
Metabolic Deregulation in Pulmonary Hypertension

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Hemodynamic Data
2.3. Estimation of Protein Expression
2.4. Double Immunofluorescence
2.5. Statistical Analysis
2.6. Gene Expression Data
2.7. Transcriptomic Analyses
2.7.1. Significant Regulation
2.7.2. Measures of Individual Genes’ Contributions to the Transcriptomic Alteration
3. Results
3.1. Significant Increase of the RVSP and RV/LV Weight Ratio in All Three PAH Models
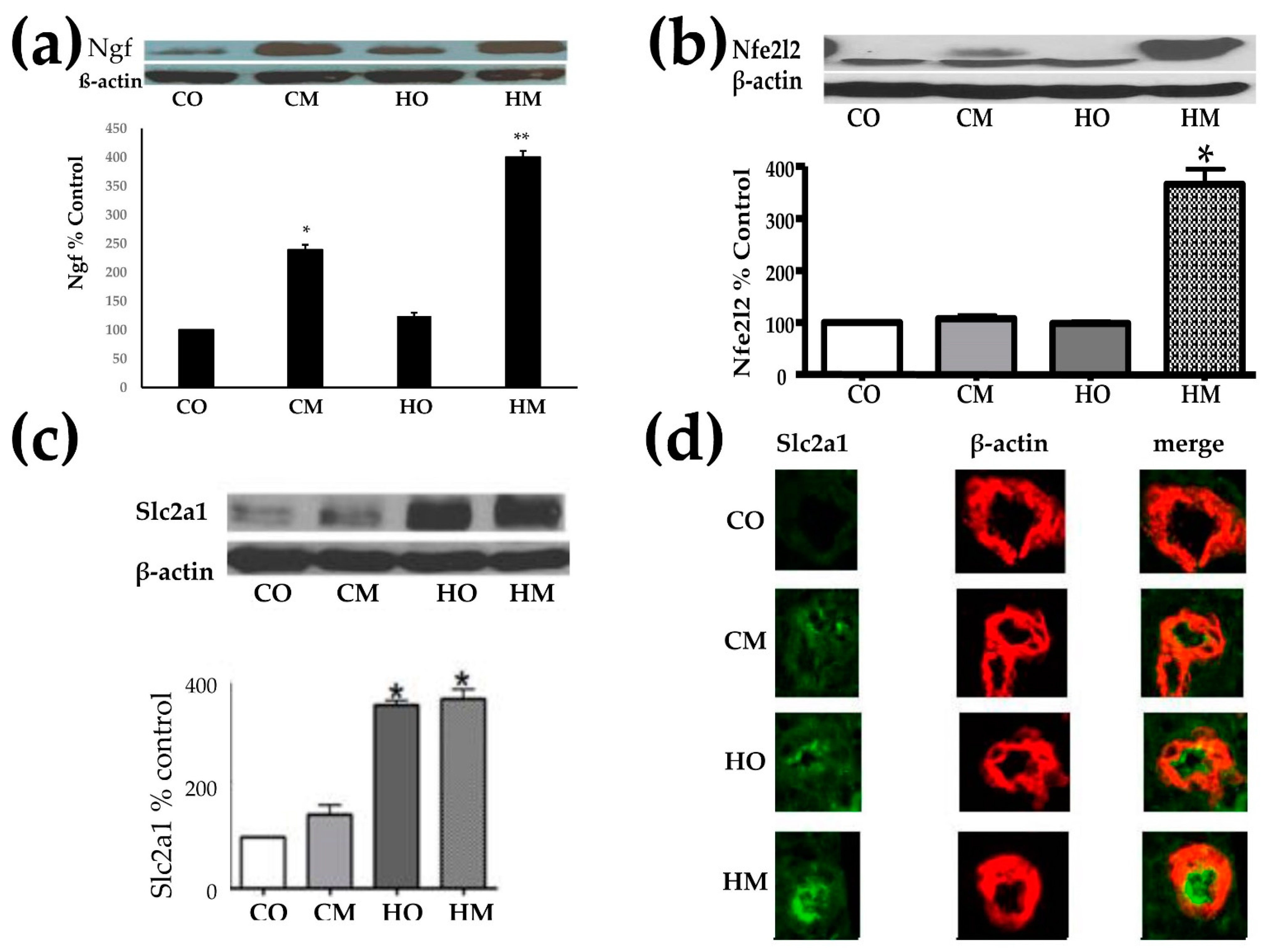
3.2. Pulmonary Arterial Hypertension Increased the Expression of the Nerve Growth Factor (Ngf), Nuclear Factor Erythroid-Derived 2-Like 2 (Nfe2l2), and Glucose Transporter Solute Carrier Family 2 (Slc2a1)
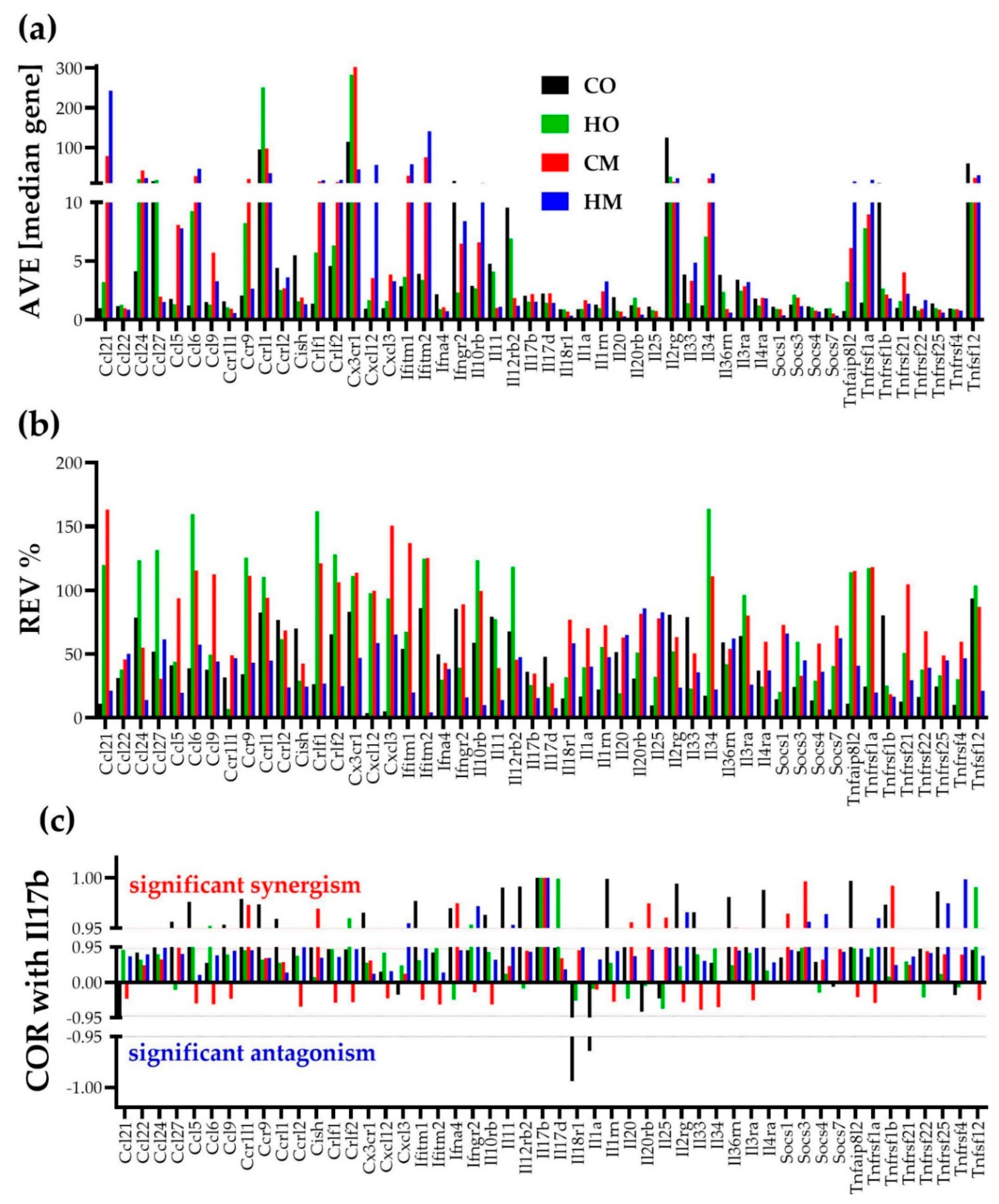
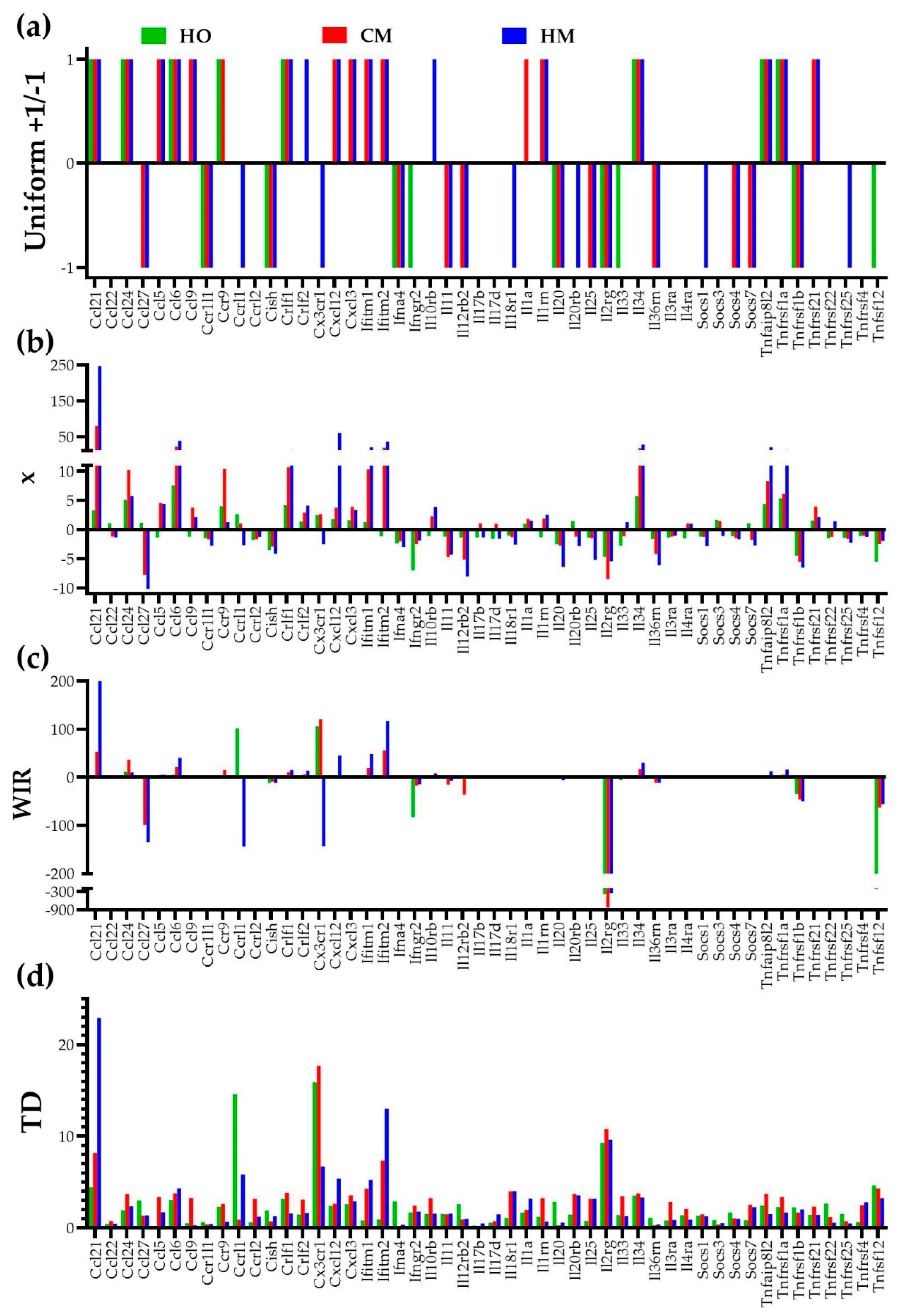
3.3. Independent Transcriptomic Characteristics
3.4. Measures of Transcriptomic Alterations
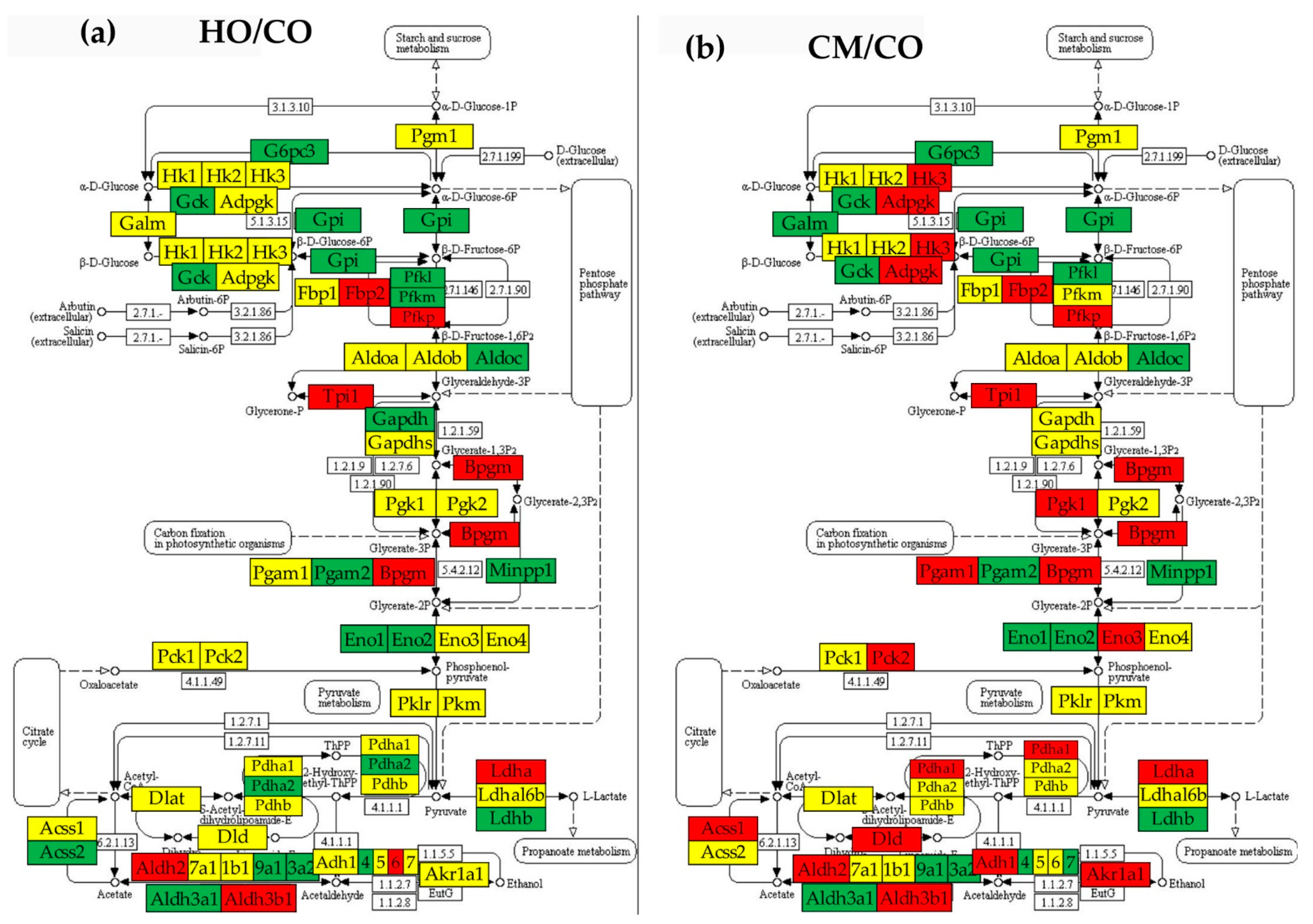
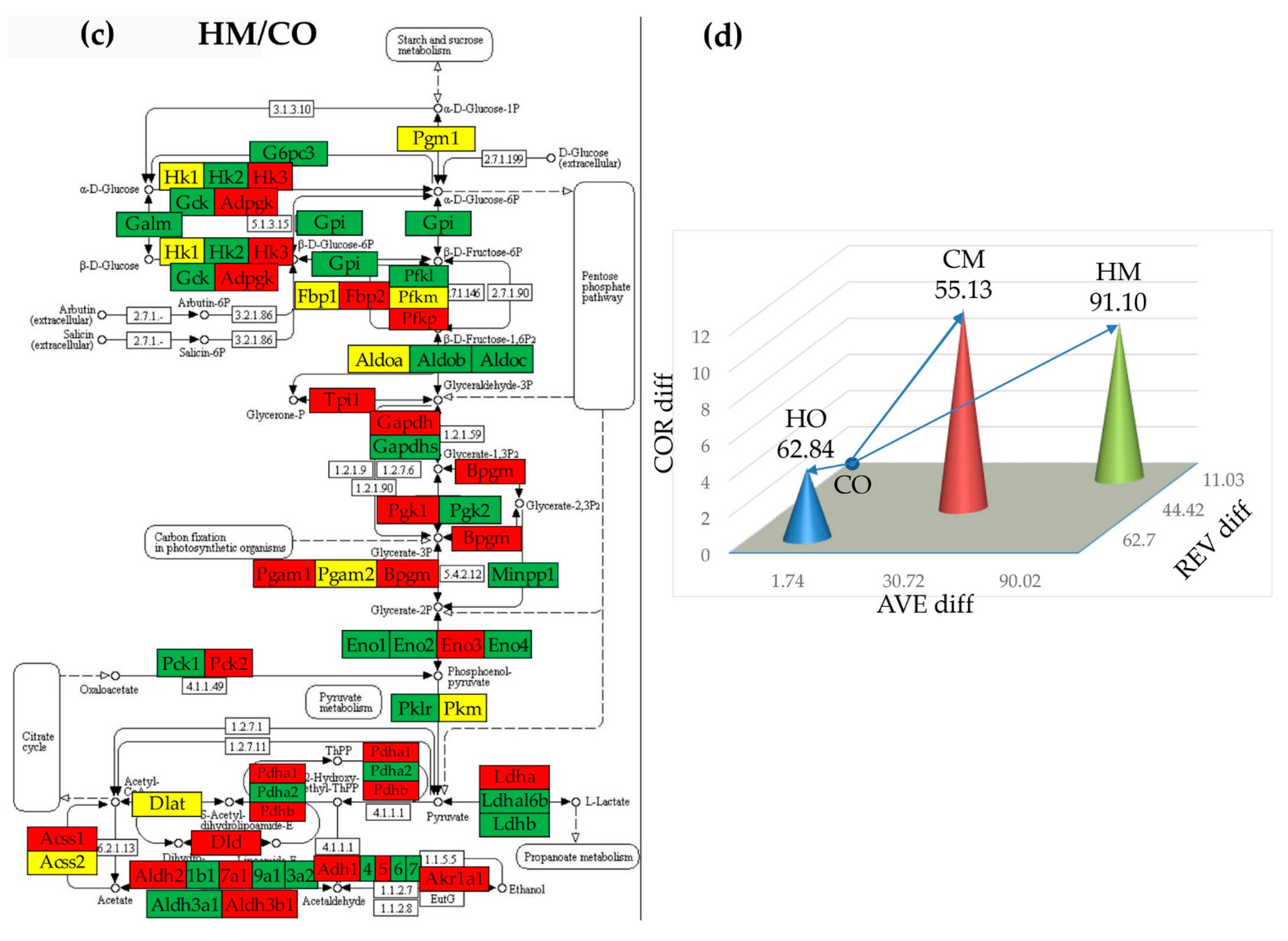
3.5. Regulation of Glycolysis/Gluconeogenesis
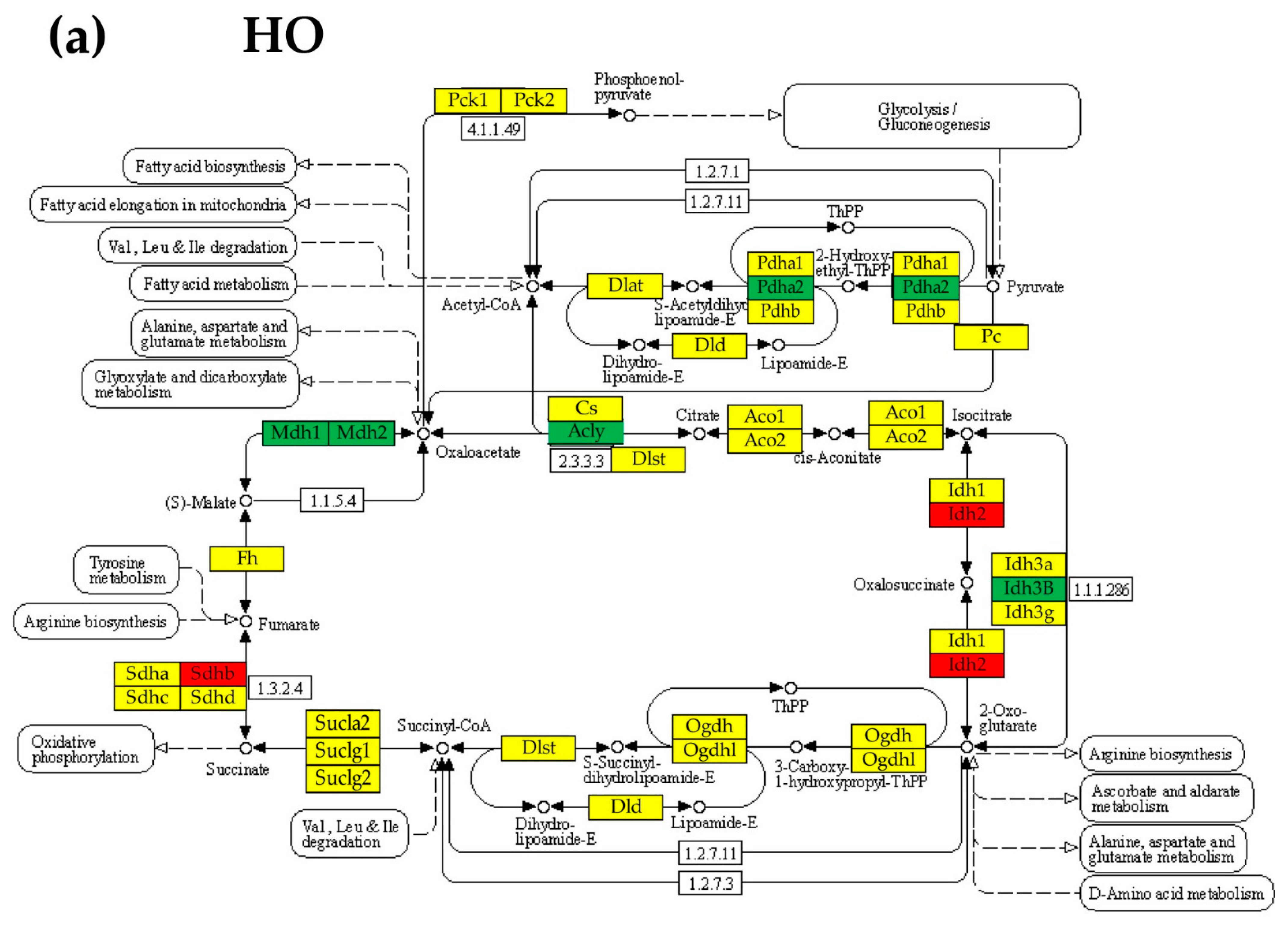
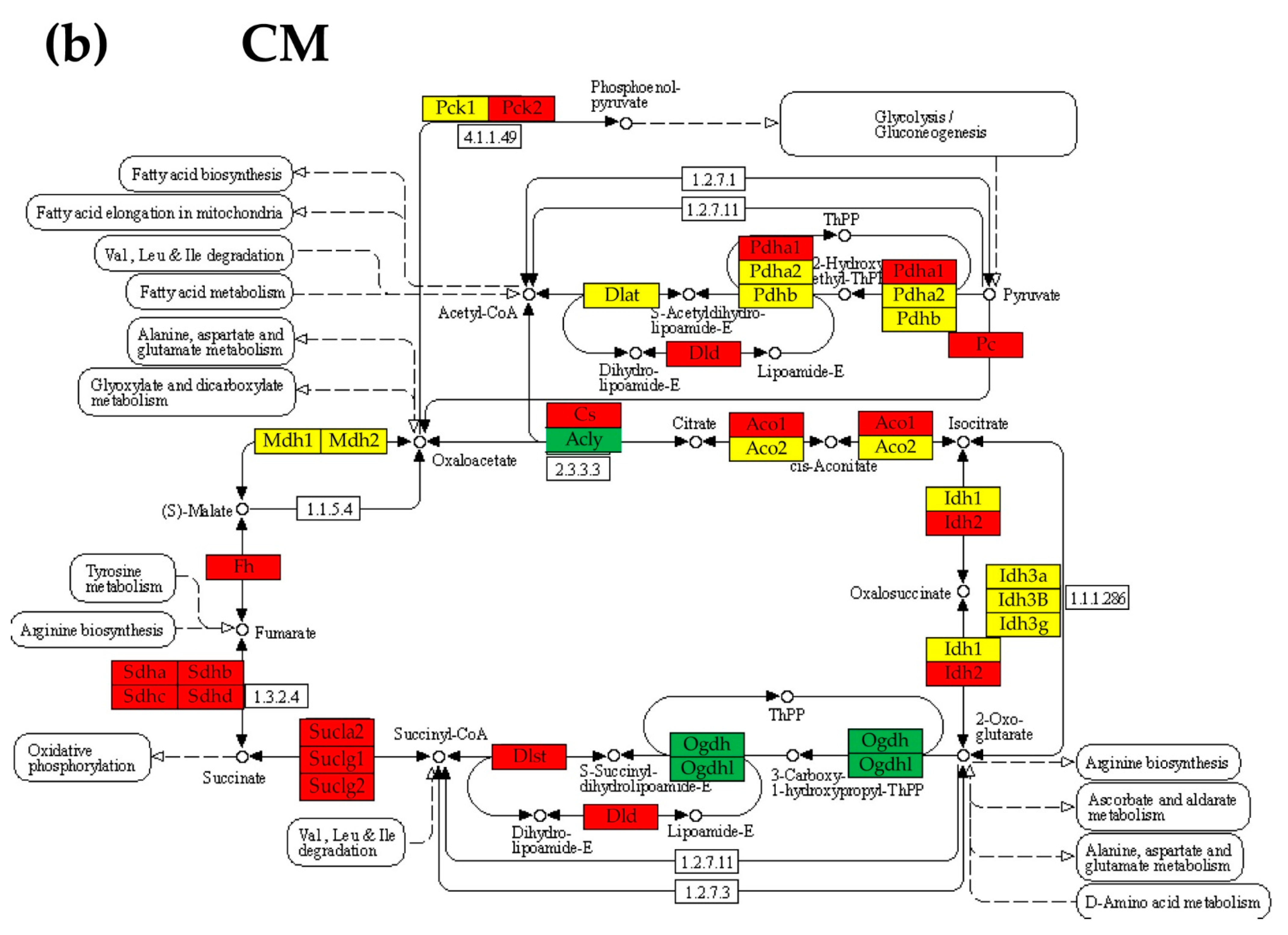
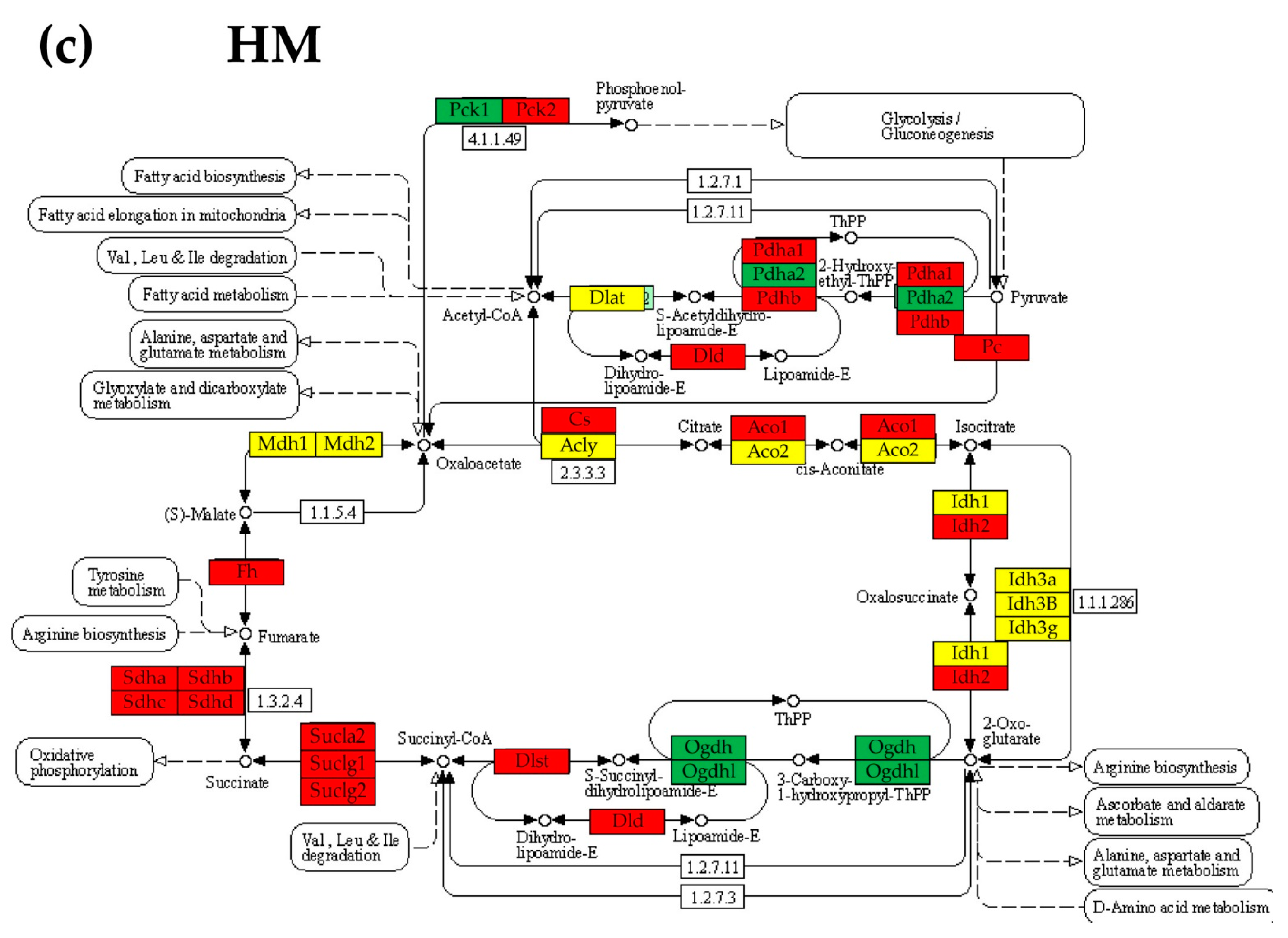
3.6. Regulation of the Citrate (TCA) Cycle
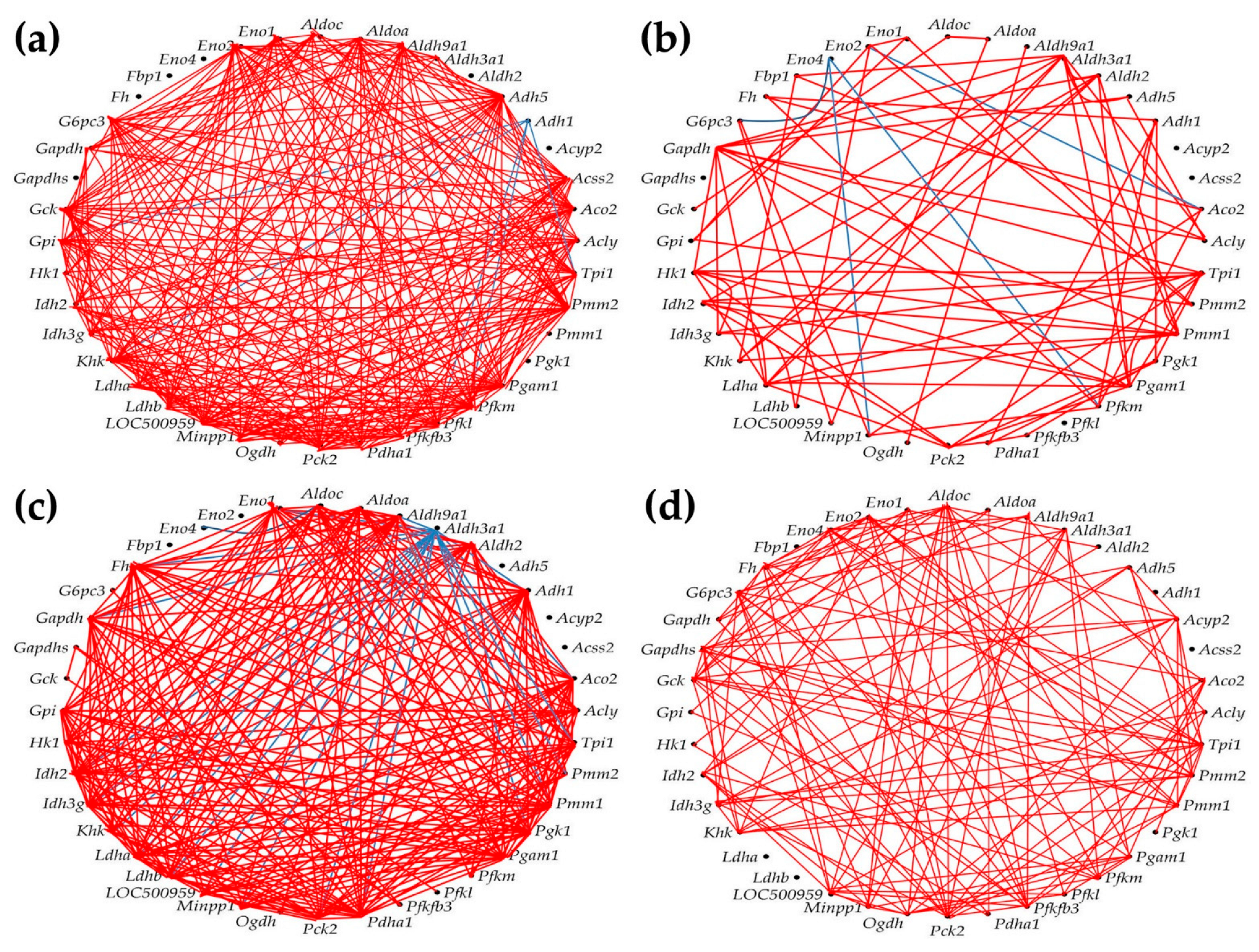
3.7. Remodeling of the Metabolic Genes Network
3.8. Regulation of Key Genes That Control Mitochondrial Membrane Potential and May Be Responsible for PAH Channelopathies
4. Discussion
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- D’Alonzo, G.E.; Barst, R.J.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Kernis, J.T.; et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann. Intern. Med. 1991, 115, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Shah, S.J.; Rich, S.; Tian, L.; Archer, S.L.; Gomberg-Maitland, M. Survival in pulmonary arterial hypertension: A reappraisal of the NIH risk stratification equation. Eur. Respir. J. 2010, 35, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaïci, A.; Weitzenblum, E.; Cordier, J.-F.; Chabot, F.; et al. Survival in Patients with Idiopathic, Familial, and Anorexigen-Associated Pulmonary Arterial Hypertension in the Modern Management Era. Circulation 2010, 122, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemo-dynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Gewitz, M.H. Pulmonary hypertension in infancy and childhood. Heart Dis. 2000, 2, 362–368. [Google Scholar] [PubMed]
- Huang, J.; Wolk, J.H.; Gewitz, M.H.; Mathew, R. Caveolin-1 expression during the progression of pulmonary hypertension. Exp. Biol. Med. 2012, 237, 956–965. [Google Scholar] [CrossRef]
- Huang, J.; Wolk, J.H.; Gewitz, M.H.; E Loyd, J.; West, J.; Austin, E.D.; Mathew, R. Enhanced caveolin-1 expression in smooth muscle cells: Possible prelude to neointima formation. World J. Cardiol. 2015, 7, 671–684. [Google Scholar] [CrossRef]
- Huang, J.; Mathew, R. Loss of cavin1 and expression of p-caveolin-1 in pulmonary hypertension: Possible role in neointima formation. World J. Hypertens. 2019, 9, 17–29. [Google Scholar] [CrossRef]
- Thyberg, J. Differences in Caveolae Dynamics in Vascular Smooth Muscle Cells of Different Phenotypes. Lab. Investig. 2000, 80, 915–929. [Google Scholar] [CrossRef] [Green Version]
- Ajduković, J. HIF-1—A big chapter in cancer tale. Exp. Oncol. 2016, 38, 9–12. [Google Scholar] [CrossRef]
- Ryan, J.; Dasgupta, A.; Huston, J.; Chen, K.-H.; Archer, S.L. Mitochondrial dynamics in pulmonary arterial hypertension. J. Mol. Med. 2015, 93, 229–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiroto, T.; Romero, N.; Sugiyama, T.; Sartoretto, J.L.; Kalwa, H.; Yan, Z.; Shimokawa, H.; Michel, T. Caveolin-1 is a Critical Determinant of Autophagy, Metabolic Switching, and Oxidative Stress in Vascular Endothelium. PLoS ONE 2014, 9, e87871. [Google Scholar] [CrossRef] [Green Version]
- Vallejo, J.; Hardin, C.D. Metabolic organization in vascular smooth muscle: Distribution and localization of caveolin-1 and phosphofructokinase. Am. J. Physiol. Cell Physiol. 2004, 286, C43–C54. [Google Scholar] [CrossRef]
- Xiao, H.; Wang, J.; Yan, W.; Cui, Y.; Chen, Z.; Gao, X.; Wen, X.; Chen, J. GLUT1 regulates cell glycolysis and proliferation in prostate cancer. Prostate 2018, 78, 86–94. [Google Scholar] [CrossRef]
- Huang, J.; Frid, M.; Gewitz, M.H.; Fallon, J.T.; Brown, D.; Krafsur, G.; Stenmark, K.; Mathew, R. Hypoxia-induced pulmonary hypertension and chronic lung disease: Caveolin-1 dysfunction an important underlying feature. Pulm. Circ. 2019, 9, 2045894019837876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morani, F.; Phadngam, S.; Follo, C.; Titone, R.; Aimaretti, G.; Galetto, A.; Alabiso, O.; Isidoro, C. PTEN regulates plasma membrane expression of glucose transporter 1 and glucose uptake in thyroid cancer cells. J. Mol. Endocrinol. 2014, 53, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Khalil, W.; Kahm, J.; Jessurun, J.; Kleidon, J.; Henke, C.A. Pathologic Caveolin-1 Regulation of PTEN in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2010, 176, 2626–2637. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Li, W.; Liu, H.; Zhou, J.-S.; Cao, J.-F.; Zhou, X.-B.; Choi, A.M.; Chen, Z.-H.; Shen, H.-H. Caveolin-1 Inhibits Expression of Antioxidant Enzymes through Direct Interaction with Nuclear Erythroid 2 p45-related Factor-2 (Nrf2). J. Biol. Chem. 2012, 287, 20922–20930. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Yamamoto, M. The KEAP1–NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [Green Version]
- Panda, H.; Wen, H.; Suzuki, M.; Yamamoto, M. Multifaceted Roles of the KEAP1–NRF2 System in Cancer and Inflammatory Disease Milieu. Antioxidants 2022, 11, 538. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Rada, P.; Mendiola, M.; Ortega-Molina, A.; Wojdyla, K.; Rogowska-Wrzesinska, A.; Hardisson, D.; Serrano, M.; Cuadrado, A. The PTEN/NRF2 Axis Promotes Human Carcinogenesis. Antioxid. Redox Signal. 2014, 21, 2498–2541. [Google Scholar] [CrossRef] [PubMed]
- Freund-Michel, V.; Dos Santos, M.C.; Guignabert, C.; Montani, D.; Phan, C.; Coste, F.; Tu, L.; Dubois, M.; Girerd, B.; Courtois, A.; et al. Role of Nerve Growth Factor in Development and Persistence of Experimental Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.-N.; Tan, H.-Q.; Song, A.-B.; Jiang, N.; Liu, Q.-R.; Song, T. NGF/TrkA promotes the vitality, migration and adhesion of bone marrow stromal cells in hypoxia by regulating the Nrf2 pathway. Metab. Brain Dis. 2022, 37, 2017–2026. [Google Scholar] [CrossRef]
- Mathew, R.; Huang, J.; Iacobas, S.; Iacobas, D.A. Pulmonary Hypertension Remodels the Genomic Fabrics of Major Functional Pathways. Genes 2020, 11, 126. [Google Scholar] [CrossRef] [Green Version]
- Lago-Docampo, M.; Solarat, C.; Méndez-Martínez, L.; Baloira, A.; Valverde, D. Common Variation in EDN1 Regulatory Regions Highlights the Role of PPARγ as a Key Regulator of Endothelin in vitro. Front. Cardiovasc. Med. 2022, 25, 823133. [Google Scholar] [CrossRef]
- Castillo-Galán, S.; Riquelme, B.; Iturriaga, R. Crucial Role of Stromal Interaction Molecule-Activated TRPC-ORAI Channels in Vascular Remodeling and Pulmonary Hypertension Induced by Intermittent Hypoxia. Front. Physiol. 2022, 13, 841828. [Google Scholar] [CrossRef]
- Thomas, S.; Manivannan, S.; Garg, V.; Lilly, B. Single-Cell RNA Sequencing Reveals Novel Genes Regulated by Hypoxia in the Lung Vasculature. J. Vasc. Res. 2022, 16, 163–175. [Google Scholar] [CrossRef]
- Iacobas, D.A. Biomarkers, Master Regulators and Genomic Fabric Remodeling in a Case of Papillary Thyroid Carcinoma. Genes 2020, 11, 1030. [Google Scholar] [CrossRef]
- Iacobas, S.; Amuzescu, B.; Iacobas, D.A. Transcriptomic uniqueness and commonality of the ion channels and transporters in the four heart chambers. Sci. Rep. 2021, 11, 2743. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Lee, P.R.; Cohen, J.E.; Fields, R.D. Coordinated Activity of Transcriptional Networks Responding to the Pattern of Action Potential Firing in Neurons. Genes 2019, 10, 754. [Google Scholar] [CrossRef] [Green Version]
- Victorino, P.H.; Marra, C.; Iacobas, D.A.; Iacobas, S.; Spray, D.C.; Linden, R.; Adesse, D.; Petrs-Silva, H. Retinal Genomic Fabric Remodeling after Optic Nerve Injury. Genes 2021, 12, 403. [Google Scholar] [CrossRef]
- Qiagen Ingenuity Pathway Analysis. Available online: https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ (accessed on 20 May 2023).
- DAVID Functional Annotation Bioinformatics Microarray Analysis. Available online: https://david.ncifcrf.gov (accessed on 20 May 2023).
- Kyoto Encyclopedia of Genes and Genomes. Available online: https://www.kegg.jp/kegg/pathway.html (accessed on 20 May 2023).
- Chemokine Signaling Pathway. Available online: https://www.genome.jp/kegg-bin/show_pathway?rno04062 (accessed on 1 April 2023).
- Ramirez-Carrozzi, V.; Ota, N.; Sambandam, A.; Wong, K.; Hackney, J.; Martinez-Martin, N.; Ouyang, W.; Pappu, R. Cutting Edge: IL-17B Uses IL-17RA and IL-17RB to Induce Type 2 Inflammation from Human Lymphocytes. J. Immunol. 2019, 202, 1935–1941. [Google Scholar] [CrossRef] [Green Version]
- Iacobas, S.; Iacobas, D.A. A Personalized Genomics Approach of the Prostate Cancer. Cells 2021, 10, 1644. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Wen, J.; Iacobas, S.; Putterman, C.; Schwartz, N. TWEAKing the Hippocampus: The Effects of TWEAK on the Genomic Fabric of the Hippocampus in a Neuropsychiatric Lupus Mouse Model. Genes 2021, 12, 1172. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, S.; Ede, N.; Iacobas, D.A. The Gene Master Regulators (GMR) Approach Provides Legitimate Targets for Personalized, Time-Sensitive Cancer Gene Therapy. Genes 2019, 10, 560. [Google Scholar] [CrossRef] [Green Version]
- Glycolisis/Glucogenesis Pathway. Available online: https://www.genome.jp/kegg-bin/show_pathway?rno00010 (accessed on 20 May 2023).
- Citrate Cycle (TCA Cycle)—Rattus Norvegicus (Rat). Available online: https://www.genome.jp/kegg-bin/show_pathway?rno00020 (accessed on 20 May 2023).
- Fructose and Manose Metabolism. Available online: https://www.genome.jp/kegg-bin/show_pathway?rno00051 (accessed on 20 May 2023).
- Pyruvate Metabolism. Available online: https://www.genome.jp/kegg-bin/show_pathway?rno00620 (accessed on 20 May 2023).
- Murata, T.; Lin, M.I.; Stan, R.V.; Bauer, P.M.; Yu, J.; Sessa, W.C. Genetic Evidence Supporting Caveolae Microdomain Regulation of Calcium Entry in Endothelial Cells. J. Biol. Chem. 2007, 282, 16631–16643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maqoud, F.; Scala, R.; Hoxha, M.; Zappacosta, B.; Tricarico, D. ATP-sensitive Potassium Channel Subunits in Neuroinflammation: Novel Drug Targets in Neurodegenerative Disorders. CNS Neurol. Disord. Drug Targets 2022, 21, 130–149. [Google Scholar] [CrossRef]
- Zizzo, N.; Passantino, G.; D’Alessio, R.M.; Tinelli, A.; Lopresti, G.; Patruno, R.; Tricarico, D.; Maqoud, F.; Scala, R.; Zito, F.A.; et al. Thymidine Phosphorylase Expression and Microvascular Density Correlation Analysis in Canine Mammary Tumor: Possible Prognostic Factor in Breast Cancer. Front. Veter. Sci. 2019, 6, 368. [Google Scholar] [CrossRef] [Green Version]
- Jouen-Tachoire, T.R.H.; Tucker, S.J.; Tammaro, P. Ion channels as convergence points in the pathology of pulmonary arterial hypertension. Biochem. Soc. Trans. 2021, 49, 1855–1865. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Xi, L. Theory and Applications of the (Cardio) Genomic Fabric Approach to Post-Ischemic and Hypoxia-Induced Heart Failure. J. Pers. Med. 2022, 12, 1246. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, C.; Ye, T.; Chen, X.; Liu, X.; Chen, X.; Sun, Y.; Qu, C.; Liang, J.; Shi, S.; et al. Pinocembrin ameliorates ar-rhythmias in rats with chronic ischaemic heart failure. Ann Med. 2021, 53, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-D.; Lin, Y.-H.; Liang, C.-C.; Chen, T.-C. Effects of sacral nerve stimulation on postpartum urinary retention-related changes in rat bladder. Taiwan J. Obstet. Gynecol. 2015, 54, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, Y.; Chen, T.; Lin, Y.; Gong, J.; Xu, Q.; Wang, J.; Li, J.; Meng, Y.; Li, Y.; et al. Caveolin-1 depletion attenuates hepatic fibrosis via promoting SQSTM1-mediated PFKL degradation in HSCs. Free Radic. Biol. Med. 2023, 204, 95–107. [Google Scholar] [CrossRef]
- Ghaffarian-Ensaf, R.; Shiraseb, F.; Mirzababaei, A.; Clark, C.C.T.; Mirzaei, K. Interaction between caveolin-1 polymorphism and dietary fat quality indexes on visceral adiposity index (VAI) and body adiposity index (BAI) among overweight and obese women: A cross-sectional study. BMC Med. Genom. 2022, 15, 258. [Google Scholar] [CrossRef] [PubMed]
- Ilha, M.; Martins, L.A.M.; da Silveira Moraes, K.; Dias, C.K.; Thomé, M.P.; Petry, F.; Rohden, F.; Borojevic, R.; Trindade, V.M.T.; Klamt, F.; et al. Caveolin-1 influences mitochondrial plasticity and function in hepatic stellate cell activation. Cell Biol. Int. 2022, 46, 1787–1800. [Google Scholar] [CrossRef]
- Luo, S.; Yang, M.; Zhao, H.; Han, Y.; Jiang, N.; Yang, J.; Chen, W.; Li, C.; Liu, Y.; Zhao, C.; et al. Caveolin-1 Regulates Cellular Metabolism: A Potential Therapeutic Target in Kidney Disease. Front. Pharmacol. 2021, 12, 768100. [Google Scholar] [CrossRef]
- Gevezova, M.; Minchev, D.; Pacheva, I.; Todorova, T.; Yordanova, R.; Timova, E.; Ivanov, I.; Sarafian, V. Association of NGF and Mitochondrial Respiration with Autism Spectrum Disorder. Int. J. Mol. Sci. 2022, 23, 11917. [Google Scholar] [CrossRef]
- Selvaraju, V.; Babu, J.R.; Geetha, T. Salivary Neurotrophins Brain-Derived Neurotrophic Factor and Nerve Growth Factor Associated with Childhood Obesity: A Multiplex Magnetic Luminescence Analysis. Diagnostics 2022, 12, 1130. [Google Scholar] [CrossRef]
- Weiss-Sadan, T.; Ge, M.; Hayashi, M.; Gohar, M.; Yao, C.-H.; de Groot, A.; Harry, S.; Carlin, A.; Fischer, H.; Shi, L.; et al. NRF2 activation induces NADH-reductive stress, providing a metabolic vulnerability in lung cancer. Cell Metab. 2023, 35, 487–503.e7. [Google Scholar] [CrossRef]
- Qin, Y.; Qiao, Y.; Wang, D.; Li, L.; Li, M.; Yan, G.; Tang, C. Target Nuclear Factor Erythroid 2-Related Factor 2 in Pulmonary Hypertension: Molecular Insight into Application. Oxidative Med. Cell Longev. 2022, 2022, 7845503. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, W.; Peng, H.; Xiao, Z.; Liu, J.; Zeng, Y.; Huang, T.; Song, Q.; Wang, X.; Xiao, Y. Shikonin improves pulmonary vascular remodeling in monocrotaline-induced pulmonary arterial hypertension via regulation of PKM2. Mol. Med. Rep. 2023, 27, 60. [Google Scholar] [CrossRef] [PubMed]
- Smolders, V.F.E.D.; Rodríguez, C.; Blanco, I.; Szulcek, R.; Timens, W.; Piccari, L.; Roger, Y.; Hu, X.; Morén, C.; Bonjoch, C.; et al. Metabolic profile in endothelial cells of chronic thromboembolic pulmonary hypertension and pulmonary arterial hypertension. Sci. Rep. 2022, 12, 2283. [Google Scholar] [CrossRef]
- Bekedam, F.; Goumans, M.; Bogaard, H.; de Man, F.; Llucià-Valldeperas, A. Molecular mechanisms and targets of right ventricular fibrosis in pulmonary hypertension. Pharmacol. Ther. 2023, 244, 108389. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Lee, H.; Chanda, D.; Thoudam, T.; Kang, H.-J.; Harris, R.A.; Lee, I.-K. The Role of Pyruvate Metabolism in Mitochondrial Quality Control and Inflammation. Mol. Cells 2023, 46, 259–267. [Google Scholar] [CrossRef]
- Hoffmann-Vold, A.-M.; Hesselstrand, R.; Fretheim, H.; Ueland, T.; Andreassen, A.K.; Brunborg, C.; Palchevskiy, V.; Midtvedt, Ø.; Garen, T.; Aukrust, P.; et al. CCL21 as a Potential Serum Biomarker for Pulmonary Arterial Hypertension in Systemic Sclerosis. Arthritis Rheumatol. 2018, 70, 1644–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Wei, X.; Ma, G.-G.; Zhu, F.; Chen, F.-H.; Hu, W. Questions Regarding the Value of CCL21 as a Potential Biomarker for Pulmonary Arterial Hypertension in Systemic Sclerosis: Comment on the Article by Hoffmann-Vold et al. Arthritis Rheumatol. 2019, 71, 653–654. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, X.; Wang, L.; Yang, Q.; Ma, Q.; Xu, J.; Wang, J.; Kovacs, L.; Ayon, R.J.; Liu, Z.; et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2019, 116, 13394–13403. [Google Scholar] [CrossRef] [Green Version]
- Barnett, S.D.; Buxton, I.L.O. The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 340–354. [Google Scholar] [CrossRef] [Green Version]
- Smolle, E.; Leko, P.; Stacher-Priehse, E.; Brcic, L.; El-Heliebi, A.; Hofmann, L.; Quehenberger, F.; Hrzenjak, A.; Popper, H.H.; Olschewski, H.; et al. Distribution and prognostic significance of gluconeogenesis and glycolysis in lung cancer. Mol. Oncol. 2020, 14, 2853–2867. [Google Scholar] [CrossRef]
- Pirard, M.; Achouri, Y.; Collet, J.F.; Schollen, E.; Matthijs, G.; Van Schaftingen, E. Kinetic properties and tissular distribution of mammalian phosphomannomutase isozymes. Biochem. J. 1999, 339 Pt 1, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Voulgaridou, G.-P.; Theologidis, V.; Xanthis, V.; Papagiannaki, E.; Tsochantaridis, I.; Fadouloglou, V.E.; Pappa, A. Identification of a peptide ligand for human ALDH3A1 through peptide phage display: Prediction and characterization of protein interaction sites and inhibition of ALDH3A1 enzymatic activity. Front. Mol. Biosci. 2023, 10, 1161111. [Google Scholar] [CrossRef]
- Tan, R.; Li, J.; Peng, X.; Zhu, L.; Cai, L.; Wang, T.; Su, Y.; Irani, K.; Hu, Q. GAPDH is critical for superior efficacy of female bone marrow-derived mesenchymal stem cells on pulmonary hypertension. Cardiovasc. Res. 2013, 100, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salcin, S.; Fontem, F. Recurrent SARS-CoV-2 infection resulting in acute respiratory distress syndrome and development of pulmonary hypertension: A case report. Respir. Med. Case Rep. 2021, 33, 101314. [Google Scholar] [CrossRef]
- Eroume, À.; Egom, E.E.; Shiwani, H.A.; Nouthe, B. From acute SARS-CoV-2 infection to pulmonary hypertension. Front. Physiol. 2022, 13, 1023758. [Google Scholar] [CrossRef]
- Torge, D.; Bernardi, S.; Arcangeli, M.; Bianchi, S. Histopathological Features of SARS-CoV-2 in Extrapulmonary Organ Infection: A Systematic Review of Literature. Pathogens 2022, 11, 867. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathew, R.; Iacobas, S.; Huang, J.; Iacobas, D.A. Metabolic Deregulation in Pulmonary Hypertension. Curr. Issues Mol. Biol. 2023, 45, 4850-4874. https://doi.org/10.3390/cimb45060309
Mathew R, Iacobas S, Huang J, Iacobas DA. Metabolic Deregulation in Pulmonary Hypertension. Current Issues in Molecular Biology. 2023; 45(6):4850-4874. https://doi.org/10.3390/cimb45060309
Chicago/Turabian StyleMathew, Rajamma, Sanda Iacobas, Jing Huang, and Dumitru Andrei Iacobas. 2023. "Metabolic Deregulation in Pulmonary Hypertension" Current Issues in Molecular Biology 45, no. 6: 4850-4874. https://doi.org/10.3390/cimb45060309