Metabolome and Transcriptome Association Analysis Reveals Regulation of Flavonoid Biosynthesis by Overexpression of LaMIR166a in Larix kaempferi (Lamb.) Carr


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Sample Preparation
2.3. Chromatographic Mass Spectrometry Conditions
2.4. Metabolomic and Transcriptome Data Analysis
2.5. Library preparation, RNA sequencing and De Novo Assembly
2.6. Combined Analysis of Transcriptome and Metabolome
3. Results
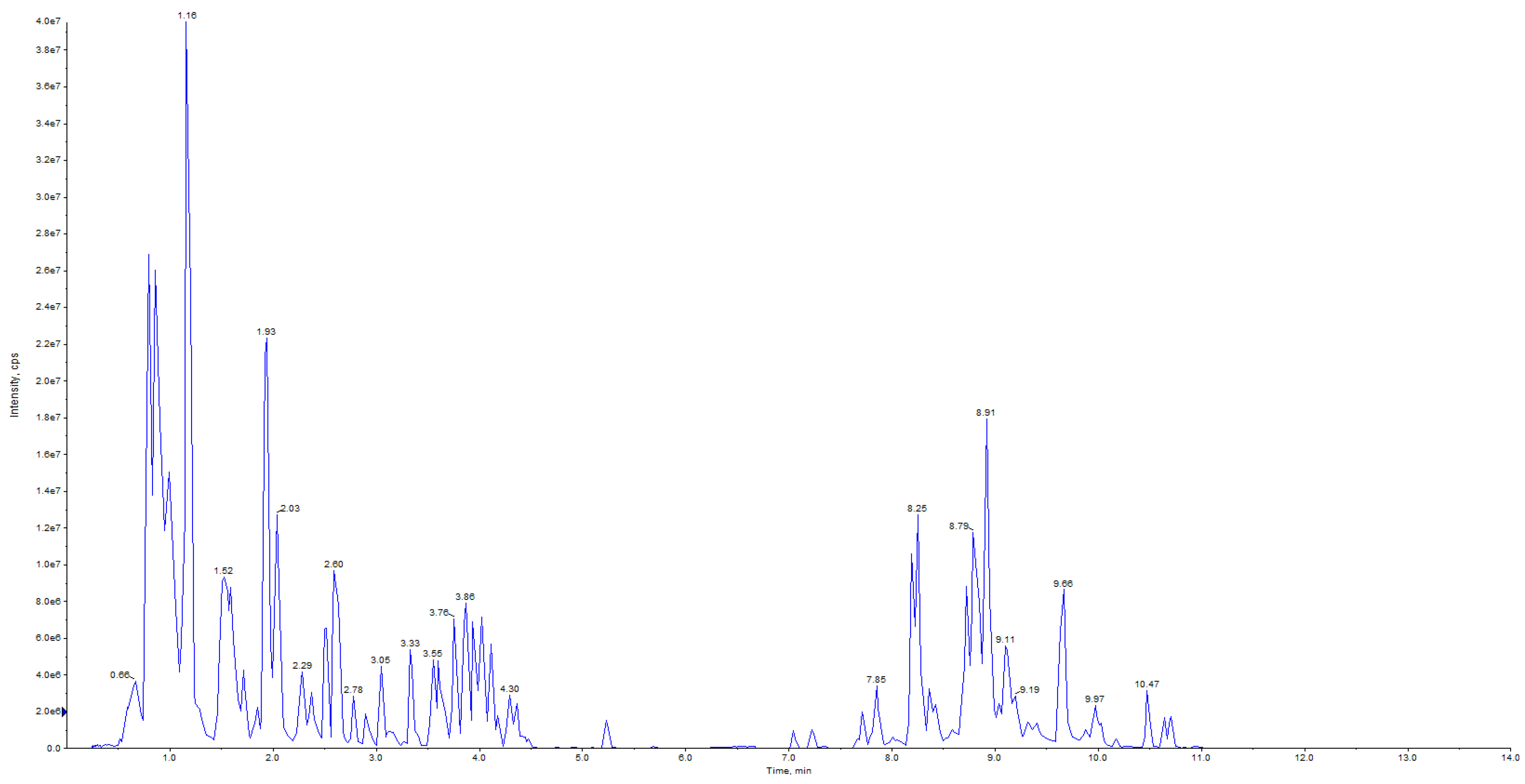
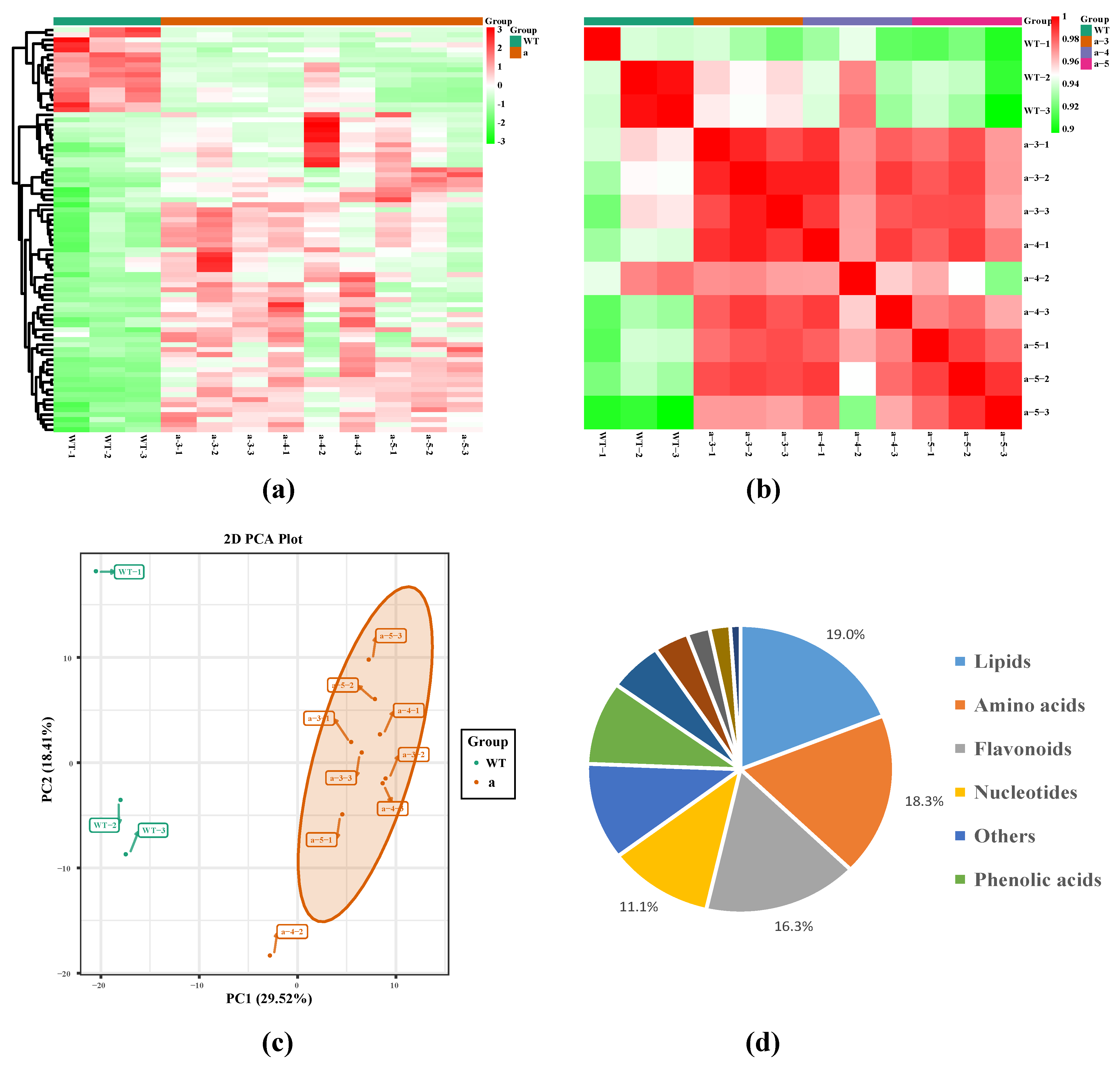
3.1. Qualitative and Quantitative Metabolomic Analysis Based on UPLC–MS/MS and Identification of Metabolites
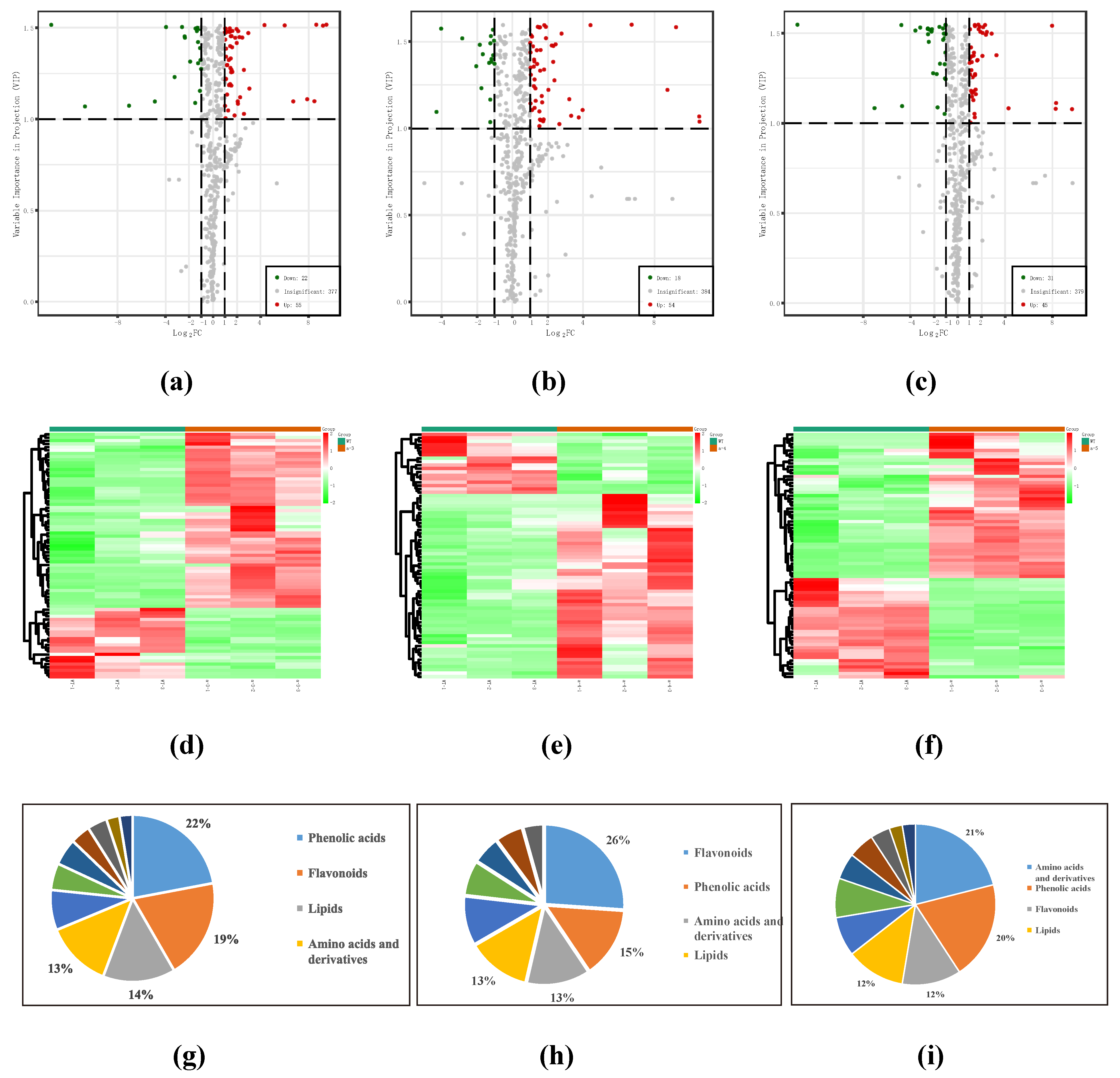
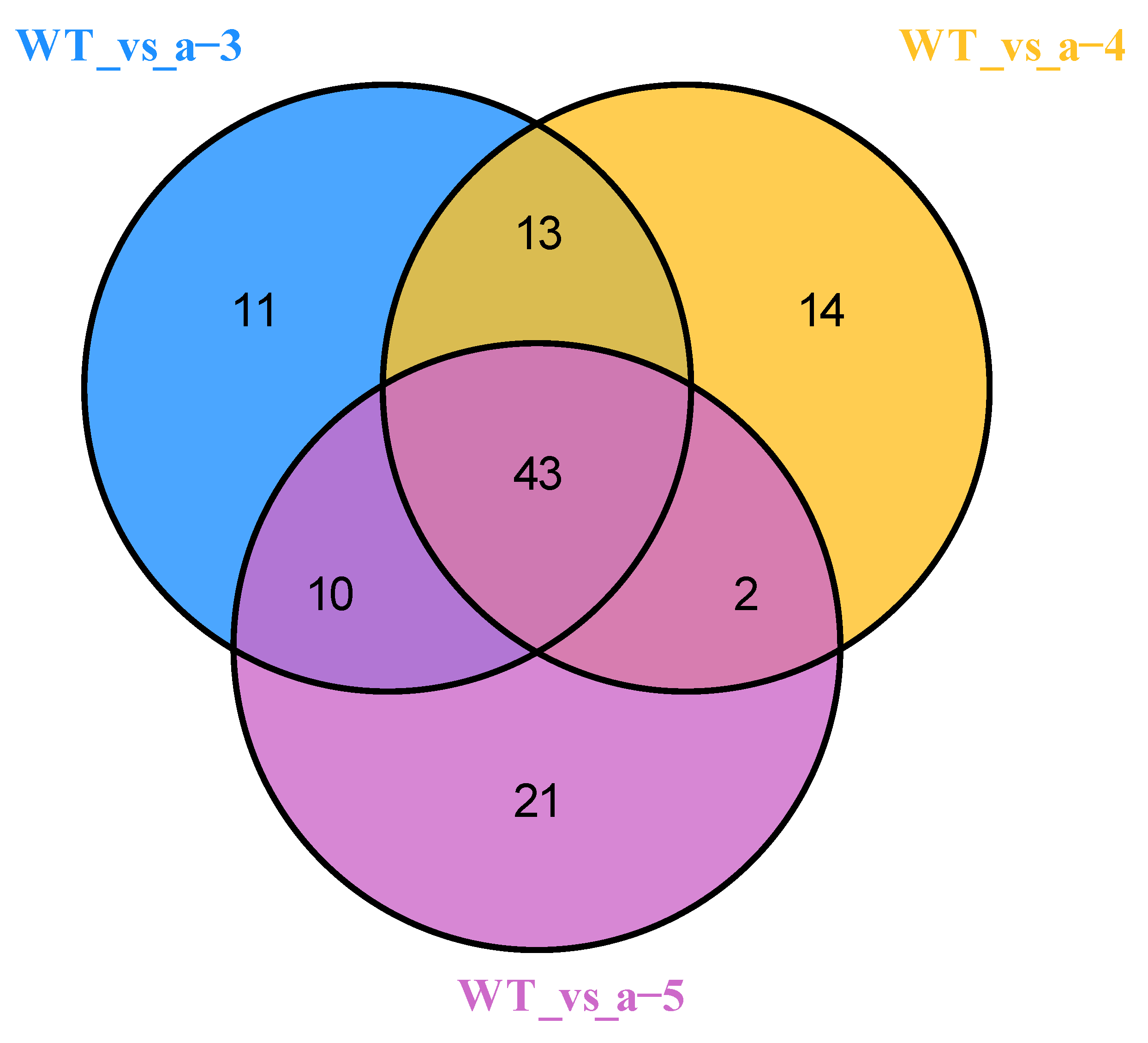
3.2. Identification of DAMs
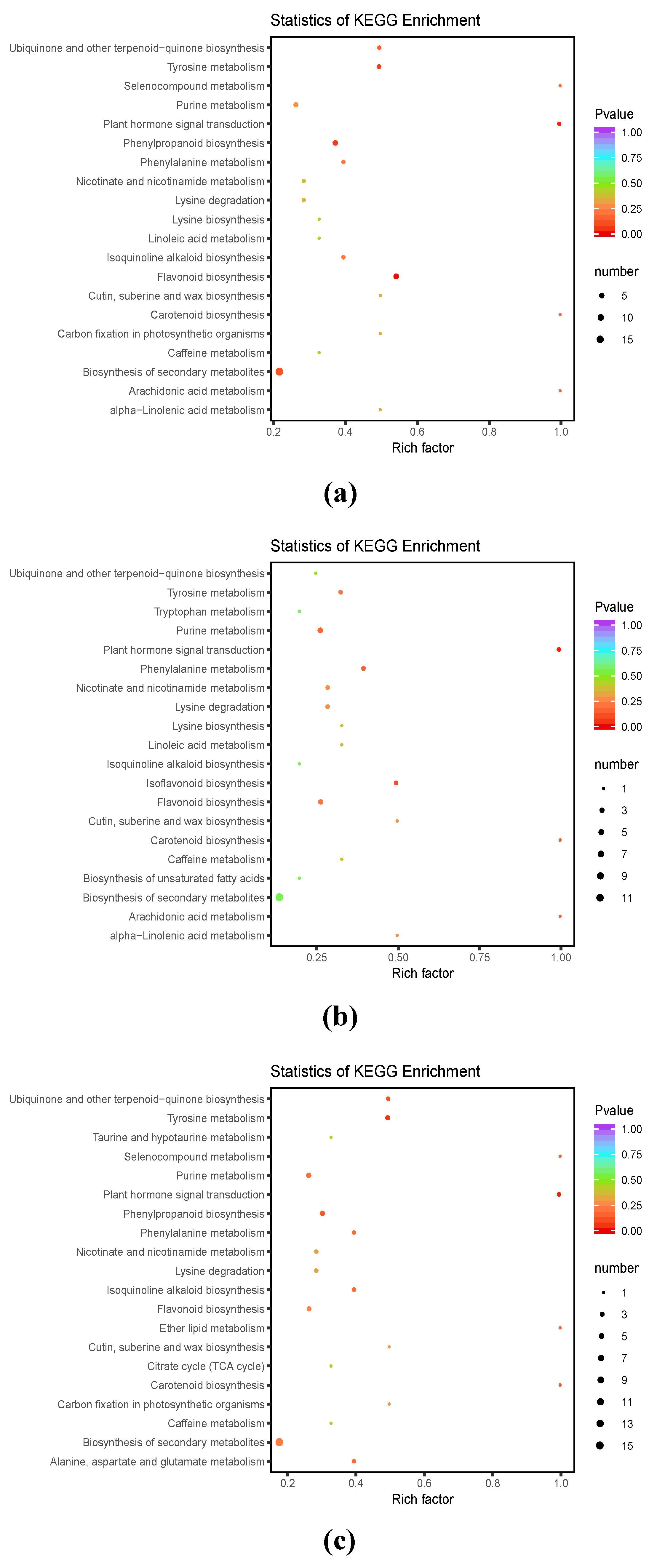
3.3. DAM Functional Annotation and KEGG Enrichment Analysis
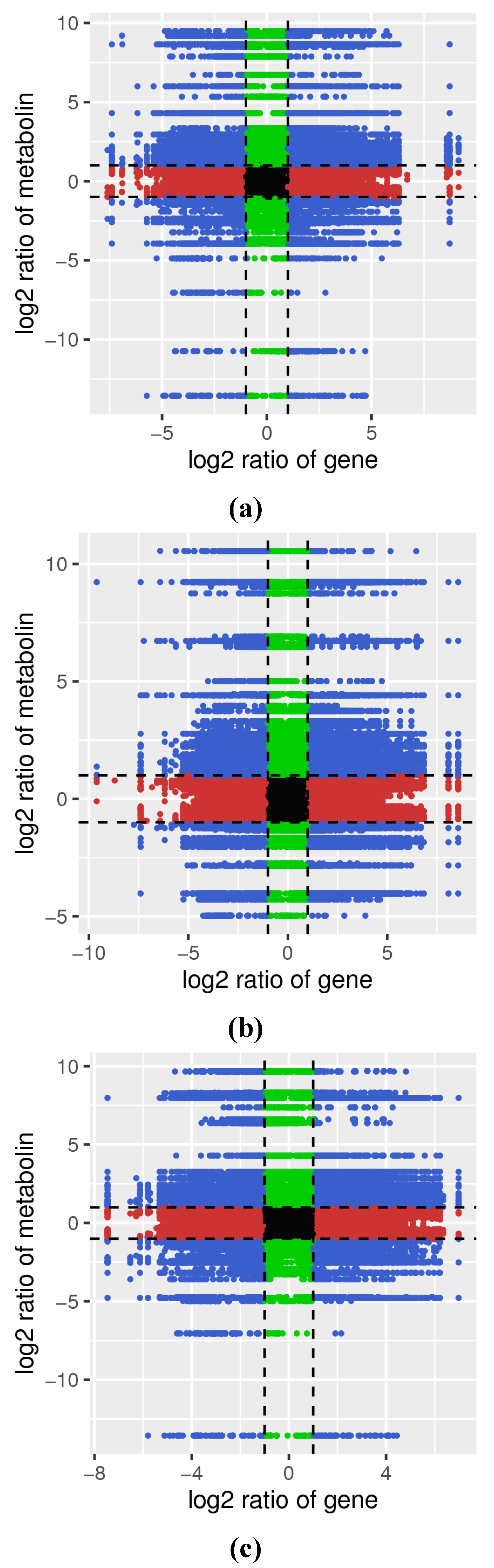
3.4. Association Analysis of Metabolome and Transcriptome Data Sets
3.4.1. KEGG Enrichment Analysis of DAMs and DEGs in L. kaempferi Embryonic Masses
3.4.2. Correlation Analysis of DAMs and DEGs
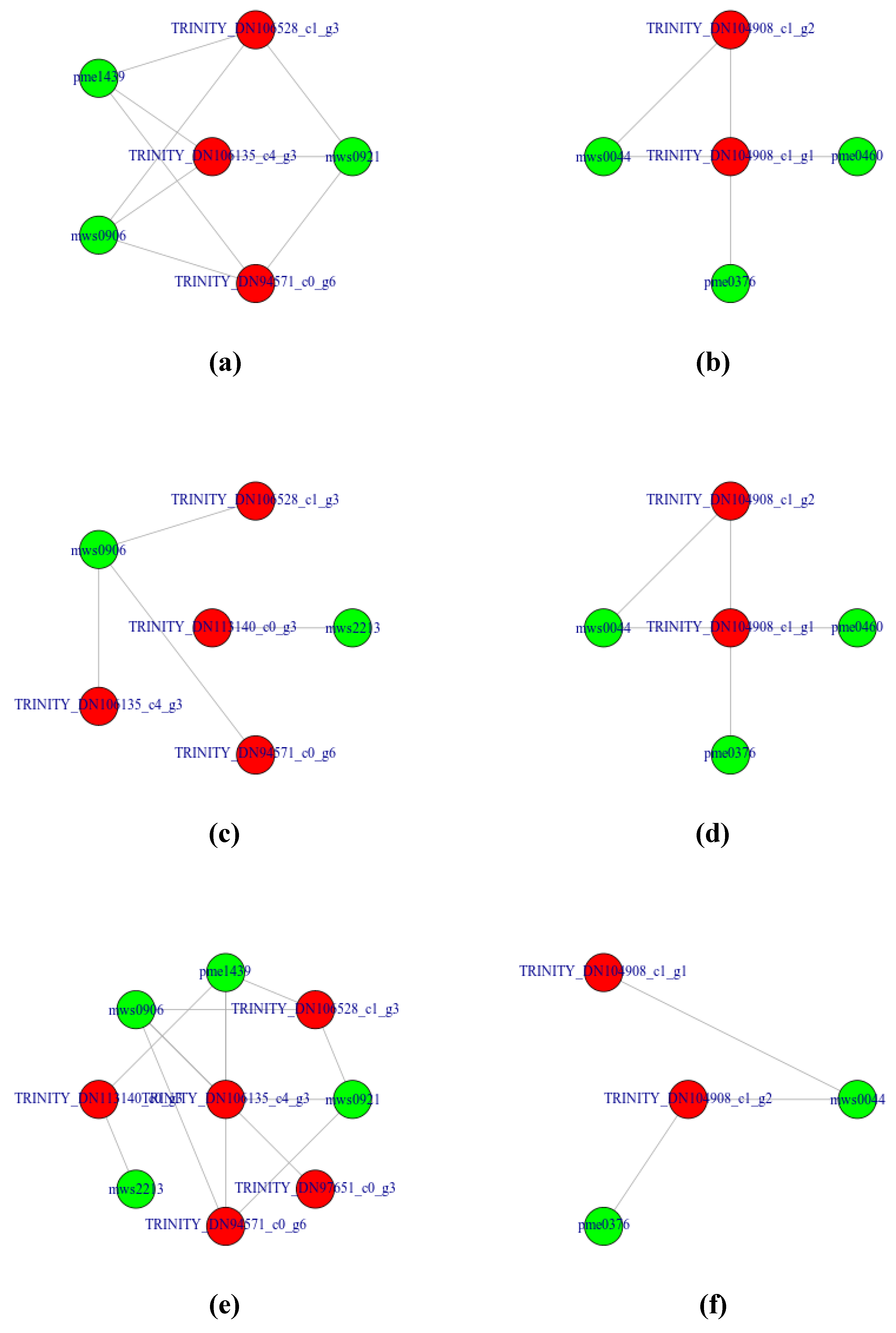
3.4.3. Representing the Transcript-Metabolite Correlation Network of DAMs and DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, P. ECOLOGY: China’s Forest Policy for the 21st Century. Science 2000, 288, 2135–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, S.; Han, S.; Wu, T.; Li, X.; Li, W.; Qi, L. Genome-wide identification of microRNAs in larch and stage-specific modulation of 11 conserved microRNAs and their targets during somatic embryogenesis. Planta 2012, 236, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Origin, Biogenesis, and Activity of Plant MicroRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Xue, L.; An, L. Functional diversity of miRNA in plants. Plant Sci. 2007, 172, 423–432. [Google Scholar] [CrossRef]
- Kidner, C.A.; Martienssen, R.A. Spatially restricted microRNA directs leaf polarity through ARGONAUTE1. Nature 2004, 428, 81–84. [Google Scholar] [CrossRef]
- Barik, S.; SarkarDas, S.; Singh, A.; Gautam, V.; Kumar, P.; Majee, M.; Sarkar, A.K. Phylogenetic analysis reveals conservation and diversification of micro RNA166 genes among diverse plant species. Genomics 2014, 103, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Merelo, P.; Ram, H.; Pia Caggiano, M.; Ohno, C.; Ott, F.; Straub, D.; Graeff, M.; Cho, S.K.; Yang, S.W.; Wenkel, S.; et al. Regulation of MIR165/166 by class II and class III homeodomain leucine zipper proteins establishes leaf polarity. Proc. Natl. Acad. Sci. USA 2016, 113, 11973–11978. [Google Scholar] [CrossRef] [Green Version]
- Rong, F.; Chen, F.; Huang, L.; Zhang, J.; Zhang, C.; Hou, D.; Cheng, Z.; Weng, Y.; Chen, P.; Li, Y. A mutation in class III homeodomain-leucine zipper (HD-ZIP III) transcription factor results in curly leaf (cul) in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2019, 132, 113–123. [Google Scholar] [CrossRef]
- Rhoades, M.W.; Reinhart, B.J.; Lim, L.P.; Burge, C.B.; Bartel, B.; Bartel, D.P. Prediction of Plant MicroRNA Targets. Cell 2002, 110, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Williams, L. Regulation of Arabidopsis shoot apical meristem and lateral organ formation by microRNA miR166g and its AtHD-ZIP target genes. Development 2005, 132, 3657–3668. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Park, C. MIR166/165 genes exhibit dynamic expression patterns in regulating shoot apical meristem and floral development in Arabidopsis. Planta 2007, 225, 1327–1338. [Google Scholar] [CrossRef]
- Boualem, A.; Laporte, P.; Jovanovic, M.; Laffont, C.; Plet, J.; Combier, J.; Niebel, A.; Crespi, M.; Frugier, F. MicroRNA166 controls root and nodule development in Medicago truncatula. Plant J. 2008, 54, 876–887. [Google Scholar] [PubMed]
- Zhang, Q.L.; Su, L.Y.; Zhang, S.T.; Xu, X.P.; Chen, X.H.; Li, X.; Jiang, M.Q.; Huang, S.Q.; Chen, Y.K.; Zhang, Z.H.; et al. Analyses of microRNA166 gene structure, expression, and function during the early stage of somatic embryogenesis in Dimocarpus longan Lour. Plant Physiol. Biochem. 2020, 147, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Hu, F.; Wang, R.; Zhou, X.; Sze, S.; Liou, L.W.; Barefoot, A.; Dickman, M.; Zhang, X. Arabidopsis Argonaute10 Specifically Sequesters miR166/165 to Regulate Shoot Apical Meristem Development. Cell 2011, 145, 242–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Lian, H.; Zhao, Q.; He, Y. MicroRNA166 Monitors SPOROCYTELESS/NOZZLE for Building of the Anther Internal Boundary. Plant Physiol. 2019, 181, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Kulcheski, F.R.; de Oliveira, L.F.; Molina, L.G.; Almerao, M.P.; Rodrigues, F.A.; Marcolino, J.; Barbosa, J.F.; Stolf-Moreira, R.; Nepomuceno, A.L.; Marcelino-Guimaraes, F.C.; et al. Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genom. 2011, 12, 307. [Google Scholar] [CrossRef] [Green Version]
- Juárez-González, V.T.; López-Ruiz, B.A.; Baldrich, P.; Luján-Soto, E.; Meyers, B.C.; Dinkova, T.D. The explant developmental stage profoundly impacts small RNA-mediated regulation at the dedifferentiation step of maize somatic embryogenesis. Sci. Rep. 2019, 9, 14511. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Lian, H.; Zhou, C.; Xu, L.; Jiao, Y.; Wang, J. A Two-Step Model for de Novo Activation of WUSCHEL during Plant Shoot Regeneration. Plant Cell 2017, 29, 1073–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Li, S.; Zhang, L.; Han, S.; Li, W.; Xu, H.; Yang, W.; Liu, Y.; Fan, Y.; Qi, L. Over-expression of miR166a inhibits cotyledon formation in somatic embryos and promotes lateral root development in seedlings of Larix leptolepis. Plant Cell Tissue Organ Cult. (PCTOC) 2016, 127, 461–473. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, L.; Li, W.; Qi, L.; Han, S. MIR166a Affects the Germination of Somatic Embryos in Larixleptolepis by Modulating IAA Biosynthesis and Signaling Genes. J. Plant Growth Regul. 2017, 36, 889–896. [Google Scholar] [CrossRef]
- Li, Z.X.; Fan, Y.R.; Dang, S.F.; Li, W.F.; Qi, L.W.; Han, S.Y. LaMIR166a-mediated auxin biosynthesis and signalling affect somatic embryogenesis in Larix leptolepis. Mol. Genet. Genom. 2018, 293, 1355–1363. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, J.; Zhang, J.; Wang, Z.; Ran, A.; Guo, H.; Wang, D.; Zhang, J. Integrated RNA-seq and sRNA-seq analysis reveals miRNA effects on secondary metabolism in Solanum tuberosum L. Mol. Genet. Genom. 2017, 292, 37–52. [Google Scholar] [CrossRef]
- Zhang, M.; Dong, Y.; Nie, L.; Lu, M.; Fu, C.; Yu, L. High-throughput sequencing reveals miRNA effects on the primary and secondary production properties in long-term subcultured Taxus cells. Front. Plant Sci. 2015, 6, 604. [Google Scholar] [CrossRef] [Green Version]
- Fiehn, O.; Kopka, J.; Dormann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, D.O.; Yang, H.; Chen, M.; Zhang, W.; Zhang, L. Metabolome and Transcriptome Sequencing Analysis Reveals Anthocyanin Metabolism in Pink Flowers of Anthocyanin-Rich Tea (Camellia sinensis). Molecules 2019, 24, 1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, Q.; Liu, Y.; Qi, Y.; Jiao, S.; Tian, F.; Jiang, L.; Wang, Y. Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower colour in grape hyacinth. J. Exp. Bot. 2014, 65, 3157–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, S.; Classen, B.; Gabius, H. Studies on Unprocessed and Acid-Treated Arabinogalactan from Larch as an Inhibitor of Glycan Binding of a Plant Toxin and Biomedically Relevant Human Lectins. Planta Med. 2015, 81, 1146–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, G.S. Larch arabinogalactan: Clinical relevance of a novel immune-enhancing polysaccharide. Altern. Med. Rev. 1999, 4, 96–103. [Google Scholar] [PubMed]
- Fuchtner, S.; Brock-Nannestad, T.; Smeds, A.; Fredriksson, M.; Pilgard, A.; Thygesen, L.G. Hydrophobic and Hydrophilic Extractives in Norway Spruce and Kurile Larch and Their Role in Brown-Rot Degradation. Front. Plant Sci. 2020, 11, 855. [Google Scholar] [CrossRef] [PubMed]
- Dziedzinski, M.; Kobus-Cisowska, J.; Szymanowska, D.; Stuper-Szablewska, K.; Baranowska, M. Identification of Polyphenols from Coniferous Shoots as Natural Antioxidants and Antimicrobial Compounds. Molecules 2020, 25, 3527. [Google Scholar] [CrossRef]
- Riikonen, J.; Kontunen-Soppela, S.; Ossipov, V.; Tervahauta, A.; Tuomainen, M.; Oksanen, E.; Vapaavuori, E.; Heinonen, J.; Kivimäenpää, M. Needle metabolome, freezing tolerance and gas exchange in Norway spruce seedlings exposed to elevated temperature and ozone concentration. Tree Physiol. 2012, 32, 1102–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Hu, C.; Hussain, S.; Tan, Q.; Wu, S.; Sun, X. Metabolomics analysis reveals potential mechanisms of tolerance to excess molybdenum in soybean seedlings. Ecotoxicol. Environ. Saf. 2018, 164, 589–596. [Google Scholar] [CrossRef]
- Zhang, M.; Li, W.; Feng, J.; Gong, Z.; Yao, Y.; Zheng, C. Integrative transcriptomics and proteomics analysis constructs a new molecular model for ovule abortion in the female-sterile line of Pinus tabuliformis Carr. Plant Sci. 2020, 294, 110462. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Jiang, B.; Zhao, B.; Mao, X.; Lu, J.; Jin, B.; Wang, L. Liquid profiling in plants: Identification and analysis of extracellular metabolites and miRNAs in pollination drops of Ginkgo biloba. Tree Physiol. 2020, 40, 1420–1436. [Google Scholar] [CrossRef]
- Liao, Q.; Du, R.; Gou, J.; Guo, L.; Shen, H.; Liu, H.; Nguyen, J.K.; Ming, R.; Yin, T.; Huang, S.; et al. The genomic architecture of the sex-determining region and sex-related metabolic variation in Ginkgobiloba. Plant J. 2020, 104, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.M.; Saldanha, L.L.; Silva, E.M.; Nogueira, F.T.S.; Dokkedal, A.L.; Santos, C.; Rolim, D.A.L.F. Effects of leaf extracts of Myrcia guianensis (Aubl.) DC. on growth and gene expression during root development of Sorghum bicolor (L.) Moench. Allelopath. J. 2015, 35, 237–248. [Google Scholar]
- Maloney, G.S.; DiNapoli, K.T.; Muday, G.K. The anthocyanin reduced Tomato Mutant Demonstrates the Role of Flavonols in Tomato Lateral Root and Root Hair Development. Plant Physiol. 2014, 166, 614–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, R.; Han, K.; Heller, W.; Albert, A.; Dobrev, P.I.; Zažímalová, E.; Schäffner, A.R. Kaempferol 3-O-rhamnoside-7-O-rhamnoside is an endogenous flavonol inhibitor of polar auxin transport in Arabidopsis shoots. New Phytol. 2013, 201, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Franco, D.M.; Silva, E.M.; Saldanha, L.L.; Adachi, S.A.; Schley, T.R.; Rodrigues, T.M.; Dokkedal, A.L.; Nogueira, F.T.; Rolim, D.A.L. Flavonoids modify root growth and modulate expression of SHORT-ROOT and HD-ZIP III. J. Plant Physiol. 2015, 188, 89–95. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Numbers | Up-Regulated | Down-Regulated |
|---|---|---|---|
| WT vs. a-3 | 77 | 55 | 22 |
| WT vs. a-4 | 72 | 54 | 18 |
| WT vs. a-5 | 76 | 45 | 31 |
| Class | Numbers of WT vs. a-3 | Percentage | Numbers of WT vs. a-4 | Percentage | Numbers of WT vs. a-5 | Percentage |
|---|---|---|---|---|---|---|
| Phenolic acids | 17 | 22.1% | 10 | 13.9% | 15 | 19.7% |
| Flavonoids | 15 | 19.5% | 18 | 25.0% | 9 | 11.8% |
| Lipids | 11 | 14.3% | 9 | 12.5% | 9 | 11.8% |
| Amino acids and derivatives | 10 | 13.0% | 9 | 12.5% | 16 | 21.1% |
| Others | 6 | 7.8% | 7 | 9.7% | 6 | 7.9% |
| Nucleotides and derivatives | 4 | 5.2% | 5 | 6.9% | 4 | 5.3% |
| Tannins | 4 | 5.2% | 3 | 4.2% | 3 | 3.9% |
| Alkaloids | 3 | 3.9% | 4 | 5.6% | 2 | 2.6% |
| Organic acids | 3 | 3.9% | 3 | 4.2% | 6 | 7.9% |
| Lignans and Coumarins | 2 | 2.6% | 4 | 5.6% | 2 | 2.6% |
| Terpenoids | 2 | 2.6% | 0 | 0.0% | 4 | 5.3% |
| Index | Compound | Class | Fold Change | Log2(FC) | Type |
|---|---|---|---|---|---|
| mws0057 | Eriodictyol 7-O-glucoside | Dihydroflavone | 2.27 | 1.18 | up |
| mws0064 | Eriodictyol | Dihydroflavone | 4.42 | 2.14 | up |
| pme0376 | Naringenin | Dihydroflavone | 4.91 | 2.30 | up |
| mws0044 | Dihydroquercetin(Taxifolin) | Dihydroflavonol | 2.69 | 1.43 | up |
| mws1361 | Astilbin | Dihydroflavonol | 3.01 | 1.59 | up |
| pmb0736 | Tricin 7-O-Glucoside | Flavonoid | 2.25 | 1.17 | up |
| HJAP006 | Syringetin-O-glucoside | Flavonoid | 2.61 | 1.39 | up |
| mws0058 | Diosmetin | Flavonoid | 2.66 | 1.41 | up |
| HJAP110 | Myricetin-O-rhamnoside | Flavonoid | 3.90 | 1.97 | up |
| HJN041 | Epicatechin glucoside | Flavonoid | 4.73 | 2.24 | up |
| mws1662 | Tectochrysin | Flavonoid | 9.08 | 3.18 | up |
| Xmyp005987 | Isorhamnetin-3-O-β-d-glucopyranoside | Flavonoid | 1.69 × 101 | 4.08 | up |
| Xmyp005746 | Isorhamnetin-3-O-rutinoside | Flavonoid | 4.18 × 102 | 8.71 | up |
| mws0913 | Kaempferol-3-O-galactoside (Trifolin) * | Flavonols | 2.29 | 1.20 | up |
| mws4183 | Quercetin-3-O-α-L-arabinopyranoside(guaijaverin) * | Flavonols | 2.77 | 1.47 | up |
| mws0061 | Quercetin-3-O-β-d-Galactoside (Hyperin) * | Flavonols | 3.38 | 1.76 | up |
| pmp001309 | 6-Hydroxykaempferol-7-O-glucoside | Flavonols | 3.50 | 1.81 | up |
| Lmdp003286 | isohyperoside | Flavonols | 3.53 | 1.82 | up |
| mws0091 | Quercetin-3-O-β-d-glucoside(Isoquercitrin) * | Flavonols | 3.76 | 1.91 | up |
| mws0856 | Spiraeoside | Flavonols | 3.79 | 1.92 | up |
| mws1329 | Quercetin-7-O-Glucoside * | Flavonols | 4.02 | 2.01 | up |
| pmp001311 | 6-Hydroxykaempferol-7,6-O-Diglucoside | Flavonols | 4.62 | 2.21 | up |
| Hmcp002316 | Isorhamnetin-3-O-arabinoside | Flavonols | 4.90 × 102 | 8.94 | up |
| mws0054 | Catechin * | Flavanols | 5.40 | 2.43 | up |
| pme0460 | Epicatechin * | Flavanols | 5.83 | 2.54 | up |
| Index | Compounds | Class | Type |
|---|---|---|---|
| mws0146 | Nicotinic Acid Methyl Ester(Methyl Nicotinate) | Alkaloids | up |
| pme2268 | Trigonelline | Alkaloids | up |
| Lmhp003064 | Cyclo(Pro-Phe) | Amino acids and derivatives | down |
| mws0193 | L-Homocitrulline | Amino acids and derivatives | up |
| mws0582 | S-(methyl)glutathione | Amino acids and derivatives | down |
| mws1401 | L-theanine | Amino acids and derivatives | down |
| pme0122 | N6-Acetyl-L-lysine | Amino acids and derivatives | up |
| pme1002 | L-Tyramine | Amino acids and derivatives | up |
| pme1228 | 5-Hydroxy-L-tryptophan | Amino acids and derivatives | up |
| pme1712 | L-Saccharopine | Amino acids and derivatives | up |
| Hmcp002316 | Isorhamnetin-3-O-arabinoside | Flavonoids | up |
| mws0044 | Dihydroquercetin(Taxifolin) | Flavonoids | up |
| ws4183 | Quercetin-3-O-α-L-arabinopyranoside(guaijaverin) | Flavonoids | up |
| pme0376 | Naringenin | Flavonoids | up |
| pmp001311 | 6-Hydroxykaempferol-7,6-O-Diglucoside | Flavonoids | up |
| Xmyp005746 | Isorhamnetin-3-O-rutinoside | Flavonoids | up |
| Xmyp005987 | Isorhamnetin-3-O-β-d-glucopyranoside | Flavonoids | up |
| Hmgn004139 | isohydroxymatairesinol | Lignans and Coumarins | up |
| mws5039 | Esculin Hydrate | Lignans and Coumarins | down |
| pma3606 | 9-Hydroxy-10,12,15-octadecatrienoic acid | Lipids | up |
| pmb2786 | 9-HOTrE | Lipids | up |
| pmn001686 | 10,16-Dihydroxy-palmitic acid | Lipids | up |
| mws0609 | Guanosine 3′,5′-cyclic monophosphate | Nucleotides and derivatives | up |
| pme0256 | Xanthine | Nucleotides and derivatives | up |
| pme3184 | 2′-Deoxyadenosine-5′-monophosphate | Nucleotides and derivatives | down |
| Lmtn004049 | Abscisic acid | Organic acids | up |
| mws0147 | 3-Hydroxy-3-methyl butyric acid | Organic acids | up |
| mws2608 | N-Acetyl-d-galactosamine | Others | down |
| mws4174 | N-Acetyl-β-d-mannosamine | Others | down |
| pme0490 | Nicotinic acid | Others | up |
| pme1261 | Pantothenol | Others | down |
| pme2529 | 1,5-Anhydro-d-glucitol | Others | up |
| HmLn002358 | Vanillic acid-glucoside | Phenolic acids | down |
| Lmgn001670 | Salicylic acid | Phenolic acids | up |
| Lmtn003598 | 3-Prenyl-4-O-β-d-glucopyranosyloxy-4-hydroxyl-benzoic acid | Phenolic acids | down |
| mws0906 | Coniferin | Phenolic acids | up |
| mws2213 | Cinnamic acid | Phenolic acids | up |
| mws2368 | Tyrosol | Phenolic acids | up |
| pmb3107 | Glucosyringic Acid | Phenolic acids | up |
| pmn001518 | 1-O-Galloyl-β-d-glucose | Phenolic acids | down |
| Zmxn001997 | Isosalicylic acid O-glycoside | Phenolic acids | up |
| mws0836 | Procyanidin B1 | Tannins | up |
| pme0434 | Procyanidin B2 | Tannins | up |
| Index | KEGG Pathway | Compound | Class | Fold Change | Log2(FC) | Type |
|---|---|---|---|---|---|---|
| pme0376 | Flavonoid biosynthesis | Naringenin | Flavonoids | 4.62 | 2.21 | up |
| mws0044 | Dihydroquercetin(Taxifolin) | Flavonoids | 2.40 | 1.26 | up | |
| pmb3074 | 3-O-p-Coumaroyl quinic acid | Phenolic acids | 6.01 | 2.59 | up | |
| pme0460 | Epicatechin * | Flavonoids | 3.48 | 1.80 | up | |
| mws0042 | Epigallocatechin * | Flavonoids | 8.23 | 3.04 | up | |
| Lmgn001670 | Plant hormone signal transduction | Salicylic acid * | Phenolic acids | 3.03 | 1.60 | up |
| Lmtn004049 | Abscisic acid | Organic acids | 3.95 | 1.98 | up | |
| mws0921 | Phenylpropanoid biosynthesis | p-Coumaryl alcohol | Phenolic acids | 2.06 | 1.04 | up |
| mws0906 | Coniferin | Phenolic acids | 2.74 | 1.45 | up | |
| pmb3074 | 3-O-p-Coumaroyl quinic acid | Phenolic acids | 6.01 | 2.59 | up | |
| mws2213 | Cinnamic acid | Phenolic acids | 2.72 | 1.44 | up | |
| pme1439 | p-Coumaric acid | Phenolic acids | 2.02 | 1.02 | up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Li, Z.; Zhang, L.; Han, S.; Qi, L. Metabolome and Transcriptome Association Analysis Reveals Regulation of Flavonoid Biosynthesis by Overexpression of LaMIR166a in Larix kaempferi (Lamb.) Carr. Forests 2020, 11, 1367. https://doi.org/10.3390/f11121367
Fan Y, Li Z, Zhang L, Han S, Qi L. Metabolome and Transcriptome Association Analysis Reveals Regulation of Flavonoid Biosynthesis by Overexpression of LaMIR166a in Larix kaempferi (Lamb.) Carr. Forests. 2020; 11(12):1367. https://doi.org/10.3390/f11121367
Chicago/Turabian StyleFan, Yanru, Zhexin Li, Lifeng Zhang, Suying Han, and Liwang Qi. 2020. "Metabolome and Transcriptome Association Analysis Reveals Regulation of Flavonoid Biosynthesis by Overexpression of LaMIR166a in Larix kaempferi (Lamb.) Carr" Forests 11, no. 12: 1367. https://doi.org/10.3390/f11121367