xiao, K. Expression pattern of wheat miRNAs under salinity stress and prediction of salt-inducible miRNAs targets. Front. Agric. China 2011, 5, 413–422. [Google Scholar] [CrossRef]
- Eren, H.; Pekmezci, M.; Okay, S.; Turktas, M.; Inal, B.; Ilhan, E.; Atak, M.; Erayman, M.; Unver, T. Hexaploid wheat (Triticum aestivum) root mirnome analysis in response to salt stress. Ann. Appl. Biol. 2015, 167, 208–216. [Google Scholar] [CrossRef]
- Feng, K.; Nie, X.; Cui, L.; Deng, P.; Wang, M.; Song, W. Genome-wide identification and characterization of salinity stress-responsive miRNAs in wild emmer wheat (Triticum turgidum ssp. Dicoccoides). Genes 2017, 8, 156. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.-L.; Yu, Q.-Y.; Tang, L.-L.; Ji, W.; Bai, X.; Cai, H.; Liu, X.-F.; Ding, X.-D.; Zhu, Y.-M. GsSRK, a g-type lectin s-receptor-like serine/threonine protein kinase, is a positive regulator of plant tolerance to salt stress. J. Plant Physiol. 2013, 170, 505–515. [Google Scholar] [CrossRef]
- Lee, W.S.; Gudimella, R.; Wong, G.R.; Tammi, M.T.; Khalid, N.; Harikrishna, J.A. Transcripts and microRNAs responding to salt stress in Musa acuminata colla (aaa group) cv. Berangan roots. PLoS ONE 2015, 10, e0127526. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, Y. Unraveling salt stress signaling in plants. J. Integr. Plant Biol. 2018, 60, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Parihar, P.; Singh, S.; Singh, R.; Singh, V.P.; Prasad, S.M. Effect of salinity stress on plants and its tolerance strategies: A review. Environ. Sci. Pollut. Res. 2015, 22, 4056–4075. [Google Scholar] [CrossRef]
- Gao, L.; Xiang, C.-B. The genetic locus at1g73660 encodes a putative MAPKKK and negatively regulates salt tolerance in Arabidopsis. Plant Mol. Biol. 2008, 67, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, J.; Pan, C.; Guan, X.; Wang, Y.; Liu, S.; He, Y.; Chen, J.; Chen, L.; Lu, G. Genome-wide identification of MAPKK and MAPKKK gene families in tomato and transcriptional profiling analysis during development and stress response. PLoS ONE 2014, 9, e103032. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Yang, G.; Yan, J.; Pan, Y.; Nie, X. Genome-wide identification, expression profiles and regulatory network of MAPK cascade gene family in barley. BMC Genom. 2019, 20, 1–20. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample | Total Reads | N% > 10% | Low Quality | 5 Adapter Contamine | 3 Adapter Null or Insert Null | with ployA/T/G/C | Clean Reads |
|---|---|---|---|---|---|---|---|
| T1-1 | 13,212,250 (100.00%) | 34 (0.00%) | 10,535 (0.08%) | 10,746 (0.08%) | 419,301 (3.17%) | 11,549 (0.09%) | 12,760,085 (96.58%) |
| T1-2 | 14,554,682 (100.00%) | 34 (0.00%) | 10,988 (0.08%) | 15,718 (0.11%) | 422,272 (2.90%) | 16,198 (0.11%) | 14,089,472 (96.80%) |
| T1-3 | 11,047,511 (100.00%) | 33 (0.00%) | 6373 (0.06%) | 13,511 (0.12%) | 371,044 (3.36%) | 11,949 (0.11%) | 10,644,601 (96.35%) |
| T2-1 | 10,616,411 (100.00%) | 21 (0.00%) | 4386 (0.04%) | 17,123 (0.16%) | 269,497 (2.54%) | 19,629 (0.18%) | 10,305,755 (97.07%) |
| T2-2 | 14,511,597 (100.00%) | 33 (0.00%) | 7570 (0.05%) | 20,556 (0.14%) | 450,817 (3.11%) | 20,197 (0.14%) | 14,012,424 (96.56%) |
| T2-3 | 13,778,931 (100.00%) | 32 (0.00%) | 7794 (0.06%) | 17,722 (0.13%) | 784,585 (5.69%) | 30,837 (0.22%) | 12,937,961 (93.90%) |
| T3-1 | 12,036,280 (100.00%) | 28 (0.00%) | 7326 (0.06%) | 8488 (0.07%) | 369,020 (3.07%) | 18,987 (0.16%) | 11,632,431 (96.64%) |
| T3-2 | 10,291,894 (100.00%) | 10 (0.00%) | 4139 (0.04%) | 11,050 (0.11%) | 455,849 (4.43%) | 12,135 (0.12%) | 9,808,711 (95.31%) |
| T3-3 | 11,345,585 (100.00%) | 26 (0.00%) | 4628 (0.04%) | 6330 (0.06%) | 348,991 (3.08%) | 7011 (0.06%) | 10,978,599 (96.77%) |
| T4-1 | 11,909,066 (100.00%) | 36 (0.00%) | 5711 (0.05%) | 8714 (0.07%) | 407,633 (3.42%) | 10,874 (0.09%) | 11,476,098 (96.36%) |
| T4-2 | 12,833,399 (100.00%) | 32 (0.00%) | 6067 (0.05%) | 15,353 (0.12%) | 519,695 (4.05%) | 8185 (0.06%) | 12,284,067 (95.72%) |
| T4-3 | 12,115,728 (100.00%) | 35 (0.00%) | 8663 (0.07%) | 9867 (0.08%) | 474,255 (3.91%) | 14,847 (0.12%) | 11,608,061 (95.81%) |
| Types | Total | T1-1 | T1-2 | T1-3 | T2-1 | T2-2 | T2-3 | T3-1 | T3-2 | T3-3 | T4-1 | T4-2 | T4-3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mapped known miRNAs | Mature | 49 | 36 | 43 | 39 | 31 | 30 | 29 | 40 | 26 | 29 | 19 | 31 | 26 |
| Hairpin | 55 | 38 | 48 | 44 | 34 | 34 | 32 | 43 | 31 | 33 | 22 | 36 | 28 | |
| Mapped novel miRNAs | Mature | 98 | 88 | 88 | 83 | 77 | 74 | 72 | 80 | 62 | 77 | 57 | 73 | 70 |
| Hairpin | 100 | 92 | 93 | 88 | 84 | 80 | 79 | 85 | 67 | 83 | 66 | 81 | 76 | |
| Star | 68 | 44 | 42 | 40 | 37 | 25 | 30 | 39 | 17 | 40 | 16 | 32 | 18 |
| miRNA | Target | Expectation | Target Accessibility | Target Description | Inhibition | Multiplicity | |
|---|---|---|---|---|---|---|---|
| T1 vs. T2 | novel_118 | CL16860Contig1 | 4.5 | 18.248 | Putative polyprotein | Cleavage | 1 |
| novel_13 | CL15Contig4 | 4.5 | 20.381 | Probable disease resistance protein | Translation | 1 | |
| novel_16 | T3_Unigene_BMK.33047 | 4 | 18.583 | Putative truncated TIR-NBS-LRR protein | Translation | 1 | |
| novel_52 | CL10744Contig1 | 4.5 | 10.073 | Zinc finger CCCH domain-containing protein 35 | Translation | 1 | |
| novel_78 | CL25843Contig1 | 4.5 | 12.165 | Probable nucleoredoxin 1 | Translation | 1 | |
| miR160a | CL8543Contig1 | 4 | 5.964 | Chaperone protein dnaJ 11 | Translation | 1 | |
| miR396b | CL5428Contig1 | 3.5 | 22.527 | Glycosyltransferase family protein 2 | Cleavage | 1 | |
| T1 vs. T3 | novel_123 | CL27539Contig1 | 4.5 | 13.132 | RNA-binding protein 25 | Cleavage | 1 |
| novel_21 | CL18504Contig1 | 4 | 24.442 | TMV resistance protein N | Translation | 1 | |
| novel_41 | CL2111Contig1 | 5 | 12.456 | Mitogen-activated protein kinase kinase kinase | Cleavage | 1 | |
| novel_4 | CL1685Contig1 | 4.5 | 12.818 | Ethylene-responsive transcription factor RAP2-13 | Cleavage | 1 | |
| novel_77 | T2_Unigene_BMK.14386 | 4.5 | 16.719 | G-type lectin S-receptor-like serine/threonine-protein kinase | Cleavage | 1 | |
| miR156a | CL14355Contig1 | 4 | 18.821 | RNA and export factor-binding protein 2 | Cleavage | 1 | |
| miR319a | CL11314Contig1 | 4 | 17.74 | Beta-amylase 1 isoform 1 | Cleavage | 1 | |
| T1 vs. T4 | novel_100 | CL24684Contig1 | 4.5 | 17.879 | ATP synthase subunit | Cleavage | 1 |
| novel_13 | CL4989Contig1 | 4 | 20.269 | Salicylate O-methyltransferase | Cleavage | 1 | |
| novel_14 | CL1013Contig1 | 3.5 | 18.997 | Probable LRR receptor-like serine/threonine-protein kinase | Cleavage | 1 | |
| novel_29 | CL11748Contig1 | 2 | 18.906 | Glycerol-3-phosphate 2-O-acyltransferase 6 | Translation | 1 | |
| novel_2 | CL14285Contig1 | 4 | 16.055 | G-type lectin S-receptor-like serine/threonine-protein kinase | Translation | 1 | |
| novel_40 | CL1182Contig1 | 5 | 18.447 | Disease resistance RPP13-like protein 4 | Translation | 1 | |
| novel_41 | CL2111Contig1 | 5 | 12.456 | Mitogen-activated protein kinase kinase kinase | Cleavage | 1 | |
| novel_42 | CL1146Contig1 | 2.5 | 13.999 | F-box/LRR-repeat protein 17 | Cleavage | 1 | |
| novel_77 | CL1110Contig2 | 4 | 19.906 | Cysteine-rich receptor-like protein kinase | Cleavage | 1 | |
| novel_98 | CL13461Contig1 | 4 | 14.38 | Homeobox-leucine zipper protein ATHB-13 | Cleavage | 1 | |
| miR159a | CL12428Contig1 | 3 | 16.021 | Chlorophyll a-b binding protein 7 | Translation | 1 | |
| miR396a-5p | CL10009Contig1 | 5 | 20.004 | DNA replication licensing factor mcm5 | Cleavage | 1 | |
| miR396f | CL2465Contig1 | 4 | 18.647 | U-box domain-containing protein 12 | Cleavage | 1 | |
| miR399d | CL1025Contig1 | 5 | 20.819 | Tonoplast dicarboxylate transporter | Cleavage | 1 | |
| T2 vs. T3 | novel_100 | CL772Contig3 | 4.5 | 13.205 | Transcription factor MYB59 | Cleavage | 1 |
| novel_108 | CL805Contig1 | 4 | 14.766 | LRR receptor-like serine/threonine-protein kinase | Translation | 1 | |
| novel_111 | CL1347Contig2 | 3.5 | 14.988 | Subtilisin-like protease | Cleavage | 1 | |
| novel_123 | CL14243Contig1 | 4.5 | 12.566 | Trehalose-phosphatase | Translation | 1 | |
| novel_16 | CL22Contig4 | 5 | 21.524 | TMV resistance protein | Translation | 1 | |
| novel_24 | CL228Contig1 | 2.5 | 20.215 | TMV resistance protein | Cleavage | 1 | |
| novel_30 | CL14581Contig1 | 4.5 | 19.087 | Xyloglucan endotransglucosylase/hydrolase | Translation | 1 | |
| novel_41 | CL23589Contig1 | 4.5 | 13.732 | Chaperone protein dnaJ | Cleavage | 1 | |
| novel_52 | T3_Unigene_BMK.32994 | 4.5 | 23.774 | Protein LURP-one-related | Cleavage | 1 | |
| novel_77 | T2_Unigene_BMK.14386 | 4.5 | 16.719 | G-type lectin S-receptor-like serine/threonine-protein kinase | Cleavage | 1 | |
| novel_78 | CL10767Contig1 | 5 | 17.257 | BON1-associated protein | Translation | 1 | |
| novel_88 | CL15264Contig1 | 4 | 14.363 | Cysteine-rich receptor-like protein kinase | Translation | 1 | |
| novel_89 | T3_Unigene_BMK.16696 | 3 | 17.651 | Squamosa promoter-binding-like protein | Cleavage | 1 | |
| pab-miR159a | CL2378Contig1 | 3 | 16.57 | Cinnamoyl CoA reductase | Translation | 1 | |
| pab-miR319a | CL26045Contig1 | 4 | 17.17 | Disease resistance protein RPS2 | Cleavage | 1 | |
| T3 vs. T4 | novel_108 | CL1146Contig1 | 4 | 13.459 | F-box/LRR-repeat protein 17 | Translation | 1 |
| novel_13 | CL4989Contig1 | 4 | 20.269 | Salicylate O-methyltransferase | Cleavage | 1 | |
| novel_14 | CL1013Contig1 | 3.5 | 18.997 | Probable LRR receptor-like serine/threonine-protein kinase | Cleavage | 1 | |
| miR156a | CL24146Contig1 | 3 | 15.592 | Probable LRR receptor-like serine/threonine-protein kinase | Cleavage | 1 | |
| miR159a | CL12428Contig1 | 3 | 16.021 | Chlorophyll a-b binding protein 7 | Translation | 1 | |
| miR396a-5p | CL10009Contig1 | 5 | 20.004 | DNA replication licensing factor mcm5 | Cleavage | 1 | |
| miR396b | CL2465Contig1 | 4.5 | 18.647 | U-box domain-containing protein 12 | Cleavage | 1 | |
| miR396f | CL13812Contig1 | 4.5 | 16.915 | Potassium transporter 1 | Translation | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Zhang, F.; Shi, Q.; Zhang, R.; Yin, Y.; Yu, C. Identification and Characterization of Salt-Responsive MicroRNAs in Taxodium hybrid ‘Zhongshanshan 405’ by High-Throughput Sequencing. Forests 2022, 13, 1685. https://doi.org/10.3390/f13101685
Wang Z, Zhang F, Shi Q, Zhang R, Yin Y, Yu C. Identification and Characterization of Salt-Responsive MicroRNAs in Taxodium hybrid ‘Zhongshanshan 405’ by High-Throughput Sequencing. Forests. 2022; 13(10):1685. https://doi.org/10.3390/f13101685
Chicago/Turabian StyleWang, Zhiquan, Fengjiao Zhang, Qin Shi, Rui Zhang, Yunlong Yin, and Chaoguang Yu. 2022. "Identification and Characterization of Salt-Responsive MicroRNAs in Taxodium hybrid ‘Zhongshanshan 405’ by High-Throughput Sequencing" Forests 13, no. 10: 1685. https://doi.org/10.3390/f13101685