
Recent Progress on Hydrogen Storage and Production Using Chemical Hydrogen Carriers

 , , , and
, , , and
Abstract
:1. Introduction
2. Methanol
2.1. General Properties and Applications
2.2. MeOH Synthesis
2.3. Hydrogen Generation from Methanol
Process Parameters and Catalysts for MSR
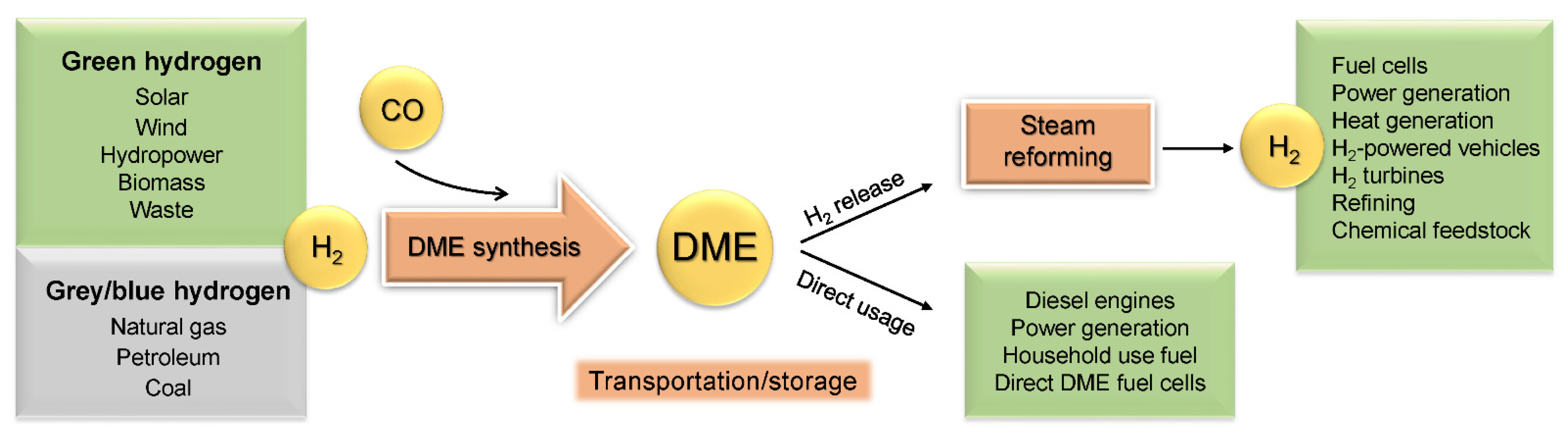
3. Dimethyl Ether
3.1. General Properties and Applications
3.2. DME Synthesis
3.3. Steam Reforming of DME
Process Parameters and Catalysts for DME SR
4. Dibenzyltoluenes
4.1. Properties of DBT
4.2. Reversible Hydrogen Storage
4.2.1. Hydrogenation Process
4.2.2. Dehydrogenation Process
5. Comparison of MeOH, DME, and DBT Properties and H2 Storage Technologies
6. Final Remarks
6.1. Energy Storage
6.2. Energy Transport
6.3. Mobility
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- European Commission. A Hydrogen Strategy for a Climate-Neutral Europe; Com(2020) 301; European Commission: Brussels, Belgium, 2020. [Google Scholar]
- Abdin, Z.; Zafaranloo, A.; Rafiee, A.; Mérida, W.; Lipiński, W.; Khalilpour, K.R. Hydrogen as an energy vector. Renew. Sustain. Energy Rev. 2020, 120, 109620. [Google Scholar] [CrossRef]
- Wolf, E. Large-Scale Hydrogen Energy Storage. In Electrochemical Energy Storage for Renewable Sources and Grid Balancing; Elsevier: Amsterdam, The Netherlands, 2015; pp. 129–142. [Google Scholar] [CrossRef]
- Jorschick, H.; Vogl, M.; Preuster, P.; Bösmann, A.; Wasserscheid, P. Hydrogenation of liquid organic hydrogen carrier systems using multicomponent gas mixtures. Int. J. Hydrogen Energy 2019, 44, 31172–31182. [Google Scholar] [CrossRef]
- Eberle, U.; Felderhoff, M.; Schüth, F. Chemical and physical solutions for hydrogen storage. Angew. Chem. Int. Ed. 2009, 48, 6608–6630. [Google Scholar] [CrossRef] [PubMed]
- Mazloomi, K.; Gomes, C. Hydrogen as an energy carrier: Prospects and challenges. Renew. Sustain. Energy Rev. 2012, 16, 3024–3033. [Google Scholar] [CrossRef]
- Kanezaki, T.; Narazaki, C.; Mine, Y.; Matsuoka, S.; Murakami, Y. Effects of hydrogen on fatigue crack growth behavior of austenitic stainless steels. Int. J. Hydrogen Energy 2008, 33, 2604–2619. [Google Scholar] [CrossRef]
- Wijayanta, A.T.; Oda, T.; Purnomo, C.W.; Kashiwagi, T.; Aziz, M. Liquid hydrogen, methylcyclohexane, and ammonia as potential hydrogen storage: Comparison review. Int. J. Hydrogen Energy 2019, 44, 15026–15044. [Google Scholar] [CrossRef]
- Aftab, A.; Hassanpouryouzband, A.; Xie, Q.; Machuca, L.L.; Sarmadivaleh, M. Toward a Fundamental Understanding of Geological Hydrogen Storage. Ind. Eng. Chem. Res. 2022, 61, 3233–3253. [Google Scholar] [CrossRef]
- Gabrielli, P.; Poluzzi, A.; Kramer, G.J.; Spiers, C.; Mazzotti, M.; Gazzani, M. Seasonal energy storage for zero-emissions multi-energy systems via underground hydrogen storage. Renew. Sustain. Energy Rev. 2020, 121, 109629. [Google Scholar] [CrossRef]
- Tarkowski, R. Underground hydrogen storage: Characteristics and prospects. Renew. Sustain. Energy Rev. 2019, 105, 86–94. [Google Scholar] [CrossRef]
- Portarapillo, M.; Di Benedetto, A. Risk assessment of the large-scale hydrogen storage in salt caverns. Energies 2021, 14, 2856. [Google Scholar] [CrossRef]
- Crotogino, F.; Schneider, G.S.; Evans, D.J. Renewable energy storage in geological formations. Proc. Inst. Mech. Eng. Part A J. Power Energy 2018, 232, 100–114. [Google Scholar] [CrossRef]
- Andersson, J.; Grönkvist, S. Large-scale storage of hydrogen. Int. J. Hydrogen Energy 2019, 44, 11901–11919. [Google Scholar] [CrossRef]
- Langmi, H.W.; Ren, J.; North, B.; Mathe, M.; Bessarabov, D. Hydrogen storage in metal-organic frameworks: A review. Electrochim. Acta 2014, 128, 368–392. [Google Scholar] [CrossRef]
- Yürüm, Y.; Taralp, A.; Veziroglu, T.N. Storage of hydrogen in nanostructured carbon materials. Int. J. Hydrogen Energy 2009, 34, 3784–3798. [Google Scholar] [CrossRef] [Green Version]
- Langmi, H.W.; Walton, A.; Al-Mamouri, M.M.; Johnson, S.R.; Book, D.; Speight, J.D.; Edwards, P.P.; Gameson, I.; Anderson, P.A.; Harris, I.R. Hydrogen adsorption in zeolites A, X, Y and RHO. J. Alloys Compd. 2003, 356–357, 710–715. [Google Scholar] [CrossRef]
- Moradi, R.; Groth, K.M. Hydrogen storage and delivery: Review of the state of the art technologies and risk and reliability analysis. Int. J. Hydrogen Energy 2019, 44, 12254–12269. [Google Scholar] [CrossRef]
- Niermann, M.; Beckendorff, A.; Kaltschmitt, M.; Bonhoff, K. Liquid Organic Hydrogen Carrier (LOHC)—Assessment based on chemical and economic properties. Int. J. Hydrogen Energy 2019, 44, 6631–6654. [Google Scholar] [CrossRef]
- Jorschick, H.; Preuster, P.; Bösmann, A.; Wasserscheid, P. Hydrogenation of aromatic and heteroaromatic compounds-a key process for future logistics of green hydrogen using liquid organic hydrogen carrier systems. Sustain. Energy Fuels 2021, 5, 1311–1346. [Google Scholar] [CrossRef]
- Makepeace, J.W.; He, T.; Weidenthaler, C.; Jensen, T.R.; Chang, F.; Vegge, T.; Ngene, P.; Kojima, Y.; de Jongh, P.E.; Chen, P.; et al. Reversible ammonia-based and liquid organic hydrogen carriers for high-density hydrogen storage: Recent progress. Int. J. Hydrogen Energy 2019, 44, 7746–7767. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kumar Parsapur, R.; Huang, K.-W. Limitations of Ammonia as a Hydrogen Energy Carrier for the Transportation Sector. ACS Energy Lett. 2021, 6, 4390–4394. [Google Scholar] [CrossRef]
- Saxena, S.; Kumar, S.; Drozd, V. A modified steam-methane-reformation reaction for hydrogen production. Int. J. Hydrogen Energy 2011, 36, 4366–4369. [Google Scholar] [CrossRef]
- Catizzone, E.; Freda, C.; Braccio, G.; Frusteri, F.; Bonura, G. Dimethyl ether as circular hydrogen carrier: Catalytic aspects of hydrogenation/dehydrogenation steps. J. Energy Chem. 2021, 58, 55–77. [Google Scholar] [CrossRef]
- Frei, M.S.; Mondelli, C.; Short, M.I.M.; Pérez-Ramírez, J. Methanol as a Hydrogen Carrier: Kinetic and Thermodynamic Drivers for its CO2-Based Synthesis and Reforming over Heterogeneous Catalysts. ChemSusChem 2020, 13, 6330–6337. [Google Scholar] [CrossRef] [PubMed]
- Eppinger, J.; Huang, K.-W. Formic Acid as a Hydrogen Energy Carrier. ACS Energy Lett. 2017, 2, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Aakko-Saksa, P.T.; Cook, C.; Kiviaho, J.; Repo, T. Liquid organic hydrogen carriers for transportation and storing of renewable energy—Review and discussion. J. Power Sources 2018, 396, 803–823. [Google Scholar] [CrossRef]
- Modisha, P.M.; Ouma, C.N.M.; Garidzirai, R.; Wasserscheid, P.; Bessarabov, D. The Prospect of Hydrogen Storage Using Liquid Organic Hydrogen Carriers. Energy Fuels 2019, 33, 2778–2796. [Google Scholar] [CrossRef]
- Shuwa, S.M.; Jibril, B.Y.; Al-Hajri, R.S. Hydrogenation of toluene on Ni-Co-Mo supported zeolite catalysts. Niger. J. Technol. 2018, 36, 1114. [Google Scholar] [CrossRef] [Green Version]
- Akamatsu, K.; Ohta, Y.; Sugawara, T.; Kanno, N.; Tonokura, K.; Hattori, T.; Nakao, S.I. Stable high-purity hydrogen production by dehydrogenation of cyclohexane using a membrane reactor with neither carrier gas nor sweep gas. J. Memb. Sci. 2009, 330, 1–4. [Google Scholar] [CrossRef]
- Hodoshima, S.; Takaiwa, S.; Shono, A.; Satoh, K.; Saito, Y. Hydrogen storage by decalin/naphthalene pair and hydrogen supply to fuel cells by use of superheated liquid-film-type catalysis. Appl. Catal. A Gen. 2005, 283, 235–242. [Google Scholar] [CrossRef]
- Ali, A.; Kumar, G.U.; Lee, H.J. Investigation of hydrogenation of Dibenzyltoluene as liquid organic hydrogen carrier. Mater. Today Proc. 2021, 45, 1123–1127. [Google Scholar] [CrossRef]
- Markiewicz, M.; Zhang, Y.Q.; Empl, M.T.; Lykaki, M.; Thöming, J.; Steinberg, P.; Stolte, S. Hazard assessment of quinaldine-, alkylcarbazole-, benzene- and toluene-based liquid organic hydrogen carrier (LOHCs) systems. Energy Environ. Sci. 2019, 12, 366–383. [Google Scholar] [CrossRef]
- Peters, W.; Eypasch, M.; Frank, T.; Schwerdtfeger, J.; Körner, C.; Bösmann, A.; Wasserscheid, P. Efficient hydrogen release from perhydro-N-ethylcarbazole using catalyst-coated metallic structures produced by selective electron beam melting. Energy Environ. Sci. 2015, 8, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Permissible Exposure Limits, United State Department of Labor, Occupational Safety and Health Administration. Available online: https://www.osha.gov/annotated-pels/table-z-1 (accessed on 15 June 2022).
- Bertau, M.; Offermanns, H.; Plass, L.; Schmidt, F.; Wernicke, H.-J. Methanol: The Basic Chemical and Energy Feedstock of the Future; Springer: Heidelberg, Germany; New York, NY, USA; Dordrecht, The Netherlands; London, UK, 2014; Volume 41, ISBN 9783642397080. [Google Scholar]
- Dieterich, V.; Buttler, A.; Hanel, A.; Spliethoff, H.; Fendt, S. Power-to-liquid via synthesis of methanol, DME or Fischer–Tropsch-fuels: A review. Energy Environ. Sci. 2020, 13, 3207–3252. [Google Scholar] [CrossRef]
- Garcia, G.; Arriola, E.; Chen, W.H.; De Luna, M.D. A comprehensive review of hydrogen production from methanol thermochemical conversion for sustainability. Energy 2021, 217, 119384. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency Methanol. Available online: https://iris.epa.gov/ChemicalLanding/&substance_nmbr=305 (accessed on 15 June 2022).
- Tian, Z.; Wang, Y.; Zhen, X.; Liu, Z. The effect of methanol production and application in internal combustion engines on emissions in the context of carbon neutrality: A review. Fuel 2022, 320, 123902. [Google Scholar] [CrossRef]
- Verhelst, S.; Turner, J.W.; Sileghem, L.; Vancoillie, J. Methanol as a fuel for internal combustion engines. Prog. Energy Combust. Sci. 2019, 70, 43–88. [Google Scholar] [CrossRef] [Green Version]
- De Sá, M.H.; Pinto, A.M.F.R.; Oliveira, V.B. Passive direct methanol fuel cells as a sustainable alternative to batteries in hearing aid devices—An overview. Int. J. Hydrogen Energy 2022, 7, 16552–16567. [Google Scholar] [CrossRef]
- Velisala, V.; Srinivasulu, G.N.; Srinivasa, B.; Rao, K.V.K. Review on challenges of direct liquid fuel cells for portable application. World J. Eng. 2015, 12, 591–606. [Google Scholar] [CrossRef]
- Scott, K.; Xing, L. Direct Methanol Fuel Cells. In Advances in Chemical Engineering; Sundmacher, K., Ed.; Academic Press: New York, NY, USA, 2012; Volume 41, pp. 145–196. ISBN 9780123868749. [Google Scholar]
- Samimi, F.; Rahimpour, M.R. Direct Methanol Fuel Cell. In Methanol: Science and Engineering; Basile, A., Dalena, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 381–397. ISBN 9780444640109. [Google Scholar]
- Nakagawa, N.; Abdelkareem, M.A.; Sekimoto, K. Control of methanol transport and separation in a DMFC with a porous support. J. Power Sources 2006, 160, 105–115. [Google Scholar] [CrossRef]
- Alias, M.S.; Kamarudin, S.K.; Zainoodin, A.M.; Masdar, M.S. Active direct methanol fuel cell: An overview. Int. J. Hydrogen Energy 2020, 45, 19620–19641. [Google Scholar] [CrossRef]
- Baydir, E.; Aras, Ö. Methanol steam reforming in a microchannel reactor coated with spray pyrolysis method for durable Cu/ZnO nanocatalyst. J. Anal. Appl. Pyrolysis 2021, 158, 105278. [Google Scholar] [CrossRef]
- Kang, J.; Song, Y.; Kim, T.; Kim, S. Recent trends in the development of reactor systems for hydrogen production via methanol steam reforming. Int. J. Hydrogen Energy 2022, 47, 3587–3610. [Google Scholar] [CrossRef]
- Niu, J.; Liu, H.; Jin, Y.; Fan, B.; Qi, W.; Ran, J. Comprehensive review of Cu-based CO2 hydrogenation to CH3OH: Insights from experimental work and theoretical analysis. Int. J. Hydrogen Energy 2022, 47, 9183–9200. [Google Scholar] [CrossRef]
- Da Silva, M.J. Synthesis of methanol from methane: Challenges and advances on the multi-step (syngas) and one-step routes (DMTM). Fuel Process. Technol. 2016, 145, 42–61. [Google Scholar] [CrossRef]
- Ali, K.A.; Zuhairi, A.; Mohamed, A.R. Recent development in catalytic technologies for methanol synthesis from renewable sources: A critical review. Renew. Sustain. Energy Rev. 2015, 44, 508–518. [Google Scholar] [CrossRef]
- Koç, M.; Sekmen, Y.; Topgül, T.; Yücesu, H.S. The effects of ethanol-unleaded gasoline blends on engine performance and exhaust emissions in a spark-ignition engine. Renew. Energy 2009, 34, 2101–2106. [Google Scholar] [CrossRef]
- Chaichan, M.T.; Ekab, N.S.; Fayad, M.A.; Dhahad, H.A. PM and NOX emissions amelioration from the combustion of diesel/ethanol-methanol blends applying exhaust gas recirculation (EGR). In IOP Conference Series: Earth and Environmental Science; IOP Publishing: Bristol, UK, 2022; Volume 961. [Google Scholar] [CrossRef]
- Kovács, M.; Papp, M.; Zsély, I.G.; Turányi, T. Main sources of uncertainty in recent methanol/NOx combustion models. Int. J. Chem. Kinet. 2021, 53, 884–900. [Google Scholar] [CrossRef]
- Garg, N.; Sarkar, A.; Sundararaju, B. Recent developments on methanol as liquid organic hydrogen carrier in transfer hydrogenation reactions. Coord. Chem. Rev. 2021, 433, 213728. [Google Scholar] [CrossRef]
- Araiza, D.G.; Gómez-Cortés, A.; Díaz, G. Methanol decomposition over bimetallic Cu-M catalysts supported on nanoceria: Effect of the second metal on the catalytic properties. Catal. Today 2020, 356, 440–455. [Google Scholar] [CrossRef]
- Chen, W.H.; Shen, C.T.; Lin, B.J.; Liu, S.C. Hydrogen production from methanol partial oxidation over Pt/Al2O3 catalyst with low Pt content. Energy 2015, 88, 399–407. [Google Scholar] [CrossRef]
- Hohn, K.L.; Lin, Y.C. Catalytic partial oxidation of methanol and ethanol for hydrogen generation. ChemSusChem 2009, 2, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Zhao, C.; Ban, S.; Su, H.; Chen, M.; Wang, H. A hybrid fuel cell system integrated with methanol steam reformer and methanation reactor. Int. J. Hydrogen Energy 2021, 46, 2565–2576. [Google Scholar] [CrossRef]
- Yu, H.; Li, Y.; Xu, C.; Jin, F.; Ye, F.; Li, X. Distinct facets to enhance the process of hydrogen production via methanol steam reforming—A review. Energy Storage Sav. 2022, 1, 53–69. [Google Scholar] [CrossRef]
- Ma, Y.; Guan, G.; Phanthong, P.; Li, X.; Cao, J.; Hao, X.; Wang, Z.; Abudula, A. Steam reforming of methanol for hydrogen production over nanostructured wire-like molybdenum carbide catalyst. Int. J. Hydrogen Energy 2014, 39, 18803–18811. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhang, G.; Liu, H.; Shu, Q.; Zhang, Q. Improved charge transfer and morphology on Ti-modified Cu/γ-Al2O3/Al catalyst enhance the activity for methanol steam reforming. Int. J. Hydrogen Energy 2022, 47, 18294–18304. [Google Scholar] [CrossRef]
- Golmakani, A.; Fatemi, S.; Tamnanloo, J. Investigating PSA, VSA, and TSA methods in SMR unit of refineries for hydrogen production with fuel cell specification. Sep. Purif. Technol. 2017, 176, 73–91. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T.; Kallitsis, J.K.; Neophytides, S. Development of an internal reforming alcohol fuel cell: Concept, challenges and opportunities. Chem. Eng. J. 2011, 176–177, 95–101. [Google Scholar] [CrossRef]
- Tahay, P.; Khani, Y.; Jabari, M.; Bahadoran, F.; Safari, N. Highly porous monolith/TiO2 supported Cu, Cu-Ni, Ru, and Pt catalysts in methanol steam reforming process for H2 generation. Appl. Catal. A Gen. 2018, 554, 44–53. [Google Scholar] [CrossRef]
- Polierer, S.; Guse, D.; Wild, S.; Delgado, K.H.; Otto, T.N.; Zevaco, T.A.; Kind, M.; Sauer, J.; Studt, F.; Pitter, S. Enhanced direct dimethyl ether synthesis from CO2-rich syngas with Cu/ZnO/ZrO2 catalysts prepared by continuous co-precipitation. Catalysts 2020, 10, 816. [Google Scholar] [CrossRef]
- Taghizadeh, M.; Abbandanak, M.H. Production of hydrogen via methanol steam reforming over mesoporous CeO2–Cu/KIT-6 nanocatalyst: Effects of polar aprotic tetrahydrofuran solvent and ZrO2 promoter on catalytic performance. Int. J. Hydrogen Energy 2022, 47, 16362–16374. [Google Scholar] [CrossRef]
- Chen, M.; Sun, G.; Wang, Y.; Liang, D.; Li, C.; Wang, J.; Liu, Q. Steam reforming of methanol for hydrogen production over attapulgite-based zeolite-supported Cu-Zr catalyst. Fuel 2022, 314, 122733. [Google Scholar] [CrossRef]
- Shao, Z.; Shen, Q.; Ding, H.; Jiang, Y.; Li, S.; Yang, G. Synthesis, characterization, and methanol steam reforming performance for hydrogen production on perovskite-type oxides SrCo1-xCuxO3-δ. Ceram. Int. 2022, 48, 11836–11848. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhou, W.; Lan, G.; Sun, X.; Wang, X.; Jiang, C.; Li, Y. High-performance Cu/ZnO/Al2O3 catalysts for methanol steam reforming with enhanced Cu-ZnO synergy effect via magnesium assisted strategy. J. Energy Chem. 2021, 63, 550–557. [Google Scholar] [CrossRef]
- Khani, Y.; Safari, N.; Kamyar, N.; Bahadoran, F.; Torabi, M. High H2 selectivity with low coke formation for methanol steam reforming over Cu/Y1.5Ce0.84Ru0.04O4 catalyst in a microchannel plate reactor. Int. J. Hydrogen Energy 2022, 47, 971–983. [Google Scholar] [CrossRef]
- Ribeirinha, P.; Mateos-Pedrero, C.; Boaventura, M.; Sousa, J.; Mendes, A. CuO/ZnO/Ga2O3 catalyst for low temperature MSR reaction: Synthesis, characterization and kinetic model. Appl. Catal. B Environ. 2018, 221, 371–379. [Google Scholar] [CrossRef]
- Liao, M.; Qin, H.; Guo, W.; Gao, P.; Xiao, H. Porous reticular CuO/ZnO/CeO2/ZrO2 catalyst derived from polyacrylic acid hydrogel system on Al2O3 foam ceramic support for methanol steam reforming microreactor. Ceram. Int. 2021, 47, 33667–33677. [Google Scholar] [CrossRef]
- Hosseini Abbandanak, M.; Taghizadeh, M.; Fallah, N. High-purity hydrogen production by sorption-enhanced methanol steam reforming over a combination of Cu–Zn–CeO2–ZrO2/MCM-41 catalyst and (Li–Na–K) NO3·MgO adsorbent. Int. J. Hydrogen Energy 2021, 46, 7099–7112. [Google Scholar] [CrossRef]
- Khani, Y.; Bahadoran, F.; Safari, N.; Soltanali, S.; Taheri, S.A. Hydrogen production from steam reforming of methanol over Cu-based catalysts: The behavior of ZnxLaxAl1-xO4 and ZnO/La2O3/Al2O3 lined on cordierite monolith reactors. Int. J. Hydrogen Energy 2019, 44, 11824–11837. [Google Scholar] [CrossRef]
- Sanches, S.G.; Flores, J.H.; da Silva, M.I.P. Cu/ZnO and Cu/ZnO/ZrO2 catalysts used for methanol steam reforming. Mol. Catal. 2018, 454, 55–62. [Google Scholar] [CrossRef]
- Azizi, Z.; Rezaeimanesh, M.; Tohidian, T.; Rahimpour, M.R. Dimethyl ether: A review of technologies and production challenges. Chem. Eng. Process. Process Intensif. 2014, 82, 150–172. [Google Scholar] [CrossRef]
- Semelsberger, T.A.; Borup, R.L.; Greene, H.L. Dimethyl ether (DME) as an alternative fuel. J. Power Sources 2006, 156, 497–511. [Google Scholar] [CrossRef]
- Styring, P.; Dowson, G.R.M.; Tozer, I.O. Synthetic Fuels Based on Dimethyl Ether as a Future Non-Fossil Fuel for Road Transport from Sustainable Feedstocks. Front. Energy Res. 2021, 9, 663331. [Google Scholar] [CrossRef]
- Yoon, E.S.; Han, C. A Review of Sustainable Energy—Recent Development and Future Prospects of Dimethyl Ether (DME); Elsevier Inc.: Philadelphia, PA, USA, 2009; Volume 27. [Google Scholar]
- Francisco, J.S. Lifetimes and global warming potentials for dimethyl ether and for fiuorinated ethers: CH3OCF3 (E143a), CHF2OCHF2 (E134), CHF2OCF3 (E125). J. Geophys. Res. Atmos. 1998, 103, 28181–28186. [Google Scholar]
- Mollavali, M.; Yaripour, F.; Atashi, H.; Sahebdelfar, S. Intrinsic kinetics study of dimethyl ether synthesis from methanol on γ-Al2O3 catalysts. Ind. Eng. Chem. Res. 2008, 47, 7130. [Google Scholar] [CrossRef] [Green Version]
- Jamil, A.K.; Muraza, O.; Miyake, K.; Ahmed, M.H.M.; Yamani, Z.H.; Hirota, Y.; Nishiyama, N. Stable Production of Gasoline-Ranged Hydrocarbons from Dimethyl Ether over Iron-Modified ZSM-22 Zeolite. Energy Fuels 2018, 32, 11796–11801. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, Y.; Wei, F.; Wang, D.; Wang, Z. In situ synthesis of SAPO-34 crystals grown onto α-Al2O3 sphere supports as the catalyst for the fluidized bed conversion of dimethyl ether to olefins. Appl. Catal. A Gen. 2008, 341, 112–118. [Google Scholar] [CrossRef]
- Li, J.; Han, D.; He, T.; Liu, G.; Zi, Z.; Wang, Z.; Wu, J.; Wu, J. Nanocrystal H[Fe, Al]ZSM-5 zeolites with different silica-alumina composition for conversion of dimethyl ether to gasoline. Fuel Process. Technol. 2019, 191, 104–110. [Google Scholar] [CrossRef]
- Cheung, P.; Bhan, A.; Sunley, G.J.; Iglesia, E. Selective carbonylation of dimethyl ether to methyl acetate catalyzed by acidic zeolites. Angew. Chem.-Int. Ed. 2006, 45, 1617–1620. [Google Scholar] [CrossRef]
- Arcoumanis, C.; Bae, C.; Crookes, R.; Kinoshita, E. The potential of di-methyl ether (DME) as an alternative fuel for compression-ignition engines: A review. Fuel 2008, 87, 1014–1030. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; Huang, Z.; Liu, Z. Study on combustion and emission of a dimethyl ether-diesel dual-fuel premixed charge compression ignition combustion engine with LPG (liquefied petroleum gas) as ignition inhibitor. Energy 2016, 96, 278–285. [Google Scholar] [CrossRef]
- Var, D.; Do, T. Production of Clean Transportation Fuel Dimethylether by Dehydration of Methanol Over Nafion Catalyst. Gazi Univ. J. Sci. 2010, 21, 37–41. [Google Scholar]
- Kato, M.; Takeuchi, H.; Koie, K.; Sekijima, H.; Kajitani, S.; Chen, Z.L.; Hashimoto, S. A study of dimethyl ether(DME) flow in diesel nozzle. SAE Trans. 2004, 113, 77–85. [Google Scholar] [CrossRef]
- Smyrnioti, M.; Theophilos, I. Dimethyl Ether Hydrolysis overWO3/γ-Al2O3 Supported Catalysts. Catalysts 2022, 12, 396. [Google Scholar] [CrossRef]
- González-Gil, R.; Herrera, C.; Larrubia, M.Á.; Kowalik, P.; Pieta, I.S.; Alemany, L.J. Hydrogen production by steam reforming of DME over Ni-based catalysts modified with vanadium. Int. J. Hydrogen Energy 2016, 41, 19781–19788. [Google Scholar] [CrossRef]
- Takeishi, K.; Akaike, Y. Hydrogen production by dimethyl ether steam reforming over copper alumina catalysts prepared using the sol-gel method. Appl. Catal. A Gen. 2016, 510, 20–26. [Google Scholar] [CrossRef]
- Aziz, M. Liquid hydrogen: A review on liquefaction, storage, transportation, and safety. Energies 2021, 14, 5917. [Google Scholar] [CrossRef]
- Fortin, C.; Gianfolcaro, N.; Gonzalez, R.; Lohest, J.; Lonneux, A.; Kesnelle, A.; Siliki, N.; Peiffer, T.; Renson, R.; Schmitz, C. Dimethyl Ether, a Review of Production Processes and a Modeling of the Indirect Route; PROJ0012-1; Liege University: Liège, Belgium, 2020. [Google Scholar]
- Saravanan, K.; Ham, H.; Tsubaki, N.; Bae, J.W. Recent progress for direct synthesis of dimethyl ether from syngas on the heterogeneous bifunctional hybrid catalysts. Appl. Catal. B Environ. 2017, 217, 494–522. [Google Scholar] [CrossRef]
- Yoo, J.H.; Choi, H.G.; Chung, C.H.; Cho, S.M. Fuel cells using dimethyl ether. J. Power Sources 2006, 163, 103–106. [Google Scholar] [CrossRef]
- Khoshbin, R.; Haghighi, M. Direct conversion of syngas to dimethyl ether as a green fuel over ultrasound-assisted synthesized CuO-ZnO-Al2O3/HZSM-5 nanocatalyst: Effect of active phase ratio on physicochemical and catalytic properties at different process conditions. Catal. Sci. Technol. 2014, 4, 1779–1792. [Google Scholar] [CrossRef]
- Li, Q.; Wu, G.; Johnston, C.M.; Zelenay, P. Direct Dimethyl Ether Fuel Cell with Much Improved Performance. Electrocatalysis 2014, 5, 310–317. [Google Scholar] [CrossRef]
- Mevawala, C.; Jiang, Y.; Bhattacharyya, D. Plant-wide modeling and analysis of the shale gas to dimethyl ether (DME) process via direct and indirect synthesis routes. Appl. Energy 2017, 204, 163–180. [Google Scholar] [CrossRef]
- Sousa-Aguiar, E.F.; Appel, L.G.; Mota, C. Natural gas chemical transformations: The path to refining in the future. Catal. Today 2005, 101, 3–7. [Google Scholar] [CrossRef]
- Li, Y.; Wang, T.; Yin, X.; Wu, C.; Ma, L.; Li, H.; Lv, Y.; Sun, L. 100 t/a-Scale demonstration of direct dimethyl ether synthesis from corncob-derived syngas. Renew. Energy 2010, 35, 583–587. [Google Scholar] [CrossRef]
- Ogawa, T.; Inoue, N.; Shikada, T.; Inokoshi, O.; Ohno, Y. Direct Dimethyl Ether (DME) synthesis from natural gas. Stud. Surf. Sci. Catal. 2004, 147, 379–384. [Google Scholar] [CrossRef]
- An, X.; Zuo, Y.Z.; Zhang, Q.; Wang, D.Z.; Wang, J.F. Dimethyl ether synthesis from CO2 hydrogenation on a CuO-ZnO-Al2O3-ZrO2/HZSM-5 bifunctional catalyst. Ind. Eng. Chem. Res. 2008, 47, 6547–6554. [Google Scholar] [CrossRef]
- Catizzone, E.; Bonura, G.; Migliori, M.; Frusteri, F.; Giordano, G. CO2 recycling to dimethyl ether: State-of-the-art and perspectives. Molecules 2018, 23, 31. [Google Scholar] [CrossRef] [Green Version]
- Ohno, Y.; Yoshida, M.; Shikada, T.; Inokoshi, O.; Ogawa, T.; Inoue, N. New direct synthesis technology for DME (dimethyl ether) and its application technology. JFE Tech. Rep. 2006, 8, 34–40. [Google Scholar]
- Asthana, S.; Samanta, C.; Bhaumik, A.; Banerjee, B.; Voolapalli, R.K.; Saha, B. Direct synthesis of dimethyl ether from syngas over Cu-based catalysts: Enhanced selectivity in the presence of MgO. J. Catal. 2016, 334, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Katurcioglu, T.Y.; Celik, M. A Review on Synthesis of Dimethyl Ether from Syngas Over Bifunctional/Hybrid Catalysts. Glob. J. Pure Appl. Chem. Res. 2019, 7, 1–24. [Google Scholar]
- Khadzhiev, S.N.; Ezhova, N.N.; Yashina, O.V. Catalysis in the dispersed phase: Slurry technology in the synthesis of dimethyl ether (Review). Pet. Chem. 2017, 57, 553–570. [Google Scholar] [CrossRef]
- Kang, S.H.; Bae, J.W.; Jun, K.W.; Potdar, H.S. Dimethyl ether synthesis from syngas over the composite catalysts of Cu-ZnO-Al2O3/Zr-modified zeolites. Catal. Commun. 2008, 9, 2035–2039. [Google Scholar] [CrossRef]
- Bozga, G.; Apan, I.T.; Bozga, R.E. Dimethyl Ether Synthesis Catalysts, Processes and Reactors. Recent Pat. Catal. 2013, 2, 68–81. [Google Scholar] [CrossRef]
- Naik, S.P.; Ryu, T.; Bui, V.; Miller, J.D.; Drinnan, N.B.; Zmierczak, W. Synthesis of DME from CO2/H2 gas mixture. Chem. Eng. J. 2011, 167, 362–368. [Google Scholar] [CrossRef]
- Wang, S.; Mao, D.; Guo, X.; Wu, G.; Lu, G. Dimethyl ether synthesis via CO2 hydrogenation over CuO—TiO2—ZrO2/HZSM-5 bifunctional catalysts. Catal. Commun. 2009, 10, 1367–1370. [Google Scholar] [CrossRef]
- Yang, C.; Ma, Z.; Zhao, N.; Wei, W.; Hu, T.; Sun, Y. Methanol synthesis from CO2-rich syngas over a ZrO2 doped CuZnO catalyst. Catal. Today 2006, 115, 222–227. [Google Scholar] [CrossRef]
- Mondal, U.; Yadav, G.D. Perspective of dimethyl ether as fuel: Part I—Catalysis. J. CO2 Util. 2019, 32, 299–320. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Cannilla, C.; Mezzapica, A.; Spadaro, L.; Arena, F.; Frusteri, F. Catalytic behaviour of a bifunctional system for the one step synthesis of DME by CO2 hydrogenation. Catal. Today 2014, 228, 51–57. [Google Scholar] [CrossRef]
- Ohno, Y.; Yagi, H.; Inoue, N.; Okuyama, K.; Aoki, S. Slurry phase DME direct synthesis technology -100 tons/day demonstration plant operation and scale up study. Stud. Surf. Sci. Catal. 2007, 167, 403–408. [Google Scholar] [CrossRef]
- Thottan, M.K.; Chuanyi, J. Dimethyl ether steam reforming catalyst and method for producing the same. No 10/730638, 30 September 2004. [Google Scholar]
- Wang, S.; Ishihara, T.; Takita, Y. Partial oxidation of dimethyl ether over various supported metal catalysts. Appl. Catal. A Gen. 2002, 228, 167–176. [Google Scholar] [CrossRef]
- Badmaev, S.D.; Pinigina, A.E.; Snytnikov, P.V.; Sobyanin, V.A. CuCeOx/γ-Al2O3 catalyst for low temperature dimethyl ether partial oxidation to hydrogen-rich gas. Mater. Lett. 2021, 302, 130345. [Google Scholar] [CrossRef]
- Badmaev, S.D.; Akhmetov, N.O.; Belyaev, V.D.; Kulikov, A.V.; Pechenkin, A.A.; Potemkin, D.I.; Konishcheva, M.V.; Rogozhnikov, V.N.; Snytnikov, P.V.; Sobyanin, V.A. Syngas production via partial oxidation of dimethyl ether over Rh/Ce0.75Zr0.25O2 catalyst and its application for SOFC feeding. Int. J. Hydrogen Energy 2020, 45, 26188–26196. [Google Scholar] [CrossRef]
- Nilsson, M.; Pettersson, L.J.; Lindström, B. Hydrogen generation from dimethyl ether for fuel cell auxiliary power units. Energy Fuels 2006, 20, 2164–2169. [Google Scholar] [CrossRef]
- Nilsson, M.; Jansson, K.; Jozsa, P.; Pettersson, L.J. Catalytic properties of Pd supported on ZnO/ZnAl2O4/Al2O3 mixtures in dimethyl ether autothermal reforming. Appl. Catal. B Environ. 2009, 86, 18–26. [Google Scholar] [CrossRef]
- Asami, K.; Seto, K.; Saima, H.; Mogi, Y. Dry Reforming of Dimethyl Ether with Carbon Dioxide over a Cu-Containing Hybrid Catalyst. Catal. Surv. Asia 2013, 17, 14–19. [Google Scholar] [CrossRef]
- Kim, D.; Park, G.; Choi, B.; Kim, Y.B. Reaction characteristics of dimethyl ether (DME) steam reforming catalysts for hydrogen production. Int. J. Hydrogen Energy 2017, 42, 29210–29221. [Google Scholar] [CrossRef]
- Galvita, V.V.; Semin, G.L.; Belyaev, V.D.; Yurieva, T.M.; Sobyanin, V.A. Production of hydrogen from dimethyl ether. Appl. Catal. A Gen. 2001, 216, 85–90. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Tanaka, Y.; Shimoda, N.; Fukunaga, T.; Kikuchi, R.; Eguchi, K. Hydrogen production from dimethyl ether steam reforming over composite catalysts of copper ferrite spinel and alumina. Appl. Catal. B Environ. 2007, 74, 144–151. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Shimoda, N.; Fukunaga, T.; Kikuchi, R.; Eguchi, K. Cu-based spinel catalysts CuB2O4 (B = Fe, Mn, Cr, Ga, Al, Fe0.75Mn0.25) for steam reforming of dimethyl ether. Appl. Catal. A Gen. 2008, 341, 139–145. [Google Scholar] [CrossRef]
- Ledesma, C.; Ozkan, U.S.; Llorca, J. Hydrogen production by steam reforming of dimethyl ether over Pd-based catalytic monoliths. Appl. Catal. B Environ. 2011, 101, 690–697. [Google Scholar] [CrossRef]
- Wang, X.; Pan, X.; Lin, R.; Kou, S.; Zou, W.; Ma, J.X. Steam reforming of dimethyl ether over Cu-Ni/γ-Al2O3 bi-functional catalyst prepared by deposition-precipitation method. Int. J. Hydrogen Energy 2010, 35, 4060–4068. [Google Scholar] [CrossRef]
- Yoshida, H.; Iwasa, N.; Akamatsu, H.; Arai, M. Stable and selective hydrogen production through steam reforming of dimethyl ether with an Al2O3 and PdZn composite catalyst. Int. J. Hydrogen Energy 2015, 40, 5624–5627. [Google Scholar] [CrossRef]
- Lian, J.H.; Tan, H.Y.; Guo, C.Q.; Wang, Z.D.; Shi, Y.; Lu, Z.X.; Shen, L.S.; Yan, C.F. A highly active and stable Pt modified molybdenum carbide catalyst for steam reforming of dimethyl ether and the reaction pathway. Int. J. Hydrogen Energy 2020, 45, 31523–31537. [Google Scholar] [CrossRef]
- Oar-Arteta, L.; Remiro, A.; Vicente, J.; Aguayo, A.T.; Bilbao, J.; Gayubo, A.G. Stability of CuZnOAl2O3/HZSM-5 and CuFe 2O4/HZSM-5 catalysts in dimethyl ether steam reforming operating in reaction-regeneration cycles. Fuel Process. Technol. 2014, 126, 145–154. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Kikuchi, R.; Eguchi, K. Thermodynamic analysis of carbon formation boundary and reforming performance for steam reforming of dimethyl ether. J. Power Sources 2007, 164, 73–79. [Google Scholar] [CrossRef]
- Inagaki, R.; Manabe, R.; Hisai, Y.; Kamite, Y.; Yabe, T.; Ogo, S.; Sekine, Y. Steam reforming of dimethyl ether promoted by surface protonics in an electric field. Int. J. Hydrogen Energy 2018, 43, 14310–14318. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kikuchi, R.; Takeguchi, T.; Eguchi, K. Steam reforming of dimethyl ether over composite catalysts of γ-Al2O3 and Cu-based spinel. Appl. Catal. B Environ. 2005, 57, 211–222. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Shimoda, N.; Viriya-empikul, N.; Kikuchi, R.; Eguchi, K. Limiting mechanisms in catalytic steam reforming of dimethyl ether. Appl. Catal. B Environ. 2010, 97, 21–27. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Tanaka, Y.; Shimoda, N.; Fukunaga, T.; Kawashima, S.; Kikuchi, R.; Eguchi, K. Influence of solid-acid catalysts on steam reforming and hydrolysis of dimethyl ether for hydrogen production. Appl. Catal. A Gen. 2006, 304, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.S.; Song, Y.H.; Zhao, Y.H.; Liu, C.; Xiao, Y.S.; Zhang, Q.; Liu, Z.T.; Liu, Z.W. Amorphous silica-alumina composite with regulated acidity for efficient production of hydrogen via steam reforming of dimethyl ether. Catal. Today 2020, 351, 68–74. [Google Scholar] [CrossRef]
- Oar-arteta, L.; Azkoiti, M.J.; Gayubo, A.G.; Vicente, J.; Ere, J.; Olazar, M.; Bilbao, J. Applied Catalysis A: General Causes of deactivation of bifunctional catalysts made up of in DME steam reforming. Appl. Catal. A Gen. 2014, 483, 76–84. [Google Scholar] [CrossRef]
- Faungnawakij, K.; Fukunaga, T.; Kikuchi, R.; Eguchi, K. Deactivation and regeneration behaviors of copper spinel-alumina composite catalysts in steam reforming of dimethyl ether. J. Catal. 2008, 256, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Takeishi, K.; Suzuki, H. Steam reforming of dimethyl ether. Appl. Catal. A Gen. 2004, 260, 111–117. [Google Scholar] [CrossRef]
- Mathew, T.; Yamada, Y.; Ueda, A.; Shioyama, H.; Kobayashi, T. Metal oxide catalysts for DME steam reforming: Ga2O3 and Ga2O3-Al2O3 catalysts with and without copper. Appl. Catal. A Gen. 2005, 286, 11–22. [Google Scholar] [CrossRef]
- Zhang, Q.; Fan, F.; Xu, G.; Ye, D.; Wang, W.; Zhu, Z. Steam reforming of dimethyl ether over a novel anodic γ-Al2O3 supported copper bi-functional catalyst. Int. J. Hydrogen Energy 2013, 38, 10305–10314. [Google Scholar] [CrossRef]
- Brückner, N.; Obesser, K.; Bösmann, A.; Teichmann, D.; Arlt, W.; Dungs, J.; Wasserscheid, P. Evaluation of industrially applied heat-transfer fluids as liquid organic hydrogen carrier systems. ChemSusChem 2014, 7, 229–235. [Google Scholar] [CrossRef]
- Do, G.; Preuster, P.; Aslam, R.; Bösmann, A.; Müller, K.; Arlt, W.; Wasserscheid, P. Hydrogenation of the liquid organic hydrogen carrier compound dibenzyltoluene-reaction pathway determination by 1H NMR spectroscopy. React. Chem. Eng. 2016, 1, 313–320. [Google Scholar] [CrossRef] [Green Version]
- Jorschick, H.; Geißelbrecht, M.; Eßl, M.; Preuster, P.; Bösmann, A.; Wasserscheid, P. Benzyltoluene/dibenzyltoluene-based mixtures as suitable liquid organic hydrogen carrier systems for low temperature applications. Int. J. Hydrogen Energy 2020, 45, 14897–14906. [Google Scholar] [CrossRef]
- Hydrogenious Technologies GmbH. Available online: https://www.hydrogenious.net/index.php/en/hydrogen-2-2/ (accessed on 15 June 2022).
- Müller, K.; Stark, K.; Emelyanenko, V.N.; Varfolomeev, M.A.; Zaitsau, D.H.; Shoifet, E.; Schick, C.; Verevkin, S.P.; Arlt, W. Liquid Organic Hydrogen Carriers: Thermophysical and Thermochemical Studies of Benzyl- and Dibenzyl-toluene Derivatives. Ind. Eng. Chem. Res. 2015, 54, 7967–7976. [Google Scholar] [CrossRef]
- Markiewicz, M.; Zhang, Y.Q.; Bösmann, A.; Brückner, N.; Thöming, J.; Wasserscheid, P.; Stolte, S. Environmental and health impact assessment of Liquid Organic Hydrogen Carrier (LOHC) systems-challenges and preliminary results. Energy Environ. Sci. 2015, 8, 1035–1045. [Google Scholar] [CrossRef] [Green Version]
- Sisáková, K.; Podrojková, N.; Oriňaková, R.; Oriňak, A. Novel catalysts for dibenzyltoluene as a potential liquid organic hydrogen carrier use—A mini-review. Energy Fuels 2021, 35, 7608–7623. [Google Scholar] [CrossRef]
- Jorschick, H.; Bulgarin, A.; Alletsee, L.; Preuster, P.; Bösmann, A.; Wasserscheid, P. Charging a Liquid Organic Hydrogen Carrier with Wet Hydrogen from Electrolysis. ACS Sustain. Chem. Eng. 2019, 7, 4186–4194. [Google Scholar] [CrossRef]
- Dürr, S.; Müller, M.; Jorschick, H.; Helmin, M.; Bösmann, A.; Palkovits, R.; Wasserscheid, P. Carbon Dioxide-Free Hydrogen Production with Integrated Hydrogen Separation and Storage. ChemSusChem 2017, 10, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Qi, S.; Qu, J.; Che, T.; Yi, C.; Yang, B. Integration of hydrogenation and dehydrogenation based on dibenzyltoluene as liquid organic hydrogen energy carrier. Int. J. Hydrogen Energy 2019, 44, 5345–5354. [Google Scholar] [CrossRef]
- Modisha, P.; Bessarabov, D. Stress tolerance assessment of dibenzyltoluene-based liquid organic hydrogen carriers. Sustain. Energy Fuels 2020, 4, 4662–4670. [Google Scholar] [CrossRef]
- Shi, L.; Zhou, Y.; Qi, S.; Smith, K.J.; Tan, X.; Yan, J.; Yi, C. Pt Catalysts Supported on H2 and O2 Plasma-Treated Al2O3 for Hydrogenation and Dehydrogenation of the Liquid Organic Hydrogen Carrier Pair Dibenzyltoluene and Perhydrodibenzyltoluene. ACS Catal. 2020, 10, 10661–10671. [Google Scholar] [CrossRef]
- Jorschick, H.; Bösmann, A.; Preuster, P.; Wasserscheid, P. Charging a Liquid Organic Hydrogen Carrier System with H2/CO2 Gas Mixtures. ChemCatChem 2018, 10, 4329–4337. [Google Scholar] [CrossRef]
- Jorschick, H. Ein-Reaktor-Konzept und Mischgashydrierung als Verfahrensvarianten zur Effizienzsteigerung in der LOHC-basierten Wasserstoffspeicherung. Ph.D. Thesis, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany, 2019. [Google Scholar]
- Ali, A.; Rohini, A.K.; Noh, Y.S.; Moon, D.J.; Lee, H.J. Hydrogenation of dibenzyltoluene and the catalytic performance of Pt/Al2O3 with various Pt loadings for hydrogen production from perhydro-dibenzyltoluene. Int. J. Energy Res. 2022, 46, 6672–6688. [Google Scholar] [CrossRef]
- Modisha, P.; Gqogqa, P.; Garidzirai, R.; Ouma, C.N.M.; Bessarabov, D. Evaluation of catalyst activity for release of hydrogen from liquid organic hydrogen carriers. Int. J. Hydrogen Energy 2019, 44, 21926–21935. [Google Scholar] [CrossRef]
- Office of Energy Efficiency & Renewable Energy. Available online: https://www.energy.gov/eere/fuelcells/hydrogen-storage (accessed on 15 June 2022).
- Niermann, M.; Timmerberg, S.; Drünert, S.; Kaltschmitt, M. Liquid Organic Hydrogen Carriers and alternatives for international transport of renewable hydrogen. Renew. Sustain. Energy Rev. 2021, 135, 110171. [Google Scholar] [CrossRef]
- Takeishi, K. Dimethyl ether and catalyst development for production from syngas. Biofuels 2010, 1, 217–226. [Google Scholar] [CrossRef]
- Putrasari, Y.; Lim, O. Dimethyl Ether as the Next Generation Fuel to Control Nitrogen Oxides and Particulate Matter Emissions from Internal Combustion Engines: A Review. ACS Omega 2022, 7, 32–37. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Storage Method | Storage Conditions | Hydrogen Storage Capacity (% wt) | H2 Release Conditions | Explosive Limits (%vol in Air) | Toxicity | PELs * (ppm) | Price |
|---|---|---|---|---|---|---|---|
| Compressed H2 | 70 MPa | 100 | Pressure reduction | 4–75 | - | - | - |
| Liquid H2 | −253 °C | 100 | Evaporation | 4–75 | - | - | - |
| Liquid NH3 | −33.5 °C, 0.1 MPa | 17.6 | Catalytic, T > 400 °C | 15–28 | Toxic | 50 | 934 USD/t |
| Formic acid | Ambient | 4.4 | Catalytic, T > 50 °C | 18–34 | Toxic | 5 | 350 USD/t |
| Toluene–methylcyclohexane (MCH) | Ambient | 6.1 | Catalytic, T > 300 °C | 1.2–7.1 (toluene) 1.2–6.7 (MCH) | Toxic | 200 500 | 910 USD/t |
| Benzene–cyclohexane | Ambient | 7.2 | Catalytic, T > 300 °C | 1.4–8.0 (benzene) 1.3–8.4 (cyclohexane) | Toxic | 10 300 | 835 USD/t |
| N-ethylcarbazole (NEC)–perhydrocarbazole | Ambient | 5.8 | Catalytic, T > 150 °C | - | Toxic | nd | 20 USD/kg |
| Methanol Properties | |
|---|---|
| Molar mass (g/mol) | 32.04 |
| Appearance | Colorless liquid |
| Density (g/cm3) | 0.792 |
| Melting point (°C) | −98 |
| Boiling point (°C) | 64.7 |
| Flash point (°C) | 12 (closed vessel 15.6 (open vessel) |
| Autoignition temperature (°C) | 470 |
| Cetane number | 5 |
| Gravimetric hydrogen density (wt %) | 12.5 |
| Volumetric hydrogen density (kgH2/m3) | 99 |
| Toxicity | |
| • RfC (reference concentration for inhalation exposure) (mgm−3) | 2 |
| • RfD (reference dose of oral exposure) (mg/kg day) | 2 |
| BMDL05 (benchmark dose at 95% lower confidence limit) (mg/kg-day) | 43.1 |
| Process | Summary Reaction/Standard Enthalpy | Process Conditions | Advantages/Disadvantages |
|---|---|---|---|
| Methanol steam reforming (MSR) | T = 150–350 °C p = 1 atm H2O/methanol molar ratio: 1.3–5 | Low temperatures and pressure High methanol conversions High content of hydrogen in outlet steam Minimal carbon monoxide content May be performed in homogeneous and heterogeneous systems Necessity of steam preheating Necessity of wastewater management | |
| Methanol decomposition (MD) | T = 100–450 °C p = 1–6 atm | Outlet stream rich in carbon monoxide Temperature increase favors the generation of by-products (e.g., dimethyl ether, methane) | |
| Partial oxidation (POM) | T = 30–450 °C O2/methanol molar ratio: 0.3–0.8 | Reduction of heating cost due to the exothermic character of the process Excess heat may be used in other endothermic processes High content of CO as a product of the reaction POM process proceeds through the formation of methoxy and formate groups, which may be present as a by-product | |
| Autothermal reforming (ATRM) | r <0, 0.5>—ratio between O2 and MeOH in inlet stream | T = 200–550 °C O2/methanol molar ratio: 0.1–0.6 H2O/methanol molar ratio: 1.0–1.5 | Hydrogen-rich gas is generated Reduction of external heating costs due to the exothermic character of oxidation and use of released heat for reforming triggering Differences in kinetics of oxidation and methanol reforming enforce strict control of parameters and the selection of a specialized catalyst |
| Catalyst | Preparation Method | Process Conditions | Methanol Conversion (%) | Hydrogen Generation Yield | CO, CO2 Selectivity/Yield | Observations/Conclusions | Ref. |
|---|---|---|---|---|---|---|---|
| Wirelike Mo2C | Carburization from aniline at different temperatures: 675, 725, and 750 °C | GHSV = 9000 cm3g−1h−1 MeOH:H2O = 1:1 T = 200–400 °C, p = 1 atm | For the most active catalyst carburized at 675 °C | The higher the temperature of the MSR, the higher the levels of methanol conversion, however, with the rise of temperature, the content of CO and CH4. | [62] | ||
| T = 200 °C, XMeOH = 30% | CH2 = 70 mol. % | CCO = 0 mol. % | |||||
| T = 250 °C, XMeOH = 45% | CH2 = 67 mol. % | CCO = 2 mol. % | |||||
| T = 300 °C, XMeOH = 85% | CH2 = 64 mol. % | CCO = 5 mol. % | |||||
| T = 350 °C, XMeOH = 100% | CH2 = 60 mol. % | CCO = 4 mol. % | |||||
| T = 400 °C, XMeOH = 100% | CH2 = 60 mol. % | CCO = 3 mol. % | |||||
| Ti-modified Cu/y-Al2O3/Al | Impregnation of y-Al2O3/Al support with titanium and copper salts Cu/Tix (x = 0, 1.5, 1.9, 2.5, 3.2) | GHSV = 4000 cm3g−1h−1 MeOH:H2O = 1:2 T = 225–350 °C, p = 1 atm | The most active catalyst, where Cu/Ti = 1:1.9 | Ti species increases the surface are a, stabilizes the copper dispersion, enhances the adsorption of methanol, and improves copper redox performance by facilitating electron transfer from Cu to Ti. Moreover, the addition of titanium promotes the reduction of the acidity of the catalyst, thus limiting the occurrence of side reactions. | [63] | ||
| T = 225 °C, XMeOH = 67%, | CH2 = 45 molkgcat−1h−1 | SCO = 1.5% | |||||
| T = 250 °C, XMeOH = 90%, | CH2 = 62 molkgcat−1h−1 | SCO = 2.5% | |||||
| T = 275 °C, XMeOH = 95%, | CH2 = 65 molkgcat−1h−1 | SCO = 3.0% | |||||
| T = 300 °C, XMeOH = ~100%, | CH2 = 68 molkgcat−1h−1 | SCO = 4.0% | |||||
| T = 325 °C, XMeOH = ~100%, | CH2 = 70 molkgcat−1h−1 | SCO = 5.0% | |||||
| T = 350 °C, XMeOH = ~100% | CH2 = 73 molkgcat−1h−1 | SCO = 5.1% | |||||
| CeO2-Cu/KIT-6 promoted with ZrO2 | Impregnation of KIT-6 with ceria, copper, and zirconium salts. The support was prepared hydrothermally | WHSV = 2 g−1h−1 MeOH:H2O = 1:2 T = 225–350 °C | ZrO2-CeO2-Cu/KIT-6, XMeOH = 96%, CeO2-Cu/KIT-6, XMeOH = 85–92% | ZrO2-CeO2-Cu/KIT-6, SH2 = 99.8% CeO2-Cu/KIT-6, SH2 = 99.2% | ZrO2-CeO2-Cu/KIT-6, SCO = 0.7%, CeO2-Cu/KIT-6, SCO = 0.8% | Promotion with ZrO2 enhanced the performance of the catalysts by stabilizing copper dispersion. | [68] |
| Cu-M/CeO2 M = Pt, Pd, Ni | Wet impregnation of ceria commercial support | T = 100–350 °C, T = 24 h | At T = 350 °C, methanol conversion drops rapidly along with time. The higher was the content of the modifying metal (Pt, Pd, or Ni), the higher was the activity observed | Not determined | Not determined | The amount and type of the modifying metal (Pt, Pd, Ni) determined the catalytic activity. Hydrogen selectivity was improved in the case of catalysts modified with Pt or Pd. | [57] |
| Mg-promoted Cu/ZnO/Al2O3, Cu/Zn/Al/Mg = 60:30:10:x, x = 0, 3, 5, 7 | Coprecipitation from nitrate’s salts with sodium carbonate | WHSV = 3.84 g−1h−1 MeOH:H2O = 1:1 T = 200 °C, p = 0.1 MPa | Not determined | H2 STY (molKg−1h−1) | The promotion with magnesium enhanced the copper surface area and Cu–ZnO interactions. The optimum content of magnesium equaled 5%. | [71] | |
| Cu/Zn/Al/Mg0: 145 | SCO2 = 99.9% | ||||||
| Cu/Zn/Al/Mg3: 170 | SCO2 = 99.5% | ||||||
| Cu/Zn/Al/Mg5: 172 | SCO2 = 99.2% | ||||||
| Cu/Zn/Al/Mg7: 158 | SCO2 = 99.7% | ||||||
| Cu/ZnO Cu/ZnO/ZrO2 | Coprecipitation from nitrate’s salts with sodium carbonate | MeOH/H2O = 1:3 T = 250 °C, p = 1 atm | Cu/ZnO: XMeOH = 51.8%, Cu/ZnO/ZrO2: XMeOH = 88.6%, | CH2 = 0.19 molgcat−1h−1 CH2 = 12.6 molgcat−1h−1 | SCO = 4.7% SCO = traces | Zirconium component increases copper–zinc oxide microstrains, thus stabilizing dispersion. | [77] |
| DME Properties | |
|---|---|
| Molar mass (g/mol) | 46.07 |
| Appearance | colorless gas |
| Density (g/cm3) | 1.97 (1 atm, 20 °C) |
| Liquid density (g/cm3) | 0.667 (1 atm, −25 °C) |
| Melting point (°C) | −141.5 |
| Boiling point (°C) | −24.9 |
| Flash point (°C) | −41 |
| Autoignition temperature (°C) | 235–350 |
| Cetane number | 55–60 |
| Flammability limit in the air (vol %) | 3.4–17 |
| Lower heating value (MJ/kg) | 28.90 |
| Gravimetric hydrogen density (wt %) | 13 |
| Volumetric hydrogen density (kg-H2/m3) | 86.9 |
| Catalyst | Conditions | DME Conversion | H2 Production | Catalyst Stability | Ref. |
|---|---|---|---|---|---|
| CuZnOAl2O3/HZSM-5 1 | 275 °C, 1.2 atm | ΧDME = 40–44% | YH2 = 35–43% | 9% loss of ΧDME and 19% loss of YH2 after 20 h TOS | [134] |
| CuZnOAl2O3/HZSM-5 | 315 °C, 1.2 atm | ΧDME = 50–80% | YH2 = 40–75% | 38% loss of ΧDME and 47% loss of YH2 after 6 h TOS | [134] |
| CuFe2O4/HZSM-5 | 300 °C, 1.2 atm | ΧDME = 50–57% | YH2 = 45–51% | 12% loss of ΧDME and 12% loss of YH2 after 4 h TOS | [134] |
| CuFe2O4/γ-Al2O3 | 350 °C, 1 atm | ΧDME = 84–95% | VH2 = 31–35 mL/min | 12% loss of ΧDME and 11% loss of VH2 after 25 h TOS | [129] |
| CuFe1.5Mn0.50O4/γ-Al2O3 | 350 °C, 1 atm | ΧDME = 85–90% | VH2 = 28–30 mL/min | 5% loss of ΧDME and 7% loss of VH2 after 25 h TOS | [129] |
| CuAl2O4/γ-Al2O3 | 350 °C, 1 atm | ΧDME = 77–90% | VH2 = 24–31 mL/min | 14% loss of ΧDME and 23% loss of VH2 after 25 h TOS | [129] |
| CuGa2O4/γ-Al2O3 | 350 °C, 1 atm | ΧDME = 57–86% | VH2 = 20–31 mL/min | 34% loss of ΧDME and 35% loss of VH2 after 25 h TOS | [129] |
| CuCr2O4/γ-Al2O3 | 350 °C, 1 atm | ΧDME = 79–80% | VH2 = 26–27 mL/min | 1% loss of ΧDME and 4% loss of VH2 after 25 h TOS | [129] |
| CuMn2O4/γ-Al2O3 | 350 °C, 1 atm | ΧDME = 69–71% | VH2 = 23–24 mL/min | 3% loss of ΧDME and 4% loss of VH2 after 25 h TOS | [129] |
| Cu–Ni/γ-Al2O3 | 350 °C, 1 atm | ΧDME ≈ 100% | VH2 = 55–70 mmol g−1 h−1 | 0% loss of ΧDME and 21% loss of VH2 after 30 h TOS | [131] |
| Pd/ZrO2 | 360 °C, 1 atm | ΧDME ≈ 50% | YH2 ≈ 31% | n.d. | [130] |
| 420 °C, 1 atm | ΧDME ≈ 70% | YH2 ≈ 46% | n.d. | ||
| 480 °C, 1 atm | ΧDME ≈ 80% | YH2 ≈ 55% | n.d. | ||
| 550 °C, 1 atm | ΧDME ≈ 100% | YH2 ≈ 65% | n.d. | ||
| PdZn/Al2O3 | 300 °C, 1 atm | ΧDME ≈ 60% | YH2 ≈ 45% | n.d. | [132] |
| 350 °C, 1 atm | ΧDME ≈ 98% | YH2 ≈ 91% | n.d. | ||
| 400 °C, 1 atm | ΧDME ≈ 100% | YH2 ≈ 92% | no ΧDME and YH2 loss after 6 h TOS | ||
| Cu/ZnO/Al2O3 | 400 °C, 1 atm | ΧDME = 52% | YH2 = 25% | 24% loss of ΧDME and 32% loss of YH2 after 6 h TOS | [132] |
| Pt–Mo2C/Al2O3 | 300 °C, 1 atm | ΧDME = 35% | VH2 ≈ 63 mmol g−1 h−1 | n.d. | [133] |
| 350 °C, 1 atm | ΧDME ≈ 100% | VH2 ≈ 96 mmol g−1 h−1 | 20% loss of ΧDME after 50 h TOS | ||
| 400 °C, 1 atm | ΧDME ≈ 100% | VH2 ≈ 90 mmol g−1 h−1 | n.d. | ||
| 450 °C, 1 atm | ΧDME ≈ 100% | VH2 ≈ 84 mmol g−1 h−1 | n.d. | ||
| 500 °C, 1 atm | ΧDME ≈ 100% | VH2 ≈ 78 mmol g−1 h−1 | n.d. | ||
| CuFe2O4/ZSM-5 | 300 °C, 1 atm | ΧDME = 65% | VH2 = 60 mmol g−1 h−1 | n.d. | [139] |
| CuFe2O4/H-modernite | 275 °C, 1 atm | ΧDME = 70% | VH2 = 60 mmol g−1 h−1 | n.d. | [139] |
| CuFe2O4/Al2O3 | 350 °C, 1 atm | ΧDME = 70% | VH2 = 95 mmol g−1 h−1 | Stable activity for DME hydrolysis for 25 h | [139] |
| CuZnOAl2O3/ASA | 300 °C, 1 atm | ΧDME ≈ 70% | YH2 ≈ 70% | n.d. | [140] |
| 330 °C, 1 atm | ΧDME ≈ 90% | YH2 ≈ 90% | n.d. | ||
| 350 °C, 1 atm | ΧDME ≈ 100% | YH2 ≈ 100% | No ΧDME and YH2 loss after 66 h TOS |
| Acid Function | Acid Amount (umol/g) | Acid Site Strength | Optimal Temperature (°C) | DME Conversion (%) | H2 Production (mmol g−1 h−1) |
|---|---|---|---|---|---|
| Zeolite-H mordenite | 720 | Weak, strong | 250–275 | 55–70 | 50–60 |
| Zeolite-H mordenite | 260 | Strong | 250–275 | 50–70 | 40–50 |
| Zeolite-MFI(ZSM-5) | 70 | Medium | 250–300 | 30–65 | 30–60 |
| Alumina-γ-Al2O3 | 50 | Medium | 350–375 | ~70 | ~95 |
| Alumina-TA series | 182 | Weak | 350–375 | ~95 | ~70 |
| Alumina-TA series | 300 | Weak, strong | 350–375 | 90–95 | ~70 |
| Alumina-DK series | 530 | Weak, strong | 400–450 | 70–95 | 55–70 |
| Alumina-NKH series | 440 | Weak | 350–400 | ~95 | 65–70 |
| Alumina-NKH series | 250 | Weak | 375–400 | 90–95 | 60–65 |
| Alumina-NK series | 360 | Weak | 375–425 | 80–95 | 55–60 |
| Properties | Dibenzyltoluene (H0-DBT) | Perhydrodibenzyltoluene (H18-DBT) |
|---|---|---|
| Density (kg L−1) | 1.04 | 0.91 |
| Melting point (°C) | −39 | −45 |
| Boiling point (°C) | 390 | 354 |
| Ignition temperature (°C) | 450 | No data |
| Dynamic viscosity at 20 °C (mPa s) | 44.1 | 258 |
| Vapor pressure at 40 °C (Pa) | 0.07 | 0.04 |
| Hazard classes | 0.9 | No data |
| Catalyst | Conditions | Amount of the Catalyst | Degree of Hydrogen Loading (DoH) [%] | Reference |
|---|---|---|---|---|
| Ru/Al2O3 (5% wt) | 150 °C, 50 bar, 4 h | 0.25 mol. % | 45 | [146] |
| Ru/Al2O3 (0.5 % wt) | 180 °C, 30 bar, 5 h | 0.05 mol. % | 78 | [153] |
| 210 °C, 30 bar, 5 h | 81 | |||
| 240 °C, 30 bar, 5 h | 80 | |||
| Ru/Al2O3 (5 % wt) | 180 °C, 5 bar, 3 h | 0.25 mol. % | ~30 | [154] |
| 180 °C, 10 bar, 3 h | ~60 | |||
| 180 °C, 40 bar, 3 h | 100 | |||
| 180 °C, 50 bar, 100 min | ||||
| Rh/Al2O3 (5% wt) | 210 °C, 30 bar, 5 h | 0.05 mol. % | ~98 | [153] |
| Pd/Al2O3 (5% wt) | 260 °C, 30 bar, 6 h | 0.05 mol. % | ~98 | [153] |
| Pt/Al2O3 (0.3% wt) | 260 °C, 30 bar, 1 h | 0.025 mol. % | 100 | [153] |
| Pt/Al2O3 (5% wt) | 140 °C, 40 bar, 35 min | 0.3 mol. % | 100 | [155] |
| Raney-Ni | 170 °C, 9 bar, 10 h | 1 g per 10 g H0-DBT | 21 | [32] |
| NiSat 310 (50% wt Ni) | 150–170 °C, 3–15 bar | No data | 74–98 | [156] |
| Temperature (°C) | Ru/Al2O3 | Rh/Al2O3 | Pt/Al2O3 | Pd/Al2O3 | ||||
|---|---|---|---|---|---|---|---|---|
| CO2 (ppm) | CH4 (ppm) | CO2 (ppm) | CH4 (ppm) | CO2 (ppm) | CH4 (ppm) | CO2 (ppm) | CH4 (ppm) | |
| 150 | 6 | 485 | 45 | 319 | 15 | 68 | 35 | 103 |
| 180 | 8 | 1502 | 40 | 537 | 66 | 93 | 25 | 232 |
| 210 | 17 | 11,193 | 19 | 515 | 42 | 240 | 36 | 327 |
| 240 | 46 | 49,994 | 29 | 844 | 54 | 368 | 77 | 421 |
| 260 | 48 | 154 430 | 42 | 1376 | 74 | 574 | 38 | 560 |
| Catalyst | Conditions | Amount of the Catalyst | Degree of Dehydrogenation (DoD) (%) | Reference |
|---|---|---|---|---|
| Pt/Al2O3 (5% wt) | 270 °C, 3.5 h | 0.15 mol. % | 40 | [146] |
| Pt/C (5% wt) | 270 °C, 3.5 h | 0.15 mol. % | 55 | [146] |
| Pt/C (1% wt) | 270 °C, 3.5 h 290 °C, 3.5 h | 0.15 mol. % | 71 98 | [146] |
| Pt/SiO2 (1% wt) | 270 °C, 3.5 h | 0.15 mol. % | 10 | [146] |
| Pd/C (5% wt) | 270 °C, 3.5 h | 0.15 mol. % | 16 | [146] |
| Pd/Al2O3 (5% wt) | 270 °C, 3.5 h | 0.15 mol. % | 8 | [146] |
| Pt–Pd/Al2O3 (1:1% wt) | 320 °C, 80 min | 0.4 mol. % | 6 | [161] |
| MeOH | DME | H0-DBT/H18-DBT | |
|---|---|---|---|
| Physical state | Liquid | Gas 1 | Liquid |
| Density (g/cm3) | 0.79 | 1.97 2 | 1.04/0.91 |
| Melting point (°C) | −98 | −142 | −39/−45 |
| Boiling point (°C) | 65 | −25 | 390/354 |
| Flash point (°C) | 12 | −41 | 212 |
| Autoignition temperature (°C) | 470 | 235–350 | Not autoflammable |
| Toxicity | Highly toxic | Nontoxic | Nontoxic/no data |
| Gravimetric hydrogen density (wt %) | 12.5 | 13 | 6.2 |
| Volumetric hydrogen density (kg-H2/m3) | 99 | 87 | 58 |
| H2 Carrier | Hydrogenation/Synthesis | Dehydrogenation/Steam Reforming | Advantages | Disadvantages | ||
|---|---|---|---|---|---|---|
| Conditions | Catalysts | Conditions | Catalysts | |||
| MeOH | 220–230 °C, 50–100 bar | CuO/ZnO/Al2O3 (CZA) promoted with variable stabilizing additives (Cr, Zr, Mg compounds) | 200–400 °C, 1 bar | Cu-based catalysts, mostly CuO/ZnO/Al2O3 modified with various promoters (ZrO2, Ga2O3, TiO2, ZnO, CeO2, MgO) | Low synthesis and steam reforming temperatures Possibility of CO2 utilization as feedstock Low pressure of steam reforming Highest volumetric H2 density | High-pressure synthesis conditions MeOH toxicity MeOH flammability Besides CO2, CO or CH4 may be formed as a side product of steam reforming |
| DME | 240–280 °C, 30–80 bar | Bifunctional catalysts: Cu-based metallic function (mostly CZA) and alumina or zeolites as solid acid function (i.e., CZA/HZSM-5, CZA/γ-Al2O3) | 250–450 °C, 1 bar | Bifunctional catalysts: Cu-based metallic function (mostly Cu spinel) and alumina or zeolites as solid acid function (i.e., CuFe2O4/HZSM-5, CuFe2O4/γ-Al2O3) | Low synthesis and steam reforming temperatures Possibility of CO2 utilization as feedstock Low pressure of steam reforming Highest gravimetric H2 density Nontoxic | High-pressure synthesis conditions DME flammability Besides CO2, CO or CH4 may be formed as a side product of steam reforming |
| DBT | 150–260 °C, 3–50 bar | Mostly Ru, Rh, and Pt-based catalysts (i.e., Ru/Al2O3, Rh/Al2O3, Pt/Al2O3) | 270–320 °C 1 bar | Pt- and Pd-based catalysts (i.e., Pt/Al2O3, Pd/C, Pt-Pd/Al2O3) | Hydrogenation may be performed under low pressure Lower hydrogenation temperatures compared with DME and MeOH synthesis Hydrogen-rich mixtures may be used as feedstock instead of pure H2 Possibility of cyclic hydrogenation and dehydrogenation Nontoxic and nonflammable | Impurities may be present in the hydrogenated mixture (i.e., benzyltoluene, benzene, toluene) Besides gaseous by-products (CH4, CO2), compounds such as MBT, benzene, xylenes, toluene, and benzylmethylfluorenes may be formed during dehydrogenation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawelczyk, E.; Łukasik, N.; Wysocka, I.; Rogala, A.; Gębicki, J. Recent Progress on Hydrogen Storage and Production Using Chemical Hydrogen Carriers. Energies 2022, 15, 4964. https://doi.org/10.3390/en15144964
Pawelczyk E, Łukasik N, Wysocka I, Rogala A, Gębicki J. Recent Progress on Hydrogen Storage and Production Using Chemical Hydrogen Carriers. Energies. 2022; 15(14):4964. https://doi.org/10.3390/en15144964
Chicago/Turabian StylePawelczyk, Ewelina, Natalia Łukasik, Izabela Wysocka, Andrzej Rogala, and Jacek Gębicki. 2022. "Recent Progress on Hydrogen Storage and Production Using Chemical Hydrogen Carriers" Energies 15, no. 14: 4964. https://doi.org/10.3390/en15144964