The First Evaluation of Proteinase K-Resistant Prion Protein (PrPSc) in Korean Appendix Specimens



Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statements
2.2. Genomic DNA Extraction
2.3. Genetic Analysis
2.4. Protein Extraction
2.5. Proteinase K Digestion
2.6. Western Blotting
3. Results
3.1. Subjects
3.2. Investigation of Genetic Susceptibility Factors for CJD in Korean Patients with Appendectomy
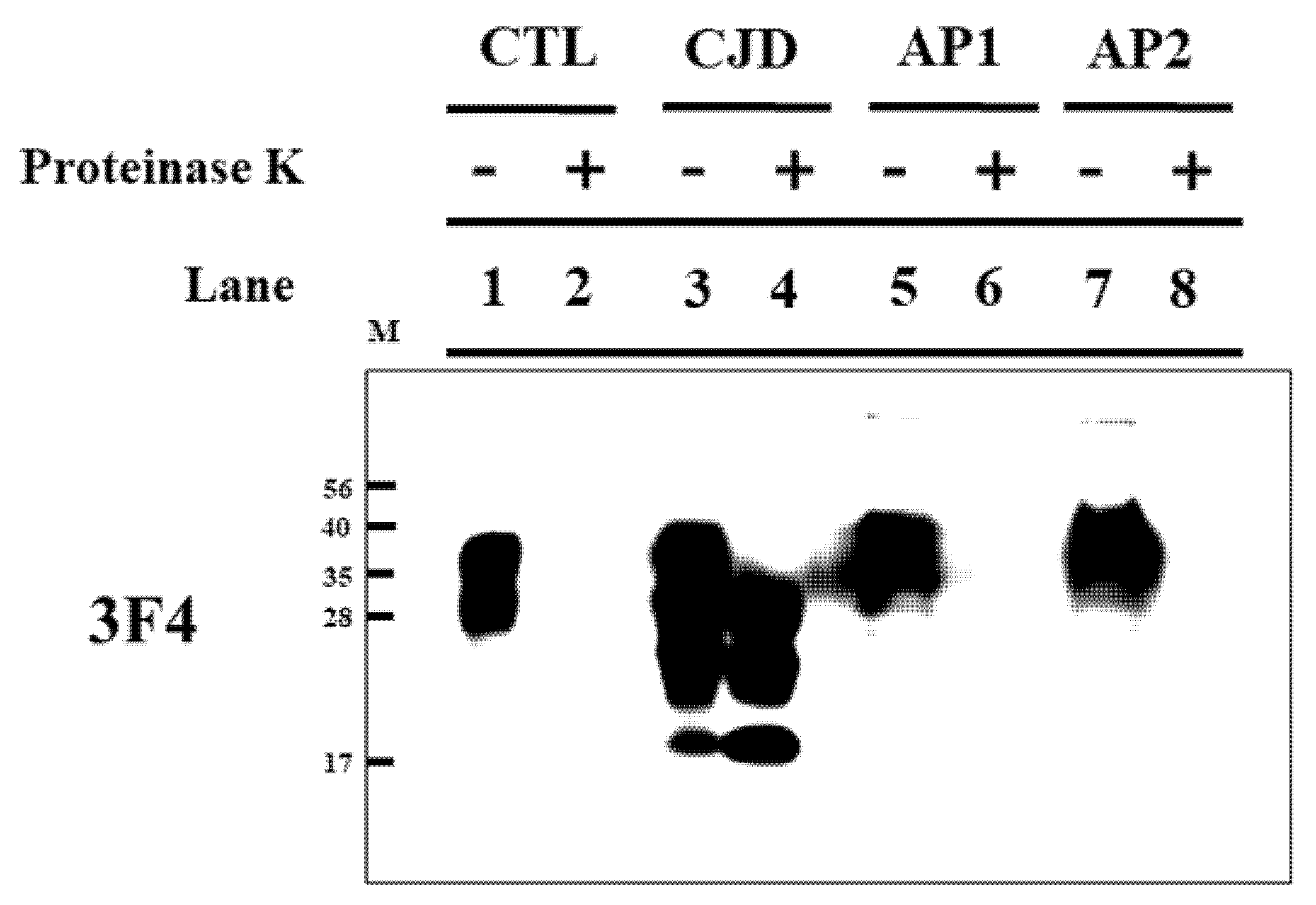
3.3. CJD Inspection in Appendices of Koreans
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [Green Version]
- Wadsworth, J.D.; Collinge, J. Update on human prion disease. Biochim. Biophys. Acta 2007, 1772, 598–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambetti, P.; Kong, Q.; Zou, W.; Parchi, P.; Chen, S.G. Sporadic and familial CJD: Classification and characterisation. Br. Med. Bull. 2003, 66, 213–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uttley, L.; Carroll, C.; Wong, R.; Hilton, D.A.; Stevenson, M. Creutzfeldt-Jakob disease: A systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Dis. 2020, 20, e2–e10. [Google Scholar] [CrossRef]
- Alzualde, A.; Moreno, F.; Martinez-Lage, P.; Ferrer, I.; Gorostidi, A.; Otaegui, D.; Blazquez, L.; Atares, B.; Cardoso, S.; Martinez de Pancorbo, M.; et al. Somatic mosaicism in a case of apparently sporadic Creutzfeldt-Jakob disease carrying a de novo D178N mutation in the PRNP gene. Am. J. Med. Genet. B Neuropsychiatric Genet. 2010, 153, 1283–1291. [Google Scholar] [CrossRef]
- Vollmert, C.; Windl, O.; Xiang, W.; Rosenberger, A.; Zerr, I.; Wichmann, H.E.; Bickeboller, H.; Illig, T.; Kretzschmar, H.A. Significant association of a M129V independent polymorphism in the 5’ UTR of the PRNP gene with sporadic Creutzfeldt-Jakob disease in a large German case-control study. J. Med. Genet. 2006, 43, e53. [Google Scholar] [CrossRef] [Green Version]
- Jeong, B.H.; Kim, Y.S. Genetic studies in human prion diseases. J. Korean Med. Sci. 2014, 29, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.G.; Chenoweth, C.E.; Sullivan, S.E. Iatrogenic Creutzfeldt-Jakob disease via surgical instruments. J. Clin. Neurosci. 2013, 20, 1207–1212. [Google Scholar] [CrossRef]
- Will, R.G.; Knight, R.S.; Ward, H.J.; Ironside, J.W. vCJD: The epidemic that never was. New variant Creutzfeldt-Jakob disease: The critique that never was. BMJ 2002, 325, 102. [Google Scholar] [CrossRef]
- Hill, A.F.; Butterworth, R.J.; Joiner, S.; Jackson, G.; Rossor, M.N.; Thomas, D.J.; Frosh, A.; Tolley, N.; Bell, J.E.; Spencer, M.; et al. Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet 1999, 353, 183–189. [Google Scholar] [CrossRef]
- Gill, O.N.; Spencer, Y.; Richard-Loendt, A.; Kelly, C.; Brown, D.; Sinka, K.; Andrews, N.; Dabaghian, R.; Simmons, M.; Edwards, P.; et al. Prevalence in Britain of abnormal prion protein in human appendices before and after exposure to the cattle BSE epizootic. Acta Neuropathol. 2020, 139, 965–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.; Jaunmuktane, Z.; Joiner, S.; Campbell, T.; Morgan, C.; Wakerley, B.; Golestani, F.; Rudge, P.; Mead, S.; Jager, H.R.; et al. Variant Creutzfeldt-Jakob Disease in a Patient with Heterozygosity at PRNP Codon 129. N. Engl. J. Med. 2017, 376, 292–294. [Google Scholar] [CrossRef] [PubMed]
- McGuire, L.I.; Peden, A.H.; Orru, C.D.; Wilham, J.M.; Appleford, N.E.; Mallinson, G.; Andrews, M.; Head, M.W.; Caughey, B.; Will, R.G.; et al. Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 2012, 72, 278–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, B.H.; Lee, K.H.; Kim, N.H.; Jin, J.K.; Kim, J.I.; Carp, R.I.; Kim, Y.S. Association of sporadic Creutzfeldt-Jakob disease with homozygous genotypes at PRNP codons 129 and 219 in the Korean population. Neurogenetics 2005, 6, 229–232. [Google Scholar] [CrossRef]
- Kim, Y.C.; Jeong, B.H. The First Meta-Analysis of the M129V Single-Nucleotide Polymorphism (SNP) of the Prion Protein Gene (PRNP) with Sporadic Creutzfeldt-Jakob Disease. Cells 2021, 10, 3132. [Google Scholar] [CrossRef]
- Heppner, F.L.; Christ, A.D.; Klein, M.A.; Prinz, M.; Fried, M.; Kraehenbuhl, J.P.; Aguzzi, A. Transepithelial prion transport by M cells. Nat. Med. 2001, 7, 976–977. [Google Scholar] [CrossRef]
- Marshall, A.; Bradford, B.M.; Clarke, A.R.; Manson, J.C.; Mabbott, N.A. Oral Prion Neuroinvasion Occurs Independently of PrP(C) Expression in the Gut Epithelium. J. Virol. 2018, 92, e01010–e01018. [Google Scholar] [CrossRef] [Green Version]
- Hilton, D.A.; Ghani, A.C.; Conyers, L.; Edwards, P.; McCardle, L.; Penney, M.; Ritchie, D.; Ironside, J.W. Accumulation of prion protein in tonsil and appendix: Review of tissue samples. BMJ 2002, 325, 633–634. [Google Scholar] [CrossRef] [Green Version]
- Watson, N.; Brandel, J.P.; Green, A.; Hermann, P.; Ladogana, A.; Lindsay, T.; Mackenzie, J.; Pocchiari, M.; Smith, C.; Zerr, I.; et al. The importance of ongoing international surveillance for Creutzfeldt-Jakob disease. Nat. Rev. Neurol. 2021, 17, 362–379. [Google Scholar] [CrossRef]
- Smith, P.G.; Bradley, R. Bovine spongiform encephalopathy (BSE) and its epidemiology. Br. Med. Bull. 2003, 66, 185–198. [Google Scholar] [CrossRef] [Green Version]
- Priemer, G.; Balkema-Buschmann, A.; Hills, B.; Groschup, M.H. Biochemical Characteristics and PrP(Sc) Distribution Pattern in the Brains of Cattle Experimentally Challenged with H-type and L-type Atypical BSE. PLoS ONE 2013, 8, e67599. [Google Scholar] [CrossRef] [PubMed]
- Biacabe, A.G.; Laplanche, J.L.; Ryder, S.; Baron, T. Distinct molecular phenotypes in bovine prion diseases. EMBO Rep. 2004, 5, 110–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casalone, C.; Zanusso, G.; Acutis, P.; Ferrari, S.; Capucci, L.; Tagliavini, F.; Monaco, S.; Caramelli, M. Identification of a second bovine amyloidotic spongiform encephalopathy: Molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc. Natl. Acad. Sci. USA 2004, 101, 3065–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, J.G.; Langeveld, J.P.; Biacabe, A.G.; Acutis, P.L.; Polak, M.P.; Gavier-Widen, D.; Buschmann, A.; Caramelli, M.; Casalone, C.; Mazza, M.; et al. Molecular discrimination of atypical bovine spongiform encephalopathy strains from a geographical region spanning a wide area in Europe. J. Clin. Microbiol. 2007, 45, 1821–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richt, J.A.; Kunkle, R.A.; Alt, D.; Nicholson, E.M.; Hamir, A.N.; Czub, S.; Kluge, J.; Davis, A.J.; Hall, S.M. Identification and characterization of two bovine spongiform encephalopathy cases diagnosed in the United States. J. Vet. Diagn. Investig. 2007, 19, 142–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakawa, Y.; Hagiwara, K.; Nohtomi, K.; Nakamura, Y.; Nishijima, M.; Higuchi, Y.; Sato, Y.; Sata, T.; Expert Committee for BSE Diagnosis; Ministry of Health; et al. Atypical proteinase K-resistant prion protein (PrPres) observed in an apparently healthy 23-month-old Holstein steer. Jpn. J. Infect. Dis. 2003, 56, 221–222. [Google Scholar]
- Biacabe, A.G.; Morignat, E.; Vulin, J.; Calavas, D.; Baron, T.G. Atypical bovine spongiform encephalopathies, France, 2001–2007. Emerg. Infect. Dis. 2008, 14, 298–300. [Google Scholar] [CrossRef]
- Moore, S.J.; West Greenlee, M.H.; Smith, J.D.; Vrentas, C.E.; Nicholson, E.M.; Greenlee, J.J. A Comparison of Classical and H-Type Bovine Spongiform Encephalopathy Associated with E211K Prion Protein Polymorphism in Wild-Type and EK211 Cattle Following Intracranial Inoculation. Front. Vet. Sci. 2016, 3, 78. [Google Scholar] [CrossRef] [Green Version]
- Richt, J.A.; Hall, S.M. BSE case associated with prion protein gene mutation. PLoS Pathog. 2008, 4, e1000156. [Google Scholar] [CrossRef] [Green Version]
- Won, S.Y.; Kim, Y.C.; Jeong, B.H. First Report of the Potential Bovine Spongiform Encephalopathy (BSE)-Related Somatic Mutation E211K of the Prion Protein Gene (PRNP) in Cattle. Int. J. Mol. Sci. 2020, 21, 4246. [Google Scholar] [CrossRef]
- Gill, O.N.; Spencer, Y.; Richard-Loendt, A.; Kelly, C.; Dabaghian, R.; Boyes, L.; Linehan, J.; Simmons, M.; Webb, P.; Bellerby, P.; et al. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: Large scale survey. BMJ 2013, 347, f5675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, D.A.; Ghani, A.C.; Conyers, L.; Edwards, P.; McCardle, L.; Ritchie, D.; Penney, M.; Hegazy, D.; Ironside, J.W. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J. Pathol. 2004, 203, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Strammiello, R.; Notari, S.; Giese, A.; Langeveld, J.P.; Ladogana, A.; Zerr, I.; Roncaroli, F.; Cras, P.; Ghetti, B.; et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: An updated classification. Acta Neuropathol. 2009, 118, 659–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; Won, S.Y.; Jeong, B.H. Altered expression of glymphatic system-related proteins in prion diseases: Implications for the role of the glymphatic system in prion diseases. Cell. Mol. Immunol. 2021, 18, 2281–2283. [Google Scholar] [CrossRef]


{kind=link}
| Characteristics | Samples | |
|---|---|---|
| Number | 86 | |
| Age (years) | Total | 44.98 ± 24.07 |
| Male | 39.95 ± 24.39 | |
| Female | 50.26 ± 22.85 | |
| Sex (n) | Male | 44 |
| Female | 42 |
| Genotype Frequency, n (%) | Allele Frequency, n (%) | ||||
|---|---|---|---|---|---|
| c.341G > T (G114V) | GG 86 (100) | GT 0 (0) | TT 0 (0) | G 172 (100) | T 0 (0) |
| c.385A > G (M129V) | AA 82 (95.35) | AG 4 (4.65) | GG 0 (0) | A 168 (97.67) | G 4 (2.33) |
| c.532G > A (D178N) | GG 86 (100) | GA 0 (0) | AA 0 (0) | G 172 (100) | A 0 (0) |
| c.538G > A (V180I) | GG 86 (100) | GA 0 (0) | AA 0 (0) | G 172 (100) | A 0 (0) |
| c.549A > G (T183A) | AA 86 (100) | AG 0 (0) | GG 0 (0) | A 172 (100) | G 0 (0) |
| c.563C > A (T188K) | CC 86 (100) | CA 0 (0) | AA 0 (0) | C 172 (100) | A 0 (0) |
| c.586G > A (E196K) | GG 86 (100) | GA 0 (0) | AA 0 (0) | G 172 (100) | A 0 (0) |
| c.598G > A (E200K) | GG 86 (100) | GA 0 (0) | AA 0 (0) | G 172 (100) | A 0 (0) |
| c.607G > A (V203I) | GG 86 (100) | GA 0 (0) | AA 0 (0) | G 172 (100) | A 0 (0) |
| c.623G > A (R208H) | GG 86 (100) | GA 0 (0) | AA 0 (0) | G 172 (100) | A 0 (0) |
| c.628G > A (V210I) | GG 86 (100) | GA 0 (0) | AA 0 (0) | G 172 (100) | A 0 (0) |
| c.631G > C (E211Q) | GG 86 (100) | GC 0 (0) | CC 0 (0) | G 172 (100) | C 0 (0) |
| c.655G > A (E219K) | GG 76 (88.37) | GA 10 (11.63) | AA 0 (0) | G 162 (94.19) | A 10 (5.81) |
| c.695T > G (M232R) | TT 86 (100) | TG 0 (0) | GG 0 (0) | T 172 (100) | G 0 (0) |
| c.712C > T (P238S) | CC 86 (100) | CT 0 (0) | TT 0 (0) | C 172 (100) | T 0 (0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Won, S.-Y.; Kim, Y.-C.; Lee, Y.-N.; Park, C.-G.; Kim, W.-Y.; Jeong, B.-H. The First Evaluation of Proteinase K-Resistant Prion Protein (PrPSc) in Korean Appendix Specimens. Medicina 2022, 58, 947. https://doi.org/10.3390/medicina58070947
Won S-Y, Kim Y-C, Lee Y-N, Park C-G, Kim W-Y, Jeong B-H. The First Evaluation of Proteinase K-Resistant Prion Protein (PrPSc) in Korean Appendix Specimens. Medicina. 2022; 58(7):947. https://doi.org/10.3390/medicina58070947
Chicago/Turabian StyleWon, Sae-Young, Yong-Chan Kim, Yu-Ni Lee, Chan-Gyun Park, Woo-Young Kim, and Byung-Hoon Jeong. 2022. "The First Evaluation of Proteinase K-Resistant Prion Protein (PrPSc) in Korean Appendix Specimens" Medicina 58, no. 7: 947. https://doi.org/10.3390/medicina58070947