
Treponema denticola Has the Potential to Cause Neurodegeneration in the Midbrain via the Periodontal Route of Infection—Narrative Review
{kind=link}
Abstract
:1. Introduction
2. T. denticola and Its Virulence Factors
3. The Mechanism of Peripheral Neurodegeneration via T. denticola
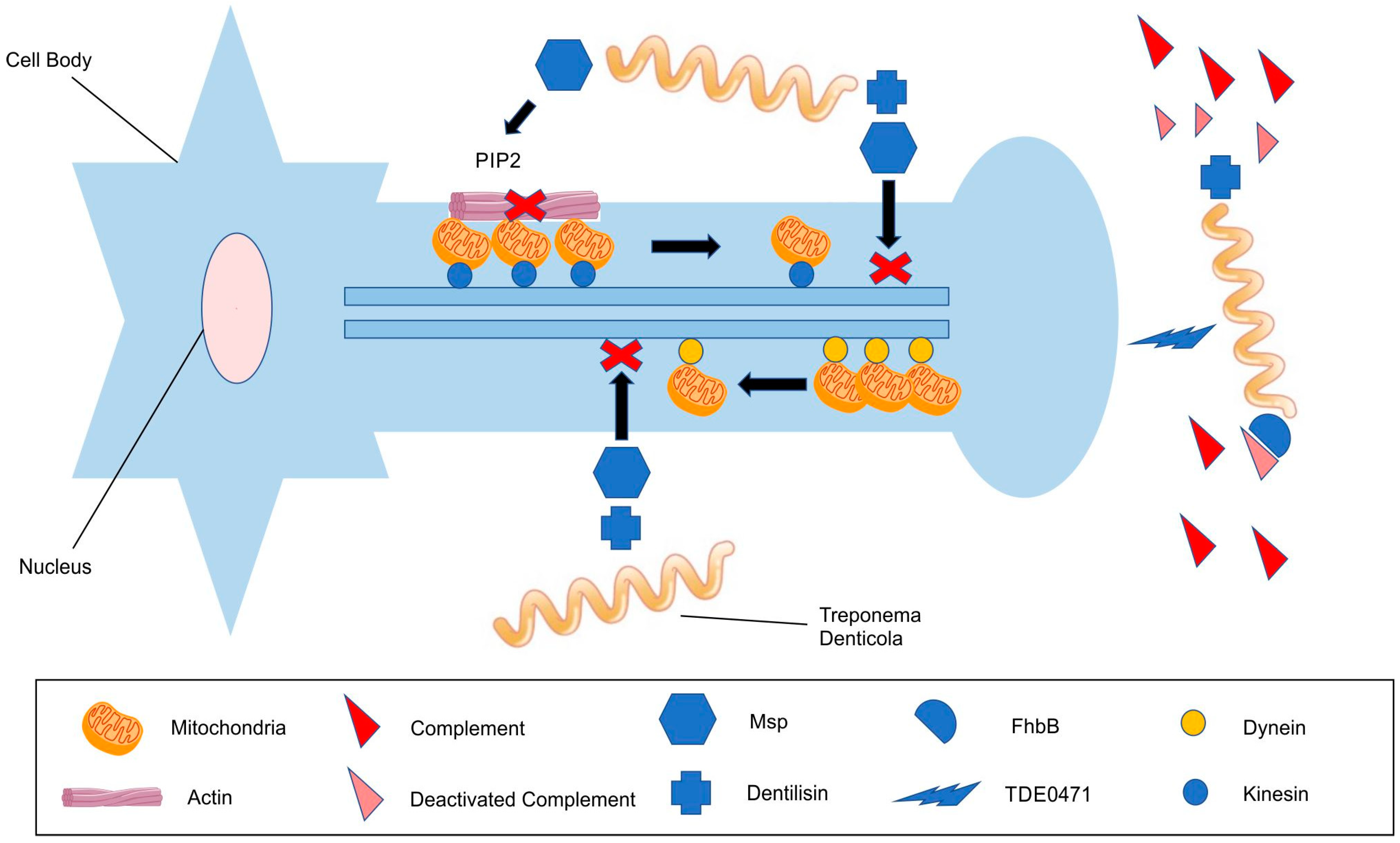
3.1. The Role of T. denticola in Peripheral Nerve Damage
3.2. The Evasion of the Complement Cascade
3.3. Cytoskeletal Impairment
4. The Role of Treponema denticola in Central Neurodegeneration
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ball, J.; Darby, I. Mental health and periodontal and peri-implant diseases. Periodontology 2000, 90, 106–124. [Google Scholar] [CrossRef]
- Delwel, S.; Binnekade, T.T.; Perez, R.S.G.M.; Hertogh, C.M.P.M.; Scherder, E.J.A.; Lobbezoo, F. Oral hygiene and oral health in older people with dementia: A comprehensive review with focus on oral soft tissues. Clin. Oral. Investig. 2018, 22, 93–108. [Google Scholar] [CrossRef] [Green Version]
- Fereshtehnejad, S.M.; Lökk, J.; Wimo, A.; Eriksdotter, M. No Significant Difference in Cognitive Decline and Mortality between Parkinson’s Disease Dementia and Dementia with Lewy Bodies: Naturalistic Longitudinal Data from the Swedish Dementia Registry. J. Park. Dis. 2018, 8, 553–561. [Google Scholar] [CrossRef]
- Panzarella, V.; Mauceri, R.; Baschi, R.; Maniscalco, L.; Campisi, G.; Monastero, R. Oral Health Status in Subjects with Amnestic Mild Cognitive Impairment and Alzheimer’s Disease: Data from the Zabút Aging Project. J. Alzheimers Dis. 2022, 87, 173–183. [Google Scholar] [CrossRef]
- Ide, M.; Harris, M.; Stevens, A.; Sussams, R.; Hopkins, V.; Culliford, D.; Fuller, J.; Ibbett, P.; Raybould, R.; Thomas, R.; et al. Periodontitis and Cognitive Decline in Alzheimer’s Disease. PLoS ONE 2016, 11, e0151081. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Oh, J.K.; Wee, J.H.; Kim, Y.H.; Byun, S.H.; Choi, H.G. Association between Tooth Loss and Alzheimer’s Disease in a Nested Case-Control Study Based on a National Health Screening Cohort. J. Clin. Med. 2021, 10, 3763. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.; Singhrao, S.K. Periodontitis to dementia or converse? Br. Dent. J. 2019, 226, 634. [Google Scholar] [CrossRef]
- Harding, A.; Singhrao, S.K. Periodontitis and Dementia: A bidirectional relationship? Br. Dent. J. 2022, 101, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Singhrao, S.K.; Harding, A.; Chukkapalli, S.; Olsen, I.; Kesavalu, L.; Crean, S. Apolipoprotein E related co-morbidities and Alzheimer’s disease. J. Alzheimers Dis. 2016, 51, 935–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, C.J.; France, J.; Crean, S.; Singhrao, S.K. The Porphyromonas gingivalis/host interactome shows enrichment in GWASdb genes related to Alzheimer’s disease, diabetes and cardiovascular diseases. Front. Aging Neurosci. 2017, 9, 408. [Google Scholar] [CrossRef] [Green Version]
- Harding, A.; Gonder, U.; Robinson, S.J.; Crean, S.; Singhrao, S.K. Exploring the association between Alzheimer’s disease, oral health, microbial endocrinology and nutrition. Front. Aging Neurosci. 2017, 9, 398. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Tomlinson, B.; Irving, D.; Blessed, G. Cell loss in the locus coeruleus in senile dementia of Alzheimer type. J. Neurol. Sci. 1981, 49, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Bondareff, W.; Mountjoy, C.Q.; Roth, M. Loss of neurons of origin of the adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology 1982, 32, 164–168. [Google Scholar] [CrossRef]
- Bondareff, W.; Mountjoy, C.Q.; Roth, M.; Rossor, M.N.; Iversen, L.L.; Reynolds, G.P. Age and histopathologic heterogeneity in Alzheimer’s disease: Evidence for subtypes. Arch. Gen. Psychiatry 1987, 44, 412–417. [Google Scholar] [CrossRef]
- German, D.C.; Manaye, K.F.; White, C.L.; Woodward, D.J.; McIntire, D.D.; Smith, W.K.; Kalaria, R.N.; Mann, D. Disease-specific patterns of locus coeruleus cell loss. Ann. Neurol. 1992, 32, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Kuramoto, E.; Dhar, A.; Wang, R.P.; Seki, H.; Iwai, H.; Yamanaka, A.; Matsumoto, S.E.; Hara, H.; Michikawa, M.; et al. Neurodegeneration of Trigeminal Mesencephalic Neurons by the Tooth Loss Triggers the Progression of Alzheimer’s Disease in 3×Tg-AD Model Mice. J. Alzheimers Dis. 2020, 76, 1443–1459. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Ryskalin, L.; Ruffoli, R.; Biagioni, F.; Limanaqi, F.; Ferrucci, M.; Busceti, C.L.; Bonuccelli, U.; Fornai, F. The neuroanatomy of the reticular nucleus locus coeruleus in Alzheimer’s disease. Front. Neuroanat. 2017, 11, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolfsson, R.; Gottfries, C.; Roos, B.; Winblad, B. Changes in the brain catecholamines in patients with dementia of Alzheimer type. Br. J. Psychiatry 1979, 135, 216–223. [Google Scholar] [CrossRef]
- Kalinin, S.; Feinstein, D.L.; Xu, H.L.; Huesa, G.; Pellegrino, D.A.; Galea, E. Degeneration of noradrenergic fibres from the locus coeruleus causes tight-junction disorganisation in the rat brain. Eur. J. Neurosci. 2006, 24, 3393–3400. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Local neuroinflammation and the progression of Alzheimer’s disease. J. Neurovirol. 2002, 8, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marien, M.R.; Colpaert, F.C.; Rosenquist, A.C. Noradrenergic mechanisms in neurodegenerative diseases: A theory. Brain Res. Rev. 2004, 45, 38–78. [Google Scholar] [CrossRef]
- Gyoneva, S.; Traynelis, S.F. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J. Biol. Chem. 2013, 288, 15291–15302. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Ramanathan, M.; Jacobs, A.H.; Dumitrescu-Ozimek, L.; Bilkei-Gorzo, A.; Debeir, T.; Sastre, M.; Galldiks, N.; Zimmer, A.; Hoehn, M.; et al. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J. Neurosci. 2006, 26, 1343–1354. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Wang, X.; Kong, W.; Jiang, Q. Tooth Loss Suppresses Hippocampal Neurogenesis and Leads to Cognitive Dysfunction in Juvenile Sprague-Dawley Rats. Front. Neurosci. 2022, 16, 839622. [Google Scholar] [CrossRef]
- Jiang, Q.S.; Liang, Z.L.; Wu, M.J.; Feng, L.; Liu, L.L.; Zhang, J.J. Reduced brain-derived neurotrophic factor expression in cortex and hippocampus involved in the learning and memory deficit in molarless SAMP8 mice. Chin. Med. J. 2011, 124, 1540–1544. [Google Scholar]
- Alvarado-Mallart, M.R.; Batini, C.; Buisseret-Delmas, C.; Corvisier, J. Trigeminal representations of the masticatory and extraocular proprioceptors as revealed by horseradish peroxidase retrograde transport. Exp. Brain Res. 1975, 23, 167–179. [Google Scholar] [CrossRef]
- Jacquin, M.F.; Zeigler, H.P. Trigeminal orosensation and ingestive behavior in the rat. Behav. Neurosci. 1983, 97, 62–97. [Google Scholar] [CrossRef]
- Shigenaga, Y.; Sera, M.; Nishimori, T.; Suemune, S.; Nishimura, M.; Yoshida, A.; Tsuru, K. The central projection of masticatory afferent fibers to the trigeminal sensory nuclear complex and upper cervical spinal cord. J. Comp. Neurol. 1988, 268, 489–507. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, Y.; Mitsuhiro, Y.; Yoshida, A.; Cao, C.Q.; Tsuru, H. Morphology of single mesencephalic trigeminal neurons innervating masseter muscle of the cat. Brain Res. 1988, 445, 392–399. [Google Scholar] [CrossRef]
- Raappana, P.; Arvidsson, J. The reaction of mesencephalic trigeminal neurons to peripheral nerve transection in the adult rat. Exp. Brain Res. 1992, 90, 567–571. [Google Scholar] [CrossRef]
- Linden, R.W.; Scott, B.J. Distribution of mesencephalic nucleus and trigeminal ganglion mechanoreceptors in the periodontal ligament of the cat. J. Physiol. 1989, 410, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Muramoto, T.; Takano, Y.; Soma, K. Time-related changes in periodontal mechanoreceptors in rat molars after the loss of occlusal stimuli. Arch. Histol. Cytol. 2000, 63, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lexomboon, D.; Trulsson, M.; Wårdh, I.; Parker, M.G. Chewing ability and tooth loss: Association with cognitive impairment in an elderly population study. J. Am. Geriatr. Soc. 2012, 60, 1951–1956. [Google Scholar] [CrossRef]
- Weijenberg, R.A.; Scherder, E.J.; Lobbezoo, F. Mastication for the mind—The relationship between mastication and cognition in ageing and dementia. Neurosci. Biobehav. Rev. 2011, 35, 483–497. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, H.; Eribe, E.R.; Singhrao, S.K.; Olsen, I. High Throughput Sequencing Detect Gingivitis and Periodontal Oral Bacteria in Alzheimer’s Disease Autopsy Brains. J. Neurosci. Res. 2019, 1, 3. [Google Scholar] [CrossRef]
- Ng, H.M.; Slakeski, N.; Butler, C.A.; Veith, P.D.; Chen, Y.Y.; Liu, S.W.; Hoffmann, B.; Dashper, S.G.; Reynolds, E.C. The Role of Treponema denticola Motility in Synergistic Biofilm Formation with Porphyromonas gingivalis. Front. Cell Infect. Microbiol. 2019, 9, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dashper, S.G.; Seers, C.A.; Tan, K.H.; Reynolds, E.C. Virulence factors of the oral spirochete Treponema denticola. J. Dent. Res. 2011, 90, 691–703. [Google Scholar] [CrossRef]
- Oakes, S.G.; Repesh, L.A.; Pozos, R.S.; Fitzgerald, T.J. Electrophysiological dysfunction and cellular disruption of sensory neurones during incubation with Treponema pallidum. Br. J. Vener. Dis. 1982, 58, 220–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foschi, F.; Izard, J.; Sasaki, H.; Sambri, V.; Prati, C.; Müller, R.; Stashenko, P. Treponema denticola in disseminating endodontic infections. J. Dent. Res. 2006, 85, 761–765. [Google Scholar] [CrossRef] [Green Version]
- Holt, S.C.; Ebersole, J.L. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: The “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontology 2000 2005, 38, 72–122. [Google Scholar] [CrossRef]
- Robertson, D.; Smith, A.J. The microbiology of the acute dental abscess. J. Med. Microbiol. 2009, 58, 155–162. [Google Scholar] [CrossRef]
- Paster, B.J.; Dewhirst, F.E. Phylogenetic foundation of spirochetes. J. Mol. Microbiol. Biotechnol. 2000, 2, 341–344. [Google Scholar]
- Seshadri, R.; Myers, G.S.; Tettelin, H.; Eisen, J.A.; Heidelberg, J.F.; Dodson, R.J.; Paulsen, I.T. Comparison of the genome of the oral pathogen Treponema denticola with other spirochete genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 5646–5651. [Google Scholar] [CrossRef] [Green Version]
- Lamont, R.J.; Jenkinson, H.F. Life below the gum line: Pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 1998, 62, 1244–1263. [Google Scholar] [CrossRef] [Green Version]
- Socransky, S.; Haffajee, A.; Cugini, M.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.J.; Dashper, S.G.; Darby, I.B.; Adams, G.G.; Hoffmann, B.; Reynolds, E.C. Progression of chronic periodontitis can be predicted by the levels of Porphyromonas gingivalis and Treponema denticola in subgingival plaque. Oral. Microbiol. Immunol. 2009, 24, 469–477. [Google Scholar] [CrossRef]
- Zijnge, V.; van Leeuwen, M.B.; Degener, J.E.; Abbas, F.; Thurnheer, T.; Gmür, R.; Harmsen, H.J. Oral biofilm architecture on natural teeth. PLoS ONE 2010, 5, e9321. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Wang, X.; Ma, Q.; Zhang, X.S.; Wood, T.K. Toxin-antitoxin systems in Escherichia coli influence biofilm formation through YjgK (TabA) and fimbriae. J. Bacteriol. 2009, 191, 1258–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct. 2009, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, H.L.; Dashper, S.G.; Catmull, D.V.; Paolini, R.A.; Cleal, S.M.; Slakeski, N.; Tan, K.H.; Reynolds, E.C. Treponema denticola biofilm-induced expression of a bacteriophage, toxin-antitoxin systems and transposases. Microbiology 2010, 156, 774–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenno, J.C.; McBride, B.C. Virulence factors of oral treponemes. Anaerobe 1998, 4, 1–17. [Google Scholar] [CrossRef]
- Ellen, R.P. Virulence determinants of oral Treponemes. In Pathogenic Treponema: Molecular and Cellular Biology; Radolf, J.D., Lukehart, S.A., Eds.; Caister Academic Press: Wymondham, UK; Norfolk, UK, 2006; pp. 357–386. [Google Scholar]
- Ohta, K.; Makinen, K.K.; Loesche, W.J. Purification and characterization of an enzyme produced by Treponema denticola capable of hydrolyzing synthetic trypsin substrates. Infect. Immun. 1986, 53, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenno, J.C.; Lee, S.Y.; Bayer, C.H.; Ning, Y. The opdB locus encodes the trypsin-like peptidase activity of Treponema denticola. Infect. Immun. 2001, 69, 6193–6200. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Fenno, J.C. Expression of Treponema denticola oligopeptidase B in Escherichia coli. Curr. Microbiol. 2004, 48, 379–382. [Google Scholar] [CrossRef] [Green Version]
- Setubal, J.C.; Reis, M.; Matsunaga, J.; Haake, D.A. Lipoprotein computational prediction in spirochaetal genomes. Microbiology 2006, 152, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Wensink, J.; Witholt, B. Outer-membrane vesicles released by normally growing Escherichia coli contain very little lipoprotein. Eur. J. Biochem. 1981, 116, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Kowashi, Y.; Demuth, D.R. Outer membrane-like vesicles secreted by Actinobacillus actinomycetemcomitans are enriched in leukotoxin. Microb. Pathog. 2002, 32, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wai, S.N.; Lindmark, B.; Söderblom, T.; Takade, A.; Westermark, M.; Oscarsson, J.; Jass, J.; Richter-Dahlfors, A.; Mizunoe, Y.; Uhlin, B.E. Vesicle-mediated export and assembly of pore-forming oligomers of the enterobacterial ClyA cytotoxin. Cell 2003, 115, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Uitto, V.J.; Grenier, D.A.N.I.E.L.; Chan, E.C.; McBride, B.C. Isolation of a chymotrypsinlike enzyme from Treponema denticola. Infect. Immun. 1988, 56, 2717–2722. [Google Scholar] [CrossRef] [Green Version]
- Grenier, D.; Uitto, V.J.; McBride, B.C. Cellular location of a Treponema denticola chymotrypsinlike protease and importance of the protease in migration through the basement membrane. Infect. Immun. 1990, 58, 347–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkinen, P.L.; Mäkinen, K.K.; Syed, S.A. Role of the chymotrypsin-like membrane-associated proteinase from Treponema denticola ATCC 35405 in inactivation of bioactive peptides. Infect. Immun. 1995, 63, 3567–3575. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, K.; Miura, T.; Kuramitsu, H.K.; Okuda, K. Characterization of the Treponema denticola prtP gene encoding a prolyl-phenylalanine-specific protease (dentilisin). Infect. Immun. 1996, 64, 5178–5186. [Google Scholar] [CrossRef] [Green Version]
- Beauséjour, A.; Deslauriers, N.; Grenier, D. Activation of the interleukin-1beta precursor by Treponema denticola: A potential role in chronic inflammatory periodontal diseases. Infect. Immun. 1997, 65, 3199–3202. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 2001, 45, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, A.; Wood, T.K. Bacterial quorum sensing: Signals, circuits, and implications for biofilms and disease. Annu. Rev. Biomed. Eng. 2008, 10, 145–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, A.M.; Jenkinson, H.F.; Woodward, M.J.; Dymock, D. Binding Properties and Adhesion-Mediating Regions of the Major Sheath Protein of Treponema denticola ATCC 35405. Infect. Immun. 2005, 73, 2891–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenno, J.C.; Tamura, M.; Hannam, P.M.; Wong, G.W.; Chan, R.A.; McBride, B.C. Identification of a Treponema denticola OppA homologue that binds host proteins present in the subgingival environment. Infect. Immun. 2000, 68, 1884–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devoe, I.W.; Gilchrist, J.E. Release of endotoxin in the form of cell wall blebs during in vitro growth of Neisseria meningitidis. J. Exp. Med. 1973, 138, 1156–1167. [Google Scholar] [CrossRef] [Green Version]
- Grenier, D.; Mayrand, D. Functional characterization of extracellular vesicles produced by Bacteroides gingivalis. Infect. Immun. 1987, 55, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimasoni, G.; McBride, B.C. Adherence of Treponema denticola to modified hydroxyapatite. J. Dent. Res. 1987, 66, 1727–1729. [Google Scholar] [CrossRef]
- Weinberg, A.; Holt, S.C. Chemical and biological activities of a 64-kilodalton outer sheath protein from Treponema denticola strains. J. Bacteriol. 1991, 173, 6935–6947. [Google Scholar] [CrossRef] [Green Version]
- Kuehn, M.J.; Kesty, N.C. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 2005, 19, 2645–2655. [Google Scholar] [CrossRef] [Green Version]
- Chi, B.; Qi, M.; Kuramitsu, H.K. Role of dentilisin in Treponema denticola epithelial cell layer penetration. Res. Microbiol. 2003, 154, 637–643. [Google Scholar] [CrossRef]
- Miyamoto, M.; Ishihara, K.; Okuda, K. The Treponema denticola surface protease dentilisin degrades interleukin-1 beta (IL-1 beta), IL-6, and tumor necrosis factor alpha. Infect. Immun. 2006, 74, 2462–2467. [Google Scholar] [CrossRef] [Green Version]
- Okuda, T.; Kimizuka, R.; Miyamoto, M.; Kato, T.; Yamada, S.; Okuda, K.; Ishihara, K. Treponema denticola induces interleukin-8 and macrophage chemoattractant protein 1 production in human umbilical vein epithelial cells. Microbes Infect. 2007, 9, 907–913. [Google Scholar] [CrossRef] [Green Version]
- Dawson, J.R.; Ellen, R.P. Tip-oriented adherence of Treponema denticola to fibronectin. Infect. Immun. 1990, 58, 3924–3928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repesh, L.A.; Fitzgerald, T.J.; Oakes, S.G.; Pozos, R.S. Scanning electron microscopy of the attachment of Treponema pallidum to nerve cells in vitro. Br. J. Vener. Dis. 1982, 58, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secher, L.; Weismann, K.; Kobayasi, T. Treponema pallidum in peripheral nerve tissue of syphilitic chancres. Acta Derm. Venereol. 1982, 62, 407–411. [Google Scholar]
- Wrzolkowa, T.; Kozakiewicz, J. Ultrastructure of vascular and connective tissue changes in primary syphilis. Br. J. Vener. Dis. 1980, 56, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovcinnikov, N.M.; Delektorskij, V.V. Further studies of the morphology of Treponema pallidum under the electron microscope. Br. J. Vener. Dis. 1969, 45, 87–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovcinnikov, N.M.; Delektorskij, V.V. Treponema pallidum in nerve fibres. Br. J. Vener. Dis. 1975, 51, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Singhrao, S.K.; Neal, J.W.; Morgan, B.P.; Gasque, P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp. Neurol. 1999, 159, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Gasque, P. Complement: A unique innate immune sensor for danger signals. Mol. Immunol. 2004, 41, 1089–1098. [Google Scholar] [CrossRef]
- Gasque, P.; Neal, J.W.; Singhrao, S.K.; McGreal, E.P.; Dean, Y.D.; Van, B.J.; Morgan, B.P. Roles of the complement system in human neurodegenerative disorders: Proinflammatory and tissue remodeling activities. Mol. Neurobiol. 2002, 25, 1–17. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell. Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Dejanovic, B.; Huntley, M.A.; De Mazière, A.; Meilandt, W.J.; Wu, T.; Srinivasan, K.; Jiang, Z.; Gandham, V.; Friedman, B.A.; Ngu, A.; et al. Changes in the synaptic proteome in tauopathy and rescue of Tau-induced synapse loss by C1q antibodies. Neuron 2018, 100, 1322–1336. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Hanson, J.E. Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell. Rep. 2019, 28, 2111–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, I.; Singhrao, S.K. Is there a link between genetic defects in the complement cascade and Porphyromonas gingivalis in Alzheimer’s disease? J. Oral. Microbiol. 2020, 12, 167648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohlen, C.J.; Friedman, B.A.; Dejanovic, B.; Sheng, M. Microglia in Brain Development, Homeostasis, and Neurodegeneration. Annu. Rev. Genet. 2019, 53, 263–288. [Google Scholar] [CrossRef]
- Lee, J.D.; Woodruff, T.M. The emerging role of complement in neuromuscular disorders. Semin. Immunopathol. 2021, 43, 817–828. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDowell, J.V.; Frederick, J.; Stamm, L.; Marconi, R.T. Identification of the gene encoding the FhbB protein of Treponema denticola, a highly unique factor H-like protein 1 binding protein. Infect. Immun. 2007, 75, 1050–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Józsi, M.; Zipfel, P.F. Factor H family proteins and human diseases. Trends Immunol. 2008, 29, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Pangburn, M.K.; Schreiber, R.D.; Müller-Eberhard, H.J. Human complement C3b inactivator: Isolation, characterization, and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution. J. Exp. Med. 1977, 146, 257–270. [Google Scholar] [CrossRef] [Green Version]
- Ruddy, S.; Austen, K.F. C3 inactivator of man, I. Hemolytic measurement by the inactivation of cell bound C3. J. Immunol. 1969, 102, 533–543. [Google Scholar] [CrossRef]
- Miller, D.P.; Bell, J.K.; McDowell, J.V.; Conrad, D.H.; Burgner, J.W.; Héroux, A.; Marconi, R.T. Structure of factor H-binding protein B (FhbB) of the periopathogen, Treponema denticola: Insights into progression of periodontal disease. J. Biol. Chem. 2012, 287, 12715–12722. [Google Scholar] [CrossRef] [Green Version]
- Kurniyati, K.; Zhang, W.; Zhang, K.; Li, C. A surface-exposed neuraminidase affects complement resistance and virulence of the oral spirochaete Treponema denticola. Mol. Microbiol. 2013, 89, 842–856. [Google Scholar] [CrossRef] [Green Version]
- Visser, M.B.; Ellen, R.P. New insights into the emerging role of oral spirochaetes in periodontal disease. Clin. Microbiol. Infect. 2011, 17, 502–512. [Google Scholar] [CrossRef] [Green Version]
- Rottner, K.; Lommel, S.; Wehland, J.; Stradal, T.E. Pathogen-induced actin filament rearrangement in infectious diseases. J. Pathol. 2004, 204, 396–406. [Google Scholar] [CrossRef]
- Stebbins, C.E. Structural insights into bacterial modulation of the host cytoskeleton. Curr. Opin. Struct. Biol. 2004, 14, 731–740. [Google Scholar] [CrossRef]
- Ellen, R.P.; Galimanas, V.B. Spirochetes at the forefront of periodontal infections. Periodontology 2000 2005, 38, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Sela, M.N. Role of Treponema denticola in periodontal diseases. Crit. Rev. Oral. Biol. Med. 2001, 12, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Ho, A.C.; Lin, J.Y.; Batista da Silva, A.P.; Glogauer, M.; Ellen, R.P. Induction of de novo subcortical actin filament assembly by Treponema denticola major outer sheath protein. Infect. Immun. 2004, 72, 3650–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhães, M.A.; Sun, C.X.; Glogauer, M.; Ellen, R.P. The major outer sheath protein of Treponema denticola selectively inhibits Rac1 activation in murine neutrophils. Cell. Microbiol. 2008, 10, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Puthengady Thomas, B.; Sun, C.X.; Bajenova, E.; Ellen, R.P.; Glogauer, M. Modulation of human neutrophil functions in vitro by Treponema denticola major outer sheath protein. Infect. Immun. 2006, 74, 1954–1957. [Google Scholar] [CrossRef] [Green Version]
- Batista da Silva, A.P.; Lee, W.; Bajenova, E.; McCulloch, C.A.; Ellen, R.P. The major outer sheath protein of Treponema denticola inhibits the binding step of collagen phagocytosis in fibroblasts. Cell. Microbiol. 2004, 6, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ko, K.S.; Kapus, A.; McCulloch, C.A.; Ellen, R.P. A spirochete surface protein uncouples store-operated calcium channels in fibroblasts: A novel cytotoxic mechanism. J. Biol. Chem. 2001, 276, 23056–23064. [Google Scholar] [CrossRef] [Green Version]
- Visser, M.B.; Koh, A.; Glogauer, M.; Ellen, R.P. Treponema denticola major outer sheath protein induces actin assembly at free barbed ends by a PIP2-dependent uncapping mechanism in fibroblasts. PLoS ONE 2011, 6, e23736. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.; Condeelis, J. The cofilin activity cycle in lamellipodia and invadopodia. J. Cell. Biochem. 2009, 108, 1252–1262. [Google Scholar] [CrossRef] [Green Version]
- Uitto, V.J.; Pan, Y.M.; Leung, W.K.; Larjava, H.; Ellen, R.P.; Finlay, B.B.; McBride, B.C. Cytopathic effects of Treponema denticola chymotrypsin-like proteinase on migrating and stratified epithelial cells. Infect. Immun. 1995, 63, 3401–3410. [Google Scholar] [CrossRef] [Green Version]
- Chevalier-Larsen, E.; Holzbaur, E.L. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 2006, 1762, 1094–1108. [Google Scholar] [CrossRef] [Green Version]
- Perlson, E.; Jeong, G.B.; Ross, J.L.; Dixit, R.; Wallace, K.E.; Kalb, R.G.; Holzbaur, E.L. A switch in retrograde signaling from survival to stress in rapid-onset neurodegeneration. J. Neurosci. 2009, 29, 9903–9917. [Google Scholar] [CrossRef] [Green Version]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell. Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown RHJr Brown, H.; Tiwari, A.; Hayward, L.; Edgar, J.; et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, O.I.; Esposito, A.; Köhler, B.; Chen, C.W.; Shen, C.P.; Wu, G.H.; Butkevich, E.; Mandalapu, S.; Wenzel, D.; Wouters, F.S.; et al. Synaptic scaffolding protein SYD-2 clusters and activates kinesin-3 UNC-104 in C. elegans. Proc. Natl. Acad. Sci. USA 2009, 106, 19605–19610. [Google Scholar] [CrossRef] [Green Version]
- Leopold, P.L.; McDowall, A.W.; Pfister, K.K.; Bloom, G.S.; Brady, S.T. Association of kinesin with characterized membrane-bounded organelles. Cell Motil. Cytoskeleton 1992, 23, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Elluru, R.G.; Bloom, G.S.; Brady, S.T. Fast axonal transport of kinesin in the rat visual system: Functionality of kinesin heavy chain isoforms. Mol. Biol. Cell 1995, 6, 21–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.R.; Holzbaur, E.L. Cytoplasmic dynein/dynactin function and dysfunction in motor neurons. Int. J. Dev. Neurosci. 2006, 24, 103–111. [Google Scholar] [CrossRef]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelières, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, Y.; Setou, M. Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat. Neurosci. 2009, 12, 559–567. [Google Scholar] [CrossRef]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Vershinin, M.; Carter, B.C.; Razafsky, D.S.; King, S.J.; Gross, S.P. Multiple-motor based transport and its regulation by Tau. Proc. Natl. Acad. Sci. USA 2007, 104, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Morfini, G.; Pigino, G.; Beffert, U.; Busciglio, J.; Brady, S.T. Fast axonal transport misregulation and Alzheimer’s disease. Neuromolecular Med. 2002, 2, 89–99. [Google Scholar] [CrossRef]
- Rintoul, G.L.; Reynolds, I.J. Mitochondrial trafficking and morphology in neuronal injury. Biochim. Biophys. Acta 2010, 1802, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F. Mitochondria and neurodegeneration. Novartis Found. Symp. 2007, 287, 183–192; discussion 192–196. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Moraes, C.T. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum. Mol. Genet. 2009, 18, 1028–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, D.T.; Reynolds, I.J. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog. Neurobiol. 2006, 80, 241–268. [Google Scholar] [CrossRef]
- Morfini, G.A.; Burns, M.R.; Stenoien, D.L.; Brady, S.T. Chapter 8—Axonal Transport. In Wayne Albers, Donald L. Price, Basic Neurochemistry, 8th ed.; Scott, T., Brady, G.J., Siegel, R., Eds.; Academic Press: New York, NY, USA, 2012; p. 146. ISBN 9780123749475. [Google Scholar] [CrossRef]
- Trushina, E.; Dyer, R.B.; Badger JD 2nd Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; Van Houten, B.; et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef] [Green Version]
- Orr, A.L.; Li, S.; Wang, C.E.; Li, H.; Wang, J.; Rong, J.; Xu, X.; Mastroberardino, P.G.; Greenamyre, J.T.; Li, X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008, 28, 2783–2792. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.L.; Cleveland, D.W. Going new places using an old MAP: Tau, microtubules and human neurodegenerative disease. Curr. Opin. Cell Biol. 2001, 13, 41–48. [Google Scholar] [CrossRef]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef] [Green Version]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef]
- Stoothoff, W.H.; Johnson, G.V. Tau phosphorylation: Physiological and pathological consequences. Biochim. Biophys. Acta 2005, 1739, 280–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang S ran Ma, T.; Miranda, R.D.; Balestra, M.E.; Mahley, R.W.; Huang, Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18694–18699. [Google Scholar] [CrossRef] [Green Version]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Miklossy, J. Alzheimer’s disease—A neurospirochetosis. Anal. Evid. Follow. Koch’s Hill’s Criteria. J. Neuroinflamm. 2011, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Riviere, G.R.; Riviere, K.H.; Smith, K.S. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral. Microbiol. Immunol. 2002, 17, 113–118. [Google Scholar] [CrossRef] [PubMed]
- De Vries, H.E.; Kuiper, J.; de Boer, A.G.; Van Berkel, T.J.; Breimer, D.D. The blood-brain barrier in neuroinflammatory diseases. Pharm. Rev. 1997, 49, 143–155. [Google Scholar] [PubMed]
- Allen, H.B. Alzheimer’s Disease: Assessing the Role of Spirochetes, Biofilms, the Immune System, and Amyloid-β with Regard to Potential Treatment and Prevention. J. Alzheimers Dis. 2016, 53, 1271–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandclément, C.; Tannières, M.; Moréra, S.; Dessaux, Y.; Faure, D. Quorum quenching: Role in nature and applied developments. FEMS Microbiol. Rev. 2016, 40, 86–116. [Google Scholar] [CrossRef] [PubMed]
- Bzdrenga, J.; Daudé, D.; Rémy, B.; Jacquet, P.; Plener, L.; Elias, M.; Chabrière, E. Biotechnological applications of quorum quenching enzymes. Chem. Biol. Interact. 2017, 267, 104–115. [Google Scholar] [CrossRef]
- Rutherford, S.T.; Bassler, B.L. Bacterial quorum sensing: Its role in virulence and possibilities for its control. Cold Spring Harb. Perspect. Med. 2012, 2, a012427. [Google Scholar] [CrossRef]
- Whiteley, M.; Diggle, S.P.; Greenberg, E.P. Progress in and promise of bacterial quorum sensing research. Nature 2017, 551, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, H.B. A Novel Approach to the Treatment and Prevention of Alzheimer’s Disease Based on the Pathology and Microbiology. J. Alzheimers Dis. 2021, 84, 61–67. [Google Scholar] [CrossRef]
- Barnhart, M.M.; Chapman, M.R. Curli biogenesis and function. Annu. Rev. Microbiol. 2006, 60, 131–147. [Google Scholar] [CrossRef] [Green Version]
- Pisani, F.; Pisani, V.; Arcangeli, F.; Harding, A.; Singhrao, S.K. The Mechanistic Pathways of Periodontal Pathogens Entering the Brain: The Potential Role of Treponema denticola in Tracing Alzheimer’s Disease Pathology. Int. J. Environ. Res. Public Health 2022, 19, 9386. [Google Scholar] [CrossRef] [PubMed]
- Pisani, F.; Pisani, V.; Arcangeli, F.; Harding, A.; Singhrao, S.K. Locus Coeruleus Dysfunction and Trigeminal Mesencephalic Nucleus Degeneration: A Cue for Periodontal Infection Mediated Damage in Alzheimer’s Disease? Int. J. Environ. Res. Public Health 2023, 20, 1007. [Google Scholar] [CrossRef] [PubMed]
- Dhar, A.; Kuramoto, E.; Fukushima, M.; Iwai, H.; Yamanaka, A.; Goto, T. The Periodontium Damage Induces Neuronal Cell Death in the Trigeminal Mesencephalic Nucleus and Neurodegeneration in the Trigeminal Motor Nucleus in C57BL/6J Mice. Acta Histochem. Cytochem. 2021, 54, 11–19. [Google Scholar] [CrossRef]
- Venter, J.M.E.; Müller, E.E.; Mahlangu, M.P.; Kularatne, R.S. Treponema pallidum Macrolide Resistance and Molecular Epidemiology in Southern Africa, 2008 to 2018. J. Clin. Microbiol. 2021, 59, e0238520. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisani, F.; Pisani, V.; Arcangeli, F.; Harding, A.; Singhrao, S.K. Treponema denticola Has the Potential to Cause Neurodegeneration in the Midbrain via the Periodontal Route of Infection—Narrative Review. Int. J. Environ. Res. Public Health 2023, 20, 6049. https://doi.org/10.3390/ijerph20116049
Pisani F, Pisani V, Arcangeli F, Harding A, Singhrao SK. Treponema denticola Has the Potential to Cause Neurodegeneration in the Midbrain via the Periodontal Route of Infection—Narrative Review. International Journal of Environmental Research and Public Health. 2023; 20(11):6049. https://doi.org/10.3390/ijerph20116049
Chicago/Turabian StylePisani, Flavio, Valerio Pisani, Francesca Arcangeli, Alice Harding, and Simarjit Kaur Singhrao. 2023. "Treponema denticola Has the Potential to Cause Neurodegeneration in the Midbrain via the Periodontal Route of Infection—Narrative Review" International Journal of Environmental Research and Public Health 20, no. 11: 6049. https://doi.org/10.3390/ijerph20116049