
Lupus, DNA Methylation, and Air Pollution: A Malicious Triad
 , and
, and
Abstract
:1. Introduction
2. The Role of Genetic Predisposition in the Pathogenesis of SLE
3. The Role of Aberrant DNA Methylation in the Pathogenesis of SLE
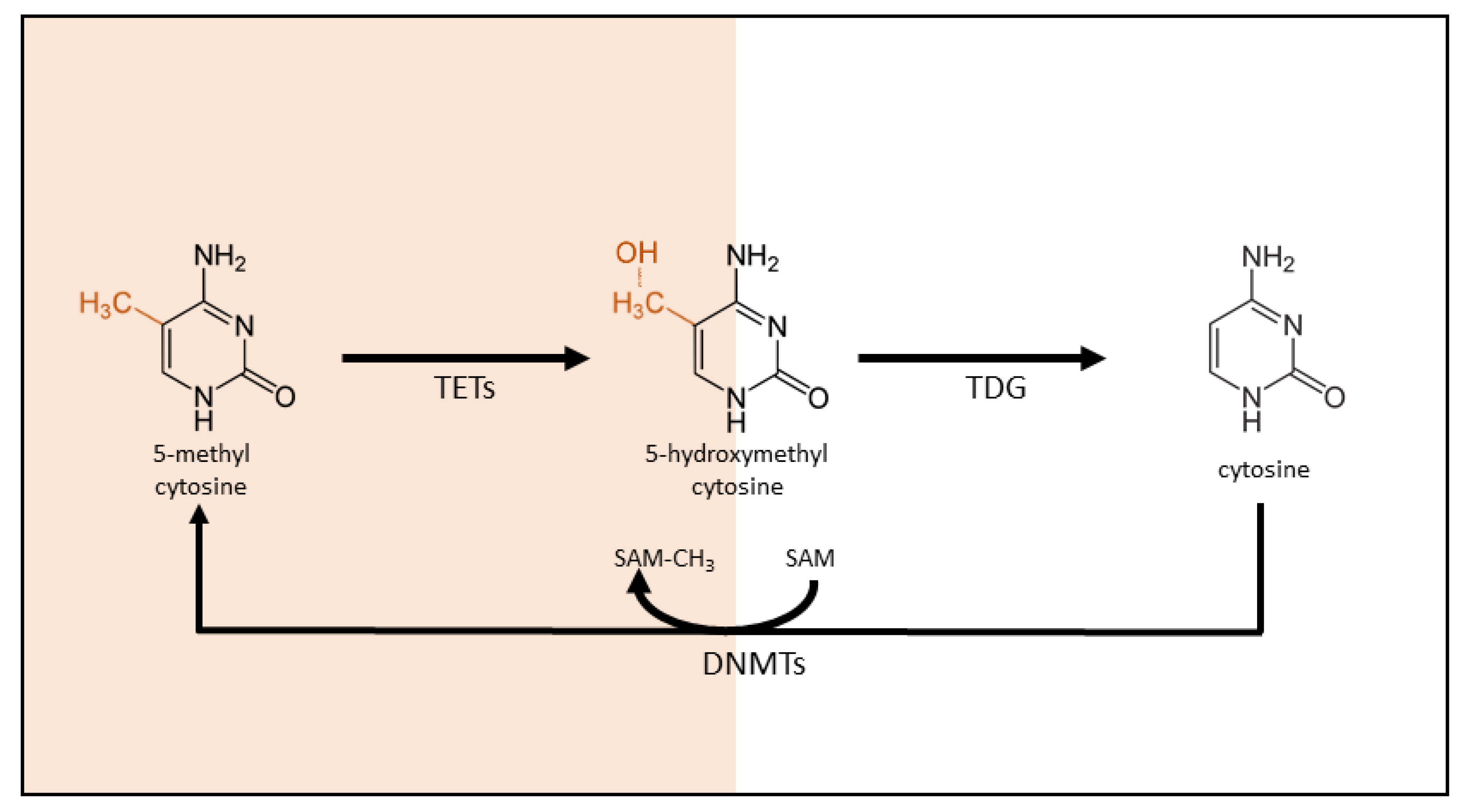
3.1. DNA Methylation: An Introduction
3.2. The Role of DNA Methylation in SLE
3.2.1. Interferon Gene Signature
3.2.2. Specific DNA Methylation in SLE T lymphocytes
3.2.3. Specific DNA Methylation in B lymphocytes
3.2.4. Specific DNA Methylation in Monocytes and Dendritic Cells
4. The Role of Air Pollution in the Pathogenesis of SLE
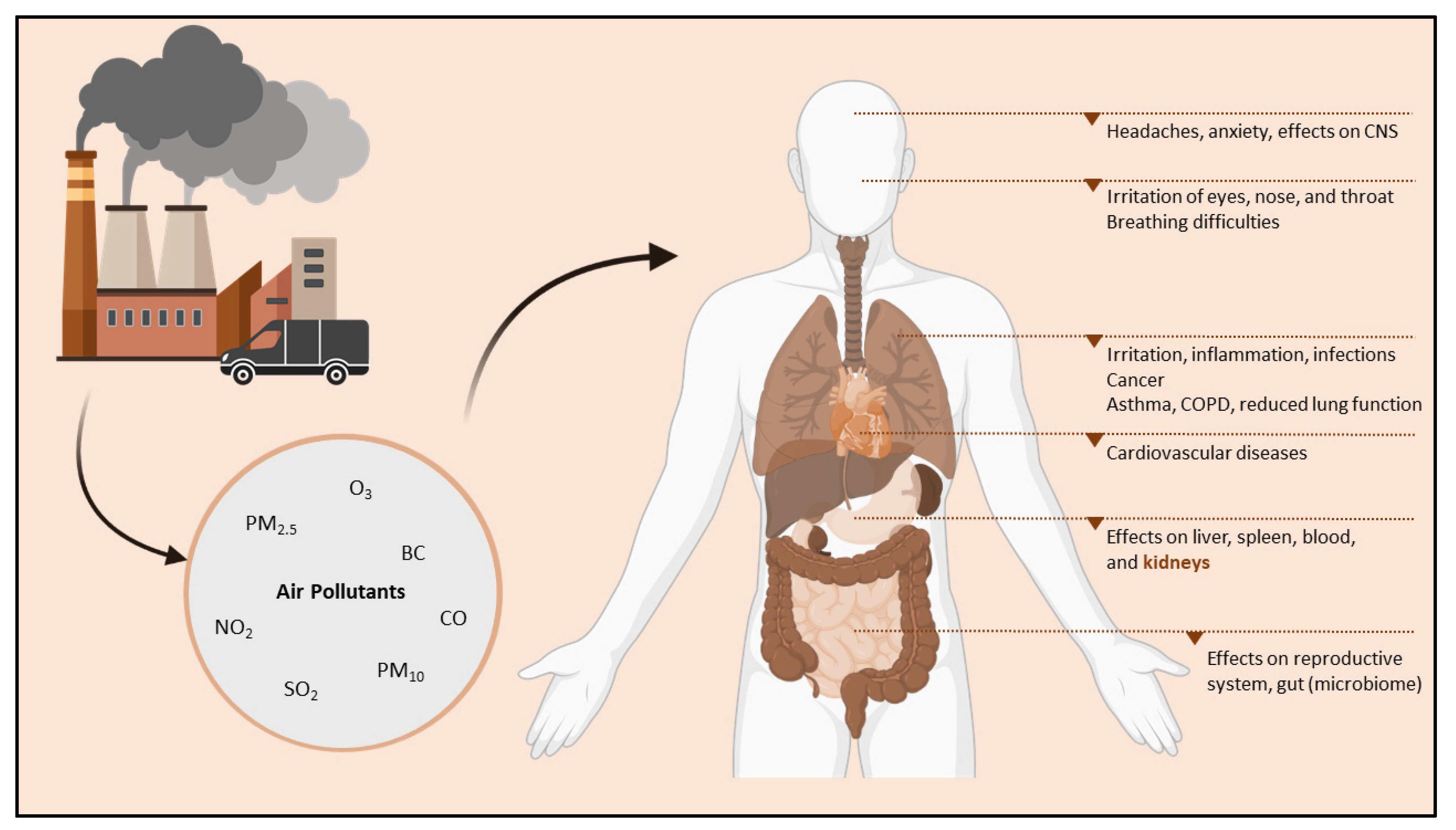
4.1. Air Pollution: An Introduction

4.2. Air Pollution and DNA Methylation
4.2.1. The Effects of Air Pollution on DNA Methylation In-Utero and during Childhood
4.2.2. The Effects of Air Pollution on DNA Methylation in Adults
4.3. Air Pollution and SLE Development

{kind=link}
{kind=link}
| Author | SLE Outcome Investigated | Environmental Factor Investigated | Main Findings |
|---|---|---|---|
| Alves et al. [157] | SLEDAI-2K score | 7-day moving average of PM2.5 exposure | A significant increase in the risk of a SLEDAI-2K score ≥8 |
| Goulart et al. [158] | Nephritis Anti-dsDNA Serum markers, i.e., C3 Urinary markers | PM2.5, NO2 exposure | PM2.5 exposure caused an increased risk of nephritis and rendered positive anti-dsDNA results PM2.5 and NO2 exposure caused a decrease in serum C3 and an increase in 24 h urinary protein |
| Conde et al. [159] | SLE development | PM10, SO2, NO2, O3, and CO exposure | All investigated environmental factors increased the risk of developing SLE during childhood |
| Mai et al. [160] | Incidence of childhood-onset SLE | PM2.5 exposure | Incidence of childhood-onset SLE increased almost 5-fold comparing the fourth quartile to the first quartile of PM2.5 exposure |
| Campos et al. [161] | Disease activity in childhood-onset SLE | PM10, SO2, NO2, O3, and CO exposure | PM10, CO, and NO2 exposure increases were associated with an increased risk of ≥8 SLEDAI-2K score, O3 and SO2 exposure had not apparent effect |
| Bernatsky et al. [162] | Systematic autoimmune rheumatic diseases (SARDs), including SLE | PM2.5 exposure | PM2.5 exposure caused a higher risk of developing SARDs, with a higher probability among females |
| Jung et al. [163] | SLE development | PM10, SO2, NO2, O3, and CO exposure | Positive associations were found for SLE development with increased NO2, CO, and PM2.5 exposure, while negative associations were demonstrated for O3 and SO2 exposure |
| Cakmak et al. [164] | Hospital admissions with SLE as the primary diagnosis | PM10, SO2, and CO exposure | All investigated environmental factors were associated with increased hospital admissions with a primary diagnosis of SLE |
| Zhao et al. [165] | Hospital admissions due to SLE Relapses of SLE | PM10, SO2, and NO2 exposure | More hospital admissions for SLE were observed with increased exposure to PM2.5, NO2, and SO2. Increased PM2.5 exposure caused an increased risk of SLE relapse High NO2 and SO2 exposure levels were associated with an increased risk of first-time admission for SLE |
| Bernatsky et al. [166] | SLEDAI-2K score Biomarkers for disease flare, i.e., urinary casts anti-dsDNA | PM2.5 exposure | Urinary casts and anti-dsDNA were associated with short-term PM2.5 variations shortly before the clinical visit(s) |
| Lanata et al. [167] | Hypomethylation in SLE | PM2.5 exposure | Three methylation sites were significantly hypomethylated in SLE patients residing close to a major highway, of which all three sites belonged to a single gene, UBE2U. |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schur, P.H.; Hahn, B.H. Epidemiology and Pathogenesis of Systemic Lupus Erythematosus; UpToDate: Waltham, MA, USA, 2011. [Google Scholar]
- Yap, D.Y.; Tang, C.S.; Ma, M.K.; Lam, M.F.; Chan, T.M. Survival analysis and causes of mortality in patients with lupus nephritis. Nephrol. Dial. Transplant. 2012, 27, 3248–3254. [Google Scholar] [CrossRef] [Green Version]
- Mccarty, D.J.; Manzi, S.; Medsger, T.A., Jr.; Ramsey-Goldman, R.; Laporte, R.E.; Kwoh, C.K. Incidence of systemic lupus erythematosus race and gender differences. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 1995, 38, 1260–1270. [Google Scholar] [CrossRef]
- Fortuna, G.; Brennan, M.T. Systemic lupus erythematosus: Epidemiology, pathophysiology, manifestations, and management. Dent. Clin. 2013, 57, 631–655. [Google Scholar]
- Blanco, P.; Palucka, A.K.; Gill, M.; Pascual, V.; Banchereau, J. Induction of Dendritic Cell Differentiation by IFN-α in Systemic Lupus Erythematosus. Science 2001, 294, 1540–1543. [Google Scholar] [CrossRef]
- Bronson, P.G.; Chaivorapol, C.; Ortmann, W.; Behrens, T.W.; Graham, R.R. The genetics of type I interferon in systemic lupus erythematosus. Curr. Opin. Immunol. 2012, 24, 530–537. [Google Scholar] [CrossRef]
- Mathian, A.; Gallegos, M.; Pascual, V.; Banchereau, J.; Koutouzov, S. Interferon-α induces unabated production of short-lived plasma cells in pre-autoimmune lupus-prone (NZB× NZW) F1 mice but not in BALB/c mice. Eur. J. Immunol. 2011, 41, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Shlomchik, M.J.; Craft, J.E.; Mamula, M.J. From T to B and back again: Positive feedback in systemic autoimmune disease. Nat. Rev. Immunol. 2001, 1, 147–153. [Google Scholar] [CrossRef]
- Taylor, K.E.; Chung, S.A.; Graham, R.R.; Ortmann, W.A.; Lee, A.T.; Langefeld, C.D.; Jacob, C.O.; Kamboh, M.I.; Alarcón-Riquelme, M.E.; Tsao, B.P.; et al. Risk Alleles for Systemic Lupus Erythematosus in a Large Case-Control Collection and Associations with Clinical Subphenotypes. PLoS Genet. 2011, 7, e1001311. [Google Scholar] [CrossRef] [Green Version]
- Rekvig, O.P.; van der Vlag, J. The pathogenesis and diagnosis of systemic lupus erythematosus: Still not resolved. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2014; Volume 36, pp. 301–311. [Google Scholar]
- Nagy, G.; Koncz, A.; Perl, A. T- and B-Cell Abnormalities in Systemic Lupus Erythematosus. Crit. Rev. Immunol. 2005, 25, 123–140. [Google Scholar] [CrossRef]
- Wu, H.; Fu, S.; Zhao, M.; Lu, L.; Lu, Q. Dysregulation of Cell Death and Its Epigenetic Mechanisms in Systemic Lupus Erythematosus. Molecules 2016, 22, 30. [Google Scholar] [CrossRef] [Green Version]
- Hedrich, C.M. Epigenetics in SLE. Curr. Rheumatol. Rep. 2017, 19, 58. [Google Scholar] [CrossRef]
- Kuo, C.-F.; Grainge, M.J.; Valdes, A.M.; See, L.-C.; Luo, S.-F.; Yu, K.-H.; Zhang, W.; Doherty, M. Familial Aggregation of Systemic Lupus Erythematosus and Coaggregation of Autoimmune Diseases in Affected Families. JAMA Intern. Med. 2015, 175, 1518–1526. [Google Scholar] [CrossRef]
- Deafen, D.; Escalante, A.; Weinrib, L.; Horwitz, D.; Bachman, B.; Roy-Burman, P.; Mack, T.M. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 1992, 35, 311–318. [Google Scholar]
- Belot, A.; Cimaz, R. Monogenic forms of systemic lupus erythematosus: New insights into SLE pathogenesis. Pediatr. Rheumatol. 2012, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Gaipl, U.S.; Voll, R.E.; Sheriff, A.; Franz, S.; Kalden, J.R.; Herrmann, M. Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun. Rev. 2005, 4, 189–194. [Google Scholar] [CrossRef]
- Banchereau, J.; Pascual, V. Type I Interferon in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Immunity 2006, 25, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Costa-Reis, P.; Sullivan, K.E. Genetics and Epigenetics of Systemic Lupus Erythematosus. Curr. Rheumatol. Rep. 2013, 15, 369. [Google Scholar] [CrossRef]
- Armstrong, D.L.; Zidovetzki, R.; E Alarcón-Riquelme, M.; Tsao, B.P.; A Criswell, L.; Kimberly, R.P.; Harley, J.B.; Sivils, K.L.; Vyse, T.J.; Gaffney, P.M.; et al. GWAS identifies novel SLE susceptibility genes and explains the association of the HLA region. Genes Immun. 2014, 15, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Tsao, B.P. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat. Rev. Rheumatol. 2010, 6, 683–692. [Google Scholar] [CrossRef]
- Absher, D.M.; Li, X.; Waite, L.L.; Gibson, A.; Roberts, K.; Edberg, J.; Chatham, W.W.; Kimberly, R. Genome-Wide DNA Methylation Analysis of Systemic Lupus Erythematosus Reveals Persistent Hypomethylation of Interferon Genes and Compositional Changes to CD4+ T-cell Populations. PLoS Genet. 2013, 9, e1003678. [Google Scholar] [CrossRef] [Green Version]
- Coit, P.; Jeffries, M.; Altorok, N.; Dozmorov, M.; Koelsch, K.A.; Wren, J.D.; Merrill, J.T.; McCune, W.J.; Sawalha, A.H. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naïve CD4+ T cells from lupus patients. J. Autoimmun. 2013, 43, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Ulff-Møller, C.J.; Asmar, F.; Liu, Y.; Svendsen, A.J.; Busato, F.; Grønbæk, K.; Tost, J.; Jacobsen, S. Twin DNA Methylation Profiling Reveals Flare-Dependent Interferon Signature and B Cell Promoter Hypermethylation in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2018, 70, 878–890. [Google Scholar] [CrossRef]
- Dieudonné, Y.; Gies, V.; Guffroy, A.; Keime, C.; Bird, A.K.; Liesveld, J.; Barnas, J.; Poindron, V.; Douiri, N.; Soulas-Sprauel, P.; et al. Transitional B cells in quiescent SLE: An early checkpoint imprinted by IFN. J. Autoimmun. 2019, 102, 150–158. [Google Scholar] [CrossRef]
- Hu, N.; Qiu, X.; Luo, Y.; Yuan, J.; Li, Y.; Lei, W.; Zhang, G.; Zhou, Y.; Su, Y.; Lu, Q. Abnormal histone modification patterns in lupus CD4+ T cells. J. Rheumatol. 2008, 35, 804–810. [Google Scholar]
- Zhao, M.; Wu, X.; Zhang, Q.; Luo, S.; Liang, G.; Su, Y.; Tan, Y.; Lu, Q. RFX1 regulates CD70 and CD11a expression in lupus T cells by recruiting the histone methyltransferase SUV39H1. Arthritis Res. Ther. 2010, 12, R227. [Google Scholar] [CrossRef] [Green Version]
- Coit, P.; Renauer, P.; Jeffries, M.A.; Merrill, J.T.; McCune, W.J.; Maksimowicz-McKinnon, K.; Sawalha, A.H. Renal involvement in lupus is characterized by unique DNA methylation changes in naïve CD4+ T cells. J. Autoimmun. 2015, 61, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhao, M.; Sawalha, A.H.; Richardson, B.; Lu, Q. Impaired DNA methylation and its mechanisms in CD4+ T cells of systemic lupus erythematosus. J. Autoimmun. 2013, 41, 92–99. [Google Scholar] [CrossRef]
- Garaud, S.; Le Dantec, C.; Jousse-Joulin, S.; Hanrotel-Saliou, C.; Saraux, A.; Mageed, R.A.; Youinou, P.; Renaudineau, Y. IL-6 Modulates CD5 Expression in B Cells from Patients with Lupus by Regulating DNA Methylation. J. Immunol. 2009, 182, 5623–5632. [Google Scholar] [CrossRef] [Green Version]
- Ospelt, C. Epigenetic biomarkers in rheumatology-the future? Swiss Med. Wkly. 2016, 146, w14312. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Heylen, L.; Thienpont, B.; Naesens, M.; Lambrechts, D.; Sprangers, B. The Emerging Role of DNA Methylation in Kidney Transplantation: A Perspective. Am. J. Transplant. 2016, 16, 1070–1078. [Google Scholar] [CrossRef] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Jing, X.; Wu, X.; Yan, M.; Li, Q.; Shen, Y.; Wang, Z. DNA methylation in spermatogenesis and male infertility. Exp. Ther. Med. 2016, 12, 1973–1979. [Google Scholar] [CrossRef]
- Paluch, B.E.; Naqash, A.R.; Brumberger, Z.; Nemeth, M.J.; Griffiths, E.A. Epigenetics: A primer for clinicians. Blood Rev. 2016, 30, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Bochtler, M.; Kolano, A.; Xu, G.-L. DNA demethylation pathways: Additional players and regulators. BioEssays 2016, 39, e201600178-13. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Wang, F.; Zhang, Y.; Chen, J.; Liang, L.; Li, Y.; Zhang, M.; Yang, X.; Pang, H.; Li, Y.; et al. Hemi-methylated CpG sites connect Dnmt1-knockdown-induced and Tet1-induced DNA demethylation during somatic cell reprogramming. Cell Discov. 2019, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Long, H.; Yin, H.; Wang, L.; Gershwin, M.E.; Lu, Q. The critical role of epigenetics in systemic lupus erythematosus and autoimmunity. J. Autoimmun. 2016, 74, 118–138. [Google Scholar] [CrossRef]
- Deng, Y.; Tsao, B.P. Advances in lupus genetics and epigenetics. Curr. Opin. Rheumatol. 2014, 26, 482–492. [Google Scholar] [CrossRef]
- Richardson, B.; Ray, N.; Yung, R.; Sawalha, A.H. Murine models of lupus induced by hypomethylated T cells. Methods Mol. Med. 2004, 102, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Sawalha, A.H.; Jeffries, M. Defective DNA methylation and CD70 overexpression in CD4+ T cells in MRL/lpr lupus-prone mice. Eur. J. Immunol. 2007, 37, 1407–1413. [Google Scholar] [CrossRef]
- Sawalha, A.H.; Jeffries, M.; Webb, R.; Lu, Q.; Gorelik, G.; Ray, D.; Osban, J.; Knowlton, N.; Johnson, K.; Richardson, B. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008, 9, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javierre, B.M.; Fernandez, A.F.; Richter, J.; Al-Shahrour, F.; Martin-Subero, J.I.; Rodriguez-Ubreva, J.; Berdasco, M.; Fraga, M.F.; O’Hanlon, T.P.; Rider, L.G.; et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010, 20, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrich, C.M.; Crispin, J.; Tsokos, G.C. Epigenetic regulation of cytokine expression in systemic lupus erythematosus with special focus on T cells. Autoimmunity 2014, 47, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, C.M.; Tsokos, G.C. Epigenetic mechanisms in systemic lupus erythematosus and other autoimmune diseases. Trends Mol. Med. 2011, 17, 714–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedeschi, S.K.; Bermas, B.; Costenbader, K.H. Sexual disparities in the incidence and course of SLE and RA. Clin. Immunol. 2013, 149, 211–218. [Google Scholar] [CrossRef]
- Moulton, V.R.; Holcomb, D.R.; Zajdel, M.C.; Tsokos, G.C. Estrogen Upregulates Cyclic AMP Response Element Modulator α Expression and Downregulates Interleukin-2 Production by Human T Lymphocytes. Mol. Med. 2012, 18, 370–378. [Google Scholar] [CrossRef]
- Liu, H.-W.; Lin, H.-L.; Yen, J.-H.; Tsai, W.-C.; Chiou, S.-S.; Chang, J.-G.; Ou, T.-T.; Wu, C.-C.; Chao, N.-C. Demethylation within the proximal promoter region of human estrogen receptor alpha gene correlates with its enhanced expression: Implications for female bias in lupus. Mol. Immunol. 2014, 61, 28–37. [Google Scholar] [CrossRef]
- Elbagir, S.; Sohrabian, A.; Elshafie, A.I.; Elagib, E.M.; Mohammed, N.E.A.; Nur, M.A.M.; Svenungsson, E.; Gunnarsson, I.; Rönnelid, J. Accumulation of antinuclear associated antibodies in circulating immune complexes is more prominent in SLE patients from Sudan than Sweden. Sci. Rep. 2020, 10, 21126. [Google Scholar] [CrossRef]
- Felux, J.; Erbacher, A.; Breckler, M.; Hervé, R.; Lemeiter, D.; Mannherz, H.G.; Decker, P. Deoxyribonuclease 1-Mediated Clearance of Circulating Chromatin Prevents from Immune Cell Activation and Pro-inflammatory Cytokine Production, a Phenomenon Amplified by Low Trap1 Activity: Consequences for Systemic Lupus Erythematosus. Front. Immunol. 2021, 12, 613597. [Google Scholar] [CrossRef]
- Hamilton, A.J.; Hsu, H.-C.; Mountz, J.D. Role of production of type I interferons by B cells in the mechanisms and pathogenesis of systemic lupus erythematosus. Discov. Med. 2018, 25, 21–29. [Google Scholar]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rönnblom, L.; Pascual, V. The innate immune system in SLE: Type I interferons and dendritic cells. Lupus 2008, 17, 394–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald-Bocarsly, P.; Dai, J.; Singh, S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008, 19, 3–19. [Google Scholar] [CrossRef]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct Effects of Type I Interferons on Cells of the Immune System. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef] [Green Version]
- Hooks, J.J.; Moutsopoulos, H.M.; Geis, S.A.; Stahl, N.I.; Decker, J.L.; Notkins, A.L. Immune Interferon in the Circulation of Patients with Autoimmune Disease. N. Engl. J. Med. 1979, 301, 5–8. [Google Scholar] [CrossRef]
- Rönnblom, L. The type I interferon system in the etiopathogenesis of autoimmune diseases. Upsala J. Med. Sci. 2011, 116, 227–237. [Google Scholar] [CrossRef]
- Chung, S.A.; Nititham, J.; Elboudwarej, E.; Quach, H.L.; Taylor, K.E.; Barcellos, L.F.; Criswell, L.A. Genome-Wide Assessment of Differential DNA Methylation Associated with Autoantibody Production in Systemic Lupus Erythematosus. PLoS ONE 2015, 10, e0129813. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.; George, N.I.; Green-Knox, B.; Treadwell, E.L.; Word, B.; Yim, S.; Lyn-Cook, B. Epigenome-wide association study of peripheral blood mononuclear cells in systemic lupus erythematosus: Identifying DNA methylation signatures associated with interferon-related genes based on ethnicity and SLEDAI. J. Autoimmun. 2018, 96, 147–157. [Google Scholar] [CrossRef]
- Yeung, K.S.; Chung, B.H.-Y.; Choufani, S.; Mok, M.Y.; Wong, W.L.; Mak, C.C.Y.; Yang, W.; Lee, P.P.W.; Wong, W.H.S.; Chen, Y.-A.; et al. Genome-Wide DNA Methylation Analysis of Chinese Patients with Systemic Lupus Erythematosus Identified Hypomethylation in Genes Related to the Type I Interferon Pathway. PLoS ONE 2017, 12, e0169553. [Google Scholar] [CrossRef] [Green Version]
- Imgenberg-Kreuz, J.; Almlöf, J.C.; Leonard, D.; Alexsson, A.; Nordmark, G.; Eloranta, M.-L.; Rantapää-Dahlqvist, S.; A Bengtsson, A.; Jönsen, A.; Padyukov, L.; et al. DNA methylation mapping identifies gene regulatory effects in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 736–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Pu, W.; Guo, S.; Jin, L.; He, D.; Wang, J. Genome-Wide DNA Methylation Profiles Reveal Common Epigenetic Patterns of Interferon-Related Genes in Multiple Autoimmune Diseases. Front. Genet. 2019, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Mi, W.; Luo, H.; Chen, T.; Liu, S.; Raman, I.; Zuo, X.; Li, Q.-Z. Whole-genome transcription and DNA methylation analysis of peripheral blood mononuclear cells identified aberrant gene regulation pathways in systemic lupus erythematosus. Arthritis Res. Ther. 2016, 18, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef]
- Agarwal, S.; Rao, A. Modulation of Chromatin Structure Regulates Cytokine Gene Expression during T Cell Differentiation. Immunity 1998, 9, 765–775. [Google Scholar] [CrossRef] [Green Version]
- Lal, G.; Zhang, N.; Van Der Touw, W.; Ding, Y.; Ju, W.; Bottinger, E.P.; Reid, S.P.; Levy, D.E.; Bromberg, J.S. Epigenetic Regulation of Foxp3 Expression in Regulatory T Cells by DNA Methylation. J. Immunol. 2008, 182, 259–273. [Google Scholar] [CrossRef] [Green Version]
- Del Prete, G. The Concept of Type-1 and Type-2 Helper T Cells and Their Cytokines in Humans. Int. Rev. Immunol. 1998, 16, 427–455. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, Z.; Chang, C.; Lu, L.; Lau, C.S.; Lu, Q. Th9 cells and IL-9 in autoimmune disorders: Pathogenesis and therapeutic potentials. Hum. Immunol. 2017, 78, 120–128. [Google Scholar] [CrossRef]
- Pernis, A.B. Th17 cells in rheumatoid arthritis and systemic lupus erythematosus. J. Intern. Med. 2009, 265, 644–652. [Google Scholar] [CrossRef]
- Azizi, G.; Simhag, A.; El Rouby, N.M.M.; Mirshafiey, A. Th22 Cells Contribution in Immunopathogenesis of Rheumatic Diseases. Iran. J. Allergy Asthma Immunol. 2015, 14, 246–254. [Google Scholar]
- Nakayamada, S.; Tanaka, Y. Clinical relevance of T follicular helper cells in systemic lupus erythematosus. Expert Rev. Clin. Immunol. 2021, 17, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K. Regulatory T cells in systemic lupus erythematosus. Eur. J. Immunol. 2014, 45, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chang, C.; Lu, Q. Epigenetics of CD4+ T cells in autoimmune diseases. Curr. Opin. Rheumatol. 2017, 29, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Y.; Richardson, B. Decreased DNA methyltransferase levels contribute to abnormal gene expression in “senescent” CD4+ CD28− T cells. Clin. Immunol. 2009, 132, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Tang, J.; Gao, F.; Wu, X.; Liang, Y.; Yin, H.; Lu, Q. Hypomethylation of IL10 and IL13 promoters in CD4+ T cells of patients with systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 2010, 931018. [Google Scholar] [CrossRef]
- Rauen, T.; Hedrich, C.; Tenbrock, K.; Tsokos, G.C. cAMP responsive element modulator: A critical regulator of cytokine production. Trends Mol. Med. 2013, 19, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Richardson, B.C.; Scheinbart, L.; Strahler, J.R.; Gross, L.; Hanash, S.; Johnson, M.A. Evidence for impaired t cell dna methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Care Res. 1990, 33, 1665–1673. [Google Scholar] [CrossRef]
- Corvetta, A.; Della Bitta, R.; Luchetti, M.M.; Pomponio, G. 5-Methylcytosine content of DNA in blood, synovial mononuclear cells and synovial tissue from patients affected by autoimmune rheumatic diseases. J. Chromatogr. B Biomed. Sci. Appl. 1991, 566, 481–491. [Google Scholar] [CrossRef]
- Renauer, P.; Coit, P.; A Jeffries, M.; Merrill, J.T.; McCune, W.J.; Maksimowicz-McKinnon, K.; Sawalha, A.H. DNA methylation patterns in naïve CD4+ T cells identify epigenetic susceptibility loci for malar rash and discoid rash in systemic lupus erythematosus. Lupus Sci. Med. 2015, 2, e000101. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Wu, A.; Tesmer, L.; Ray, N.; Yousif, D.; Richardson, B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J. Immunol. 2007, 179, 6352–6358. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.-H.; Zhu, X.-H.; Liang, J.; Yang, Y.-S.; Wang, S.-S.; Shi, W.-M.; Xu, J.-H. Associations between aberrant DNA methylation and transcript levels of DNMT1 and MBD2 in CD4+ T cells from patients with systemic lupus erythematosus. Australas. J. Dermatol. 2012, 54, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, Q. DNA methylation in T cells from idiopathic lupus and drug-induced lupus patients. Autoimmun. Rev. 2008, 7, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Luo, Y.; Lei, W.; Luo, Y.; Yan, K.; Zhao, S.; Lu, Q. Abnormal DNA methylation in CD4+ T cells from patients with systemic lupus erythematosus, systemic sclerosis, and dermatomyositis. Scand. J. Rheumatol. 2009, 38, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, S.; Luo, S.; Wu, H.; Tang, M.; Cheng, W.; Zhang, Q.; Zhang, P.; Yu, X.; Xia, Y.; et al. DNA methylation and mRNA and microRNA expression of SLE CD4+ T cells correlate with disease phenotype. J. Autoimmun. 2014, 54, 127–136. [Google Scholar] [CrossRef]
- Jeffries, M.; Dozmorov, M.; Tang, Y.; Merrill, J.T.; Wren, J.; Sawalha, A.H. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics 2011, 6, 593–601. [Google Scholar] [CrossRef]
- Garaud, S.; Youinou, P.; Renaudineau, Y. DNA methylation and B-cell autoreactivity. In Epigenetic Contributions in Autoimmune Disease; Springer: Berlin/Heidelberg, Germany, 2011; pp. 50–60. [Google Scholar]
- Orlanski, S.; Labi, V.; Reizel, Y.; Spiro, A.; Lichtenstein, M.; Levin-Klein, R.; Koralov, S.B.; Skversky, Y.; Rajewsky, K.; Cedar, H.; et al. Tissue-specific DNA demethylation is required for proper B-cell differentiation and function. Proc. Natl. Acad. Sci. USA 2016, 113, 5018–5023. [Google Scholar] [CrossRef] [Green Version]
- Barwick, B.G.; Scharer, C.D.; Martinez, R.J.; Price, M.J.; Wein, A.N.; Haines, R.R.; Bally, A.P.R.; Kohlmeier, J.E.; Boss, J.M. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat. Commun. 2018, 9, 1900. [Google Scholar] [CrossRef] [Green Version]
- Danbara, M.; Kameyama, K.; Higashihara, M.; Takagaki, Y. DNA methylation dominates transcriptional silencing of Pax5 in terminally differentiated B cell lines. Mol. Immunol. 2002, 38, 1161–1166. [Google Scholar] [CrossRef]
- Scharer, C.D.; Blalock, E.L.; Mi, T.; Barwick, B.G.; Jenks, S.A.; Deguchi, T.; Cashman, K.S.; Neary, B.E.; Patterson, D.; Hicks, S.L.; et al. Epigenetic programming underpins B cell dysfunction in human SLE. Nat. Immunol. 2019, 20, 1071–1082. [Google Scholar] [CrossRef]
- Mahajan, V.S.; Mattoo, H.; Na Sun, N.; Viswanadham, V.; Yuen, G.J.; Allard-Chamard, H.; Ahmad, M.; Murphy, S.J.H.; Cariappa, A.; Tuncay, Y.; et al. B1a and B2 cells are characterized by distinct CpG modification states at DNMT3A-maintained enhancers. Nat. Commun. 2021, 12, 2208. [Google Scholar] [CrossRef]
- Youinou, P.; Renaudineau, Y. CD5 expression in B cells from patients with systemic lupus erythematosus. Crit. Rev. Immunol. 2011, 31, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Fali, T.; Le Dantec, C.; Thabet, Y.; Jousse, S.; Hanrotel, C.; Youinou, P.; Renaudineau, Y. DNA methylation modulates HRES1/p28 expression in B cells from patients with Lupus. Autoimmunity 2014, 47, 265–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illei, G.G.; Shirota, Y.; Yarboro, C.H.; Daruwalla, J.; Tackey, E.; Takada, K.; Lipsky, P.E. Tocilizumab in systemic lupus erythematosus: Data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2010, 62, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.J.; Strand, V.; Merrill, J.T.; Popa, S.; Spindler, A.J.; Eimon, A.; Petri, M.; Smolen, J.S.; Wajdula, J.; Christensen, J.; et al. Efficacy and safety of an interleukin 6 monoclonal antibody for the treatment of systemic lupus erythematosus: A phase II dose-ranging randomised controlled trial. Ann. Rheum. Dis. 2016, 76, 534–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tackey, E.; E Lipsky, P.; Illei, G.G. Rationale for interleukin-6 blockade in systemic lupus erythematosus. Lupus 2004, 13, 339–343. [Google Scholar] [CrossRef]
- Li, Y.; Lee, P.Y.; Reeves, W.H. Monocyte and Macrophage Abnormalities in Systemic Lupus Erythematosus. Arch. Immunol. Ther. Exp. 2010, 58, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Biermann, M.H.; Veissi, S.; Maueröder, C.; Chaurio, R.; Berens, C.; Herrmann, M.; E Munoz, L. The role of dead cell clearance in the etiology and pathogenesis of systemic lupus erythematosus: Dendritic cells as potential targets. Expert Rev. Clin. Immunol. 2014, 10, 1151–1164. [Google Scholar] [CrossRef]
- Kis-Toth, K.; Tsokos, G.C. Dendritic cell function in lupus: Independent contributors or victims of aberrant immune regulation. Autoimmunity 2010, 43, 121–130. [Google Scholar] [CrossRef]
- Anderson, J.O.; Thundiyil, J.G.; Stolbach, A. Clearing the Air: A Review of the Effects of Particulate Matter Air Pollution on Human Health. J. Med. Toxicol. 2011, 8, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Niranjan, R.; Thakur, A.K. The Toxicological Mechanisms of Environmental Soot (Black Carbon) and Carbon Black: Focus on Oxidative Stress and Inflammatory Pathways. Front. Immunol. 2017, 8, 763. [Google Scholar] [CrossRef]
- Panis, L.I.; Provost, E.B.; Cox, B.; Louwies, T.; Laeremans, M.; Standaert, A.; Dons, E.; Holmstock, L.; Nawrot, T.; De Boever, P. Short-term air pollution exposure decreases lung function: A repeated measures study in healthy adults. Environ. Health 2017, 16, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrot, T.S.; Alfaro-Moreno, E.; Nemery, B. Update in Occupational and Environmental Respiratory Disease 2007. Am. J. Respir. Crit. Care Med. 2008, 177, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Louwies, T.; Panis, L.I.; Kicinski, M.; De Boever, P.; Nawrot, T. Retinal Microvascular Responses to Short-Term Changes in Particulate Air Pollution in Healthy Adults. Environ. Health Perspect. 2013, 121, 1011–1016. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, S.; Al-Kindi, S.G.; Brook, R.D. Air pollution and cardiovascular disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2018, 72, 2054–2070. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, E.; Bélair, M.A.; Do, M.T.; Stieb, D.M.; Hystad, P.; van Donkelaar, A.; Walker, M. Maternal exposure to ambient air pollution and risk of early childhood cancers: A population-based study in Ontario, Canada. Environ. Int. 2017, 100, 139–147. [Google Scholar] [CrossRef]
- Monks, P.; Granier, C.; Fuzzi, S.; Stohl, A.; Williams, M.; Akimoto, H.; Amann, M.; Baklanov, A.; Baltensperger, U.; Bey, I.; et al. Atmospheric composition change—Global and regional air quality. Atmos. Environ. 2009, 43, 5268–5350. [Google Scholar] [CrossRef] [Green Version]
- Janssen, B.G.; Godderis, L.; Pieters, N.; Poels, K.; Kiciński, M.; Cuypers, A.; Fierens, F.; Penders, J.; Plusquin, M.; Gyselaers, W.; et al. Placental DNA hypomethylation in association with particulate air pollution in early life. Part. Fibre Toxicol. 2013, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Ning, Z.; Sioutas, C. Atmospheric Processes Influencing Aerosols Generated by Combustion and the Inference of Their Impact on Public Exposure: A Review. Aerosol Air Qual. Res. 2010, 10, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Wyzga, R.; Rohr, A. Long-term particulate matter exposure: Attributing health effects to individual PM components. J. Air Waste Manag. Assoc. 2015, 65, 523–543. [Google Scholar] [CrossRef] [Green Version]
- Nemmar, A.; Al-Maskari, S.; Ali, B.H.; Al-Amri, I. Cardiovascular and lung inflammatory effects induced by systemically administered diesel exhaust particles in rats. Am. J. Physiol. Cell. Mol. Physiol. 2007, 292, L664–L670. [Google Scholar] [CrossRef]
- Nemmar, A.; Hamoir, J.; Nemery, B.; Gustin, P. Evaluation of particle translocation across the alveolo-capillary barrier in isolated perfused rabbit lung model. Toxicology 2005, 208, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Colasanti, T.; Fiorito, S.; Alessandri, C.; Serafino, A.; Andreola, F.; Barbati, C.; Morello, F.; Alfè, M.; Di Blasio, G.; Gargiulo, V.; et al. Diesel exhaust particles induce autophagy and citrullination in Normal Human Bronchial Epithelial cells. Cell Death Dis. 2018, 9, 1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sopori, M.L. Effects of cigarette smoke on the immune system. Nat. Rev. Immunol. 2002, 2, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, J.; Zhang, Y.; Zhang, G.; Kang, Y.; Chen, A.; Feng, X.; Shao, L. The toxicity of silica nanoparticles to the immune system. Nanomedicine 2018, 13, 1939–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, T.; Tang, M. Biological effects of airborne fine particulate matter (PM2. 5) exposure on pulmonary immune system. Environ. Toxicol. Pharmacol. 2018, 60, 95–201. [Google Scholar] [CrossRef]
- Gangamma, S. Airborne Particulate Matter and Innate Immunity Activation. Environ. Sci. Technol. 2012, 46, 10879–10880. [Google Scholar] [CrossRef]
- Li, N.; Wang, M.; Oberley, T.D.; Sempf, J.M.; Nel, A.E. Comparison of the Pro-Oxidative and Proinflammatory Effects of Organic Diesel Exhaust Particle Chemicals in Bronchial Epithelial Cells and Macrophages. J. Immunol. 2002, 169, 4531–4541. [Google Scholar] [CrossRef] [Green Version]
- Nel, A.; Xia, T.; Mädler, L.; Li, N. Toxic Potential of Materials at the Nanolevel. Science 2006, 311, 622–627. [Google Scholar] [CrossRef] [Green Version]
- Bascom, R. Committee of the Environmental and Occupational Health Assembly of the American Thoracic Society Health effects of outdoor air pollution (part 1 of 2 parts). Am. J. Respir. Crit. Care Med. 1996, 153, 3–50. [Google Scholar]
- World Health Organization. Air Quality Guidelines for Europe; World Health Organization, Regional Office for Europe: Geneva, Switzerland, 2000. [Google Scholar]
- Jhun, I.; Coull, B.A.; Zanobetti, A.; Koutrakis, P. The impact of nitrogen oxides concentration decreases on ozone trends in the USA. Air Qual. Atmos. Health 2014, 8, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Rijnders, E.; Janssen, N.A.H.; Van Vliet, P.H.N.; Brunekreef, B. Personal and Outdoor Nitrogen Dioxide Concentrations in Relation to Degree of Urbanization and Traffic Density. Environ. Health Perspect. 2001, 109, 411. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Bunkar, N.; Kumar, R.; Bhargava, A.; Tiwari, R.; Chaudhury, K.; Goryacheva, I.Y.; Mishra, P.K. Air pollution associated epigenetic modifications: Transgenerational inheritance and underlying molecular mechanisms. Sci. Total Environ. 2018, 656, 760–777. [Google Scholar] [CrossRef] [PubMed]
- Ravegnini, G.; Sammarini, G.; Hrelia, P.; Angelini, S. Key Genetic and Epigenetic Mechanisms in Chemical Carcinogenesis. Toxicol. Sci. 2015, 148, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Rang, F.J.; Boonstra, J. Causes and Consequences of Age-Related Changes in DNA Methylation: A Role for ROS? Biology 2014, 3, 403–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baccarelli, A.; Wright, R.O.; Bollati, V.; Tarantini, L.; Litonjua, A.A.; Suh, H.H.; Zanobetti, A.; Sparrow, D.; Vokonas, P.S.; Schwartz, J. Rapid DNA Methylation Changes after Exposure to Traffic Particles. Am. J. Respir. Crit. Care Med. 2009, 179, 572–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tellez-Plaza, M.; Tang, W.-Y.; Shang, Y.; Umans, J.G.; Francesconi, K.A.; Goessler, W.; Ledesma, M.; León, M.; Laclaustra, M.; Pollak, J.; et al. Association of Global DNA Methylation and Global DNA Hydroxymethylation with Metals and Other Exposures in Human Blood DNA Samples. Environ. Health Perspect. 2014, 122, 946–954. [Google Scholar] [CrossRef]
- Okamoto, Y.; Iwai-Shimada, M.; Nakai, K.; Tatsuta, N.; Mori, Y.; Aoki, A.; Kojima, N.; Takada, T.; Satoh, H.; Jinno, H. Global DNA Methylation in Cord Blood as a Biomarker for Prenatal Lead and Antimony Exposures. Toxics 2022, 10, 157. [Google Scholar] [CrossRef]
- Cowley, M.; Skaar, D.A.; Jima, D.D.; Maguire, R.L.; Hudson, K.M.; Park, S.S.; Sorrow, P.; Hoyo, C. Effects of Cadmium Exposure on DNA Methylation at Imprinting Control Regions and Genome-Wide in Mothers and Newborn Children. Environ. Health Perspect. 2018, 126, 037003. [Google Scholar] [CrossRef]
- Janssen, B.G.; Madhloum, N.; Gyselaers, W.; Bijnens, E.; Clemente, D.B.; Cox, B.; Nawrot, T.S. Cohort profile: The ENVIRonmental influence ON early AGEing (ENVIR ON AGE): A birth cohort study. Int. J. Epidemiol. 2017, 46, 1386–1387. [Google Scholar] [CrossRef]
- Janssen, B.G.; Byun, H.-M.; Gyselaers, W.; Lefebvre, W.; Baccarelli, A.A.; Nawrot, T.S. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: An ENVIR ON AGE birth cohort study. Epigenetics 2015, 10, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Vos, S.; Nawrot, T.S.; Martens, D.S.; Byun, H.-M.; Janssen, B.G. Mitochondrial DNA methylation in placental tissue: A proof of concept study by means of prenatal environmental stressors. Epigenetics 2020, 16, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Neven, K.Y.; Saenen, N.D.; Tarantini, L.; Janssen, B.G.; Lefebvre, W.; Vanpoucke, C.; Nawrot, T.S. Placental promoter methylation of DNA repair genes and prenatal exposure to particulate air pollution: An ENVIRONAGE cohort study. Lancet Planet. Health 2018, 2, e174–e183. [Google Scholar] [CrossRef] [Green Version]
- Saenen, N.D.; Vrijens, K.; Janssen, B.G.; Roles, H.A.; Neven, K.Y.; Berghe, W.V.; Gyselaers, W.; Vanpoucke, C.; Lefebvre, W.; De Boever, P.; et al. Lower Placental Leptin Promoter Methylation in Association with Fine Particulate Matter Air Pollution during Pregnancy and Placental Nitrosative Stress at Birth in the environ age Cohort. Environ. Health Perspect. 2017, 125, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrot, T.S.; Saenen, N.D.; Schenk, J.; Janssen, B.G.; Motta, V.; Tarantini, L.; Cox, B.; Lefebvre, W.; Vanpoucke, C.; Maggioni, C.; et al. Placental circadian pathway methylation and in utero exposure to fine particle air pollution. Environ. Int. 2018, 114, 231–241. [Google Scholar] [CrossRef]
- Maghbooli, Z.; Hossein-Nezhad, A.; Adabi, E.; Asadollah-Pour, E.; Sadeghi, M.; Mohammad-Nabi, S.; Rad, L.Z.; Hosseini, A.-A.M.; Radmehr, M.; Faghihi, F.; et al. Air pollution during pregnancy and placental adaptation in the levels of global DNA methylation. PLoS ONE 2018, 13, e0199772. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, P.; Zhou, Y.; Xia, B.; Zhu, Q.; Ge, W.; Zhang, Y. Prenatal fine particulate matter exposure, placental DNA methylation changes, and fetal growth. Environ. Int. 2021, 147, 106313. [Google Scholar] [CrossRef]
- Ladd-Acosta, C.; Feinberg, J.I.; Brown, S.C.; Lurmann, F.W.; Croen, L.A.; Hertz-Picciotto, I.; Newschaffer, C.J.; Feinberg, A.P.; Fallin, M.D.; Volk, H.E. Epigenetic marks of prenatal air pollution exposure found in multiple tissues relevant for child health. Environ. Int. 2019, 126, 363–376. [Google Scholar] [CrossRef]
- Park, J.; Kim, W.J.; Kim, J.; Jeong, C.-Y.; Park, H.; Hong, Y.-C.; Ha, M.; Kim, Y.; Won, S.; Ha, E. Prenatal Exposure to Traffic-Related Air Pollution and the DNA Methylation in Cord Blood Cells: MOCEH Study. Int. J. Environ. Res. Public Health 2022, 19, 3292. [Google Scholar] [CrossRef]
- Kingsley, S.L.; Eliot, M.N.; Whitsel, E.A.; Huang, Y.T.; Kelsey, K.T.; Marsit, C.J.; Wellenius, G.A. Maternal residential proximity to major roadways, birth weight, and placental DNA methylation. Environ. Int. 2016, 92, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Breton, C.V.; Yao, J.; Millstein, J.; Gao, L.; Siegmund, K.D.; Mack, W.; Gilliland, F.D. Prenatal air pollution exposures, DNA methyl transferase genotypes, and associations with newborn LINE1 and Alu methylation and childhood blood pressure and carotid intima-media thickness in the Children’s Health Study. Environ. Health Perspect. 2016, 124, 1905–1912. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Zhao, Y.; Liu, P.; Xia, B.; Zhu, Q.; Wang, X.; Song, Q.; Kan, H.; Zhang, Y. Exposure to particulate air pollution during early pregnancy is associated with placental DNA methylation. Sci. Total Environ. 2017, 607–608, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Gruzieva, O.; Xu, C.-J.; Breton, C.V.; Annesi-Maesano, I.; Antó, J.M.; Auffray, C.; Ballereau, S.; Bellander, T.; Bousquet, J.; Bustamante, M.; et al. Epigenome-Wide Meta-Analysis of Methylation in Children Related to Prenatal NO 2 Air Pollution Exposure. Environ. Health Perspect. 2017, 125, 104–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruzieva, O.; Xu, C.-J.; Yousefi, P.; Relton, C.; Merid, S.K.; Breton, C.V.; Gao, L.; Volk, H.E.; Feinberg, J.I.; Ladd-Acosta, C.; et al. Prenatal Particulate Air Pollution and DNA Methylation in Newborns: An Epigenome-Wide Meta-Analysis. Environ. Health Perspect. 2019, 127, 057012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaevska, E.; Fiano, V.; Asta, F.; Stafoggia, M.; Moirano, G.; Popovic, M.; Pizzi, C.; Trevisan, M.; De Marco, L.; Polidoro, S.; et al. Prenatal exposure to PM10 and changes in DNA methylation and telomere length in cord blood. Environ. Res. 2022, 209, 112717. [Google Scholar] [CrossRef]
- Abraham, E.; Rousseaux, S.; Agier, L.; Giorgis-Allemand, L.; Tost, J.; Galineau, J.; Hulin, A.; Siroux, V.; Vaiman, D.; Charles, M.-A.; et al. Pregnancy exposure to atmospheric pollution and meteorological conditions and placental DNA methylation. Environ. Int. 2018, 118, 334–347. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.-I.; Lee, S.H.; Lee, S.Y.; Kim, H.C.; Kim, H.B.; Kim, J.H.; Hong, S.J. Prenatal PM2. 5 exposure and vitamin D–associated early persistent atopic dermatitis via placental methylation. Ann. Allergy Asthma Immunol. 2020, 125, 665–673.e1. [Google Scholar] [CrossRef]
- Rider, C.F.; Carlsten, C. Air pollution and DNA methylation: Effects of exposure in humans. Clin. Epigenet. 2019, 11, 131. [Google Scholar] [CrossRef] [Green Version]
- Bind, M.-A.; Lepeule, J.; Zanobetti, A.; Gasparrini, A.; Baccarelli, A.A.; Coull, B.A.; Schwartz, J. Air pollution and gene-specific methylation in the Normative Aging Study: Association, effect modification, and mediation analysis. Epigenetics 2014, 9, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Bind, M.-A.C.; Coull, B.A.; Peters, A.; Baccarelli, A.A.; Tarantini, L.; Cantone, L.; Vokonas, P.S.; Koutrakis, P.; Schwartz, J.D. Beyond the Mean: Quantile Regression to Explore the Association of Air Pollution with Gene-Specific Methylation in the Normative Aging Study. Environ. Health Perspect. 2015, 123, 759–765. [Google Scholar] [CrossRef] [Green Version]
- De Prins, S.; Koppen, G.; Jacobs, G.; Dons, E.; Van de Mieroop, E.; Nelen, V.; Fierens, F.; Panis, L.I.; De Boever, P.; Cox, B.; et al. Influence of ambient air pollution on global DNA methylation in healthy adults: A seasonal follow-up. Environ. Int. 2013, 59, 418–424. [Google Scholar] [CrossRef]
- Mostafavi, N.; Vermeulen, R.; Ghantous, A.; Hoek, G.; Probst-Hensch, N.; Herceg, Z.; Tarallo, S.; Naccarati, A.; Kleinjans, J.C.; Imboden, M.; et al. Acute changes in DNA methylation in relation to 24 h personal air pollution exposure measurements: A panel study in four European countries. Environ. Int. 2018, 120, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Jones, M.J.; Sava, F.; Kobor, M.S.; Carlsten, C. Short-term diesel exhaust inhalation in a controlled human crossover study is associated with changes in DNA methylation of circulating mononuclear cells in asthmatics. Part. Fibre Toxicol. 2014, 11, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, A.G.F.; Giacomin, M.F.D.A.; Braga, A.L.F.; Sallum, A.M.E.; Pereira, L.A.A.; Farhat, L.C.; Strufaldi, F.L.; Lichtenfels, A.J.D.F.C.; Carvalho, T.D.S.; Nakagawa, N.K.; et al. Influence of air pollution on airway inflammation and disease activity in childhood-systemic lupus erythematosus. Clin. Rheumatol. 2017, 37, 683–690. [Google Scholar] [CrossRef]
- Goulart, M.F.G.; Alves, A.G.F.; Farhat, J.; Braga, A.L.F.; Pereira, L.A.A.; Lichtenfels, A.J.D.F.C.; Campos, L.M.D.A.; Da Silva, C.A.A.; Elias, A.M.; Farhat, S.C.L. Influence of air pollution on renal activity in patients with childhood-onset systemic lupus erythematosus. Pediatr. Nephrol. 2020, 35, 1247–1255. [Google Scholar] [CrossRef]
- Conde, P.G.; Farhat, L.C.; Braga, A.L.F.; Sallum, A.E.M.; Farhat, S.C.L.; Silva, C.A. Are prematurity and environmental factors determinants for developing childhood-onset systemic lupus erythematosus? Mod. Rheumatol. 2017, 28, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.-H.; Shih, Y.-J.; Lin, C.-L.; Wei, C.-C. Associations between Fine Particulate Matter (PM2.5) and Childhood-Onset Systemic Lupus Erythematosus. Ind. J. Pediatr. 2022, 89, 200. [Google Scholar]
- Campos, L.; Fernandes, E.; Silva, C.; Braga, A.; Sallum, A.; Farhat, S. PreS-FINAL-2297: Atmospheric pollution: Influence on disease activity in childhood-onset systemic lupus erythematosus patients. Pediatr. Rheumatol. 2013, 11, P287. [Google Scholar] [CrossRef]
- Bernatsky, S.; Smargiassi, A.; Barnabe, C.; Svenson, L.W.; Brand, A.; Martin, R.V.; Hudson, M.; Clarke, A.E.; Fortin, P.R.; van Donkelaar, A.; et al. Fine particulate air pollution and systemic autoimmune rheumatic disease in two Canadian provinces. Environ. Res. 2016, 146, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.-R.; Chung, W.-T.; Chen, W.-T.; Lee, R.-Y.; Hwang, B.-F. Long-term exposure to traffic-related air pollution and systemic lupus erythematosus in Taiwan: A cohort study. Sci. Total Environ. 2019, 668, 342–349. [Google Scholar] [CrossRef]
- Cakmak, S.; Blanco-Vidal, C.; Lukina, A.O.; Dales, R. The association between air pollution and hospitalization for patients with systemic lupus erythematosus in Chile: A daily time series analysis. Environ. Res. 2020, 192, 110469. [Google Scholar] [CrossRef]
- Zhao, C.-N.; Mei, Y.J.; Wu, G.C.; Mao, Y.M.; Wu, Q.; Dan, Y.L.; Pan, H.F. Effect of air pollution on hospital admissions for systemic lupus erythematosus in Bengbu, China: A time series study. Lupus 2019, 28, 1541–1548. [Google Scholar] [CrossRef]
- Bernatsky, S.; Fournier, M.; Pineau, C.A.; Clarke, A.E.; Vinet, E.; Smargiassi, A. Associations between Ambient Fine Particulate Levels and Disease Activity in Patients with Systemic Lupus Erythematosus (SLE). Environ. Health Perspect. 2011, 119, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanata, C.M.; Nititham, J.; Taylor, K.; Nayak, R.; Barcellos, L.; Chung, S.A.; Criswell, L.A. CE-48 Residential proximity to highways, DNA methylation and systemic lupus erythematosus. Arch. Dis. Childhood 2016, 3, 67. [Google Scholar]
- Coit, P.; Yalavarthi, S.; Ognenovski, M.; Zhao, W.; Hasni, S.; Wren, J.D.; Kaplan, M.J.; Sawalha, A.H. Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils. J. Autoimmun. 2015, 58, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocci, V. Is it true that ozone is always toxic? The end of a dogma. Toxicol. Appl. Pharmacol. 2006, 216, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Bocci, V.; Borrelli, E.; Travagli, V.; Zanardi, I. The ozone paradox: Ozone is a strong oxidant as well as a medical drug. Med. Res. Rev. 2009, 29, 646–682. [Google Scholar] [CrossRef] [PubMed]
- Calunga, J.L.; Trujillo, Y.; Menéndez, S.; Zamora, Z.; Alonso, Y.; Merino, N.; Montero, T. Ozone oxidative post-conditioning in acute renal failure. J. Pharm. Pharmacol. 2009, 61, 221–227. [Google Scholar] [CrossRef]
- Saenen, N.D.; Bové, H.; Steuwe, C.; Roeffaers, M.B.J.; Provost, E.B.; Lefebvre, W.; Vanpoucke, C.; Ameloot, M.; Nawrot, T.S. Children’s Urinary Environmental Carbon Load. A Novel Marker Reflecting Residential Ambient Air Pollution Exposure? Am. J. Respir. Crit. Care Med. 2017, 196, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, T.S.; Adcock, I. The Detrimental Health Effects of Traffic-Related Air Pollution: A Role for DNA Methylation? American Thoracic Society: New York, NY, USA, 2009; Volume 179, pp. 523–524. [Google Scholar]

| T lymphocytes (CD4+) | B lymphocytes (CD19+) | Monocytes and Neutrophils (CD14+) |
|---|---|---|
| PTPN22 (protein tyrosine phosphatase, non-receptor type 22) | BLK (B-lymphocyte kinase) | HLA-DR (human leukocyte antigen-DR isotype) |
| TNFSF4 (TNF Superfamily Member 4) | BANK1 (B cell scaffold protein with ankyrin repeats 1) | TNFα (tumor necrosis factor α) |
| STAT4 (Signal transducer and activator of transcription 4) | LYN (LYN Proto-Oncogene, Src Family Tyrosine Kinase) | ICAM1 (intercellular adhesion molecule 1) |
| CD247 (cluster of differentiation 247) | CR2 (complement receptor 2) | Fc-γ RII (Fc gamma receptors class II) |
| CD9 (cluster of differentiation 9) | NCF2 (neutrophil cytosol factor 2) | ITGAM (CD11b) (integrin subunit alfa M [cluster of differentiation 11b]) |
| MMP9 (matrix metallopeptidase 9) | IL1 (interleukin 1) | NETs (norepinephrine transporters) |
| PDGFRA (platelet-derived growth factor receptor A) | IKZF1 (IKAROS family zinc finger 1) | IFI44L (interferon-induced protein 44-like) |
| Perforin | TLR9 (toll-like receptor 9) | ADAR (adenosine deaminase RNA specific) |
| CD11a (cluster of differentiation 11a) | CD19 (cluster of differentiation 19) | RABGAP1L (RAB GTPase activating protein 1 like) |
| CD70 (cluster of differentiation 70) | ISGs (interferon-stimulated genes) | CMPK2 (cytidine/uridine monophosphate kinase 2) |
| CD40 ligand (cluster of differentiation 40 ligand) | CD5 (cluster of differentiation 5) | TREX1 (three prime repair exonuclease 1) |
| PP22A (protein phosphatase 2A) | HRES1 (HTVL-1-related endogenous sequence) | DTX3L (deltex E3 ubiquitin ligase 3L) |
| BST2 (bone marrow stromal cell antigen 2) | TNF (tumor necrosis factor) | PARP9 (poly ADP-ribose polymerase family member 9) |
| IR7 (ionotropic receptor 7) | EP300 (E1A binding protein P300) | PLSCR1 (phospholipid scramblase 1) |
| CD80 (cluster of differentiation 80) | IFI44L (interferon-induced protein 44-like) | DDX60 (DExD/H-Box helicase 60) |
| HERC5 (HECT and RLD domain containing E3 ubiquitin-protein ligase 5) | PARP9 (poly ADP ribose polymerase family member 9) | HLA-F (major histocompatibility complex, class 1, F) |
| IFI44 (interferon-induced protein 44) | IFITM1 (interferon-induced transmembrane protein 1) | TAP1 (transporter 1, ATP binding cassette subfamily B member) |
| ISG15/20 (interferon-stimulated gene 15/20) | ISG15 (interferon-stimulated gene 15) | PSMB9 (proteasome 20S subunit beta 9) |
| ITGAX (integrin subunit alpha X) | PRDM16 (PR domain containing 16) | PARP12 (poly ADP ribose polymerase family member 12) |
| PARP12 (poly ADP ribose polymerase family member 12) | RCAN3 (RCAN family member 3) | PDE7A (phosphodiesterase 7A) |
| TNK2 (tyrosine kinase non-receptor 2) | RUNX3 (runt-related transcription factor 3) | IFIT3 (interferon-induced protein with tetratricopeptide repeats 3) |
| DUSP5 (dual specificity protein phosphatase 5) | FAM167B (family with sequence similarity 167-member B) | IFIT1 (interferon-induced protein with tetratricopeptide repeats 1) |
| TET3 (TET methylcytosine dioxygenase 3) | IFI44L (interferon-induced protein 44-like) | IFITM1 (interferon-induced transmembrane protein 1) |
| INPP4A (inositol polyphosphate 4 phosphatase type I A) | PRDX6 (peroxiredoxin 6) | OAS1 (2′-5′-Oligoadenylate synthetase 1) |
| IL1RN (interleukin 1 receptor antagonist) | RABGAP1L (RAB GTPase activating protein 1 like) | NLRC5 (NLR Family CARD domain containing 5) |
| ACVR1 (activin A receptor type 1) | CMPK2 (cytidine/uridine monophosphate kinase 2) | RNF213 (ring finger protein 213) |
| EPHA4 (ephrin type A receptor 4) | RSAD2 (radical S-adenosyl methionine domain containing 2) | ZCCHC2 (zinc finger CCHC-type containing 2) |
| CCDC12 (Coiled-coil domain-containing protein 12) | EIF2AK2 (eukaryotic translation initiation factor 2 alfa kinase 2) | PRIC285 (peroxisomal proliferator-activated receptor A-interacting complex 285 kDa protein isoform 2) |
| TREX1 (Three Prime repair exonuclease 1) | VPS54 (Vacuolar protein sorting associated protein 54) | MX1 (MX dynamin-like GTPase 1) |
| UBA7 (Ubiquitin-like modifier activating enzyme 7) | REG1B (Regenerating family member 1 beta) | GGT1 (γ-glutamyltransferase 1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasking, L.; Roelens, C.; Sprangers, B.; Thienpont, B.; Nawrot, T.S.; De Vusser, K. Lupus, DNA Methylation, and Air Pollution: A Malicious Triad. Int. J. Environ. Res. Public Health 2022, 19, 15050. https://doi.org/10.3390/ijerph192215050
Rasking L, Roelens C, Sprangers B, Thienpont B, Nawrot TS, De Vusser K. Lupus, DNA Methylation, and Air Pollution: A Malicious Triad. International Journal of Environmental Research and Public Health. 2022; 19(22):15050. https://doi.org/10.3390/ijerph192215050
Chicago/Turabian StyleRasking, Leen, Céline Roelens, Ben Sprangers, Bernard Thienpont, Tim S. Nawrot, and Katrien De Vusser. 2022. "Lupus, DNA Methylation, and Air Pollution: A Malicious Triad" International Journal of Environmental Research and Public Health 19, no. 22: 15050. https://doi.org/10.3390/ijerph192215050