
Molecular Changes in Cells of Patients with Type 2 Diabetes Mellitus Depending on Changes in Glycemia Level in the Context of Lifestyle—An Overview of the Latest Scientific Discoveries
 , ,
, ,
Abstract
:1. Introduction
2. Lifestyle Factors Affecting Blood Glucose Level in the Context of Molecular Discoveries

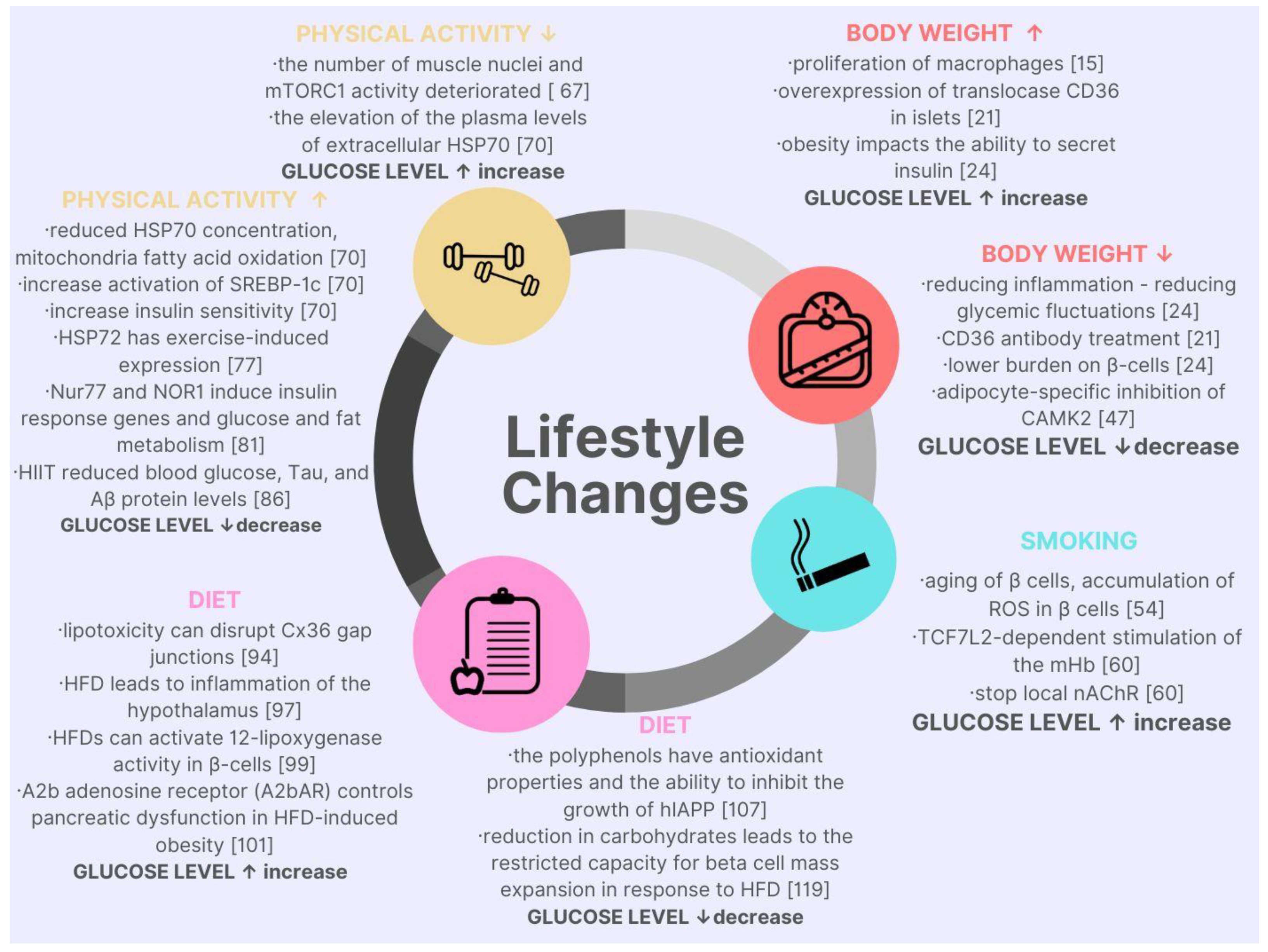{kind=link}
| Factor | Molecular Cause | Metabolic Response |
|---|---|---|
| Body mass | “macrophages impair the ability to secrete insulin by β cells” [11] | “insulin resistance leads to hyperglycemia” [12] |
| “fluctuations of blood glucose levels are promoting inflammation caused by the accumulation of circulating SFA” [13] | “lipotoxicity” [13] | |
| “translocase CD36 as a FA transporter” [14] | “a key molecule in the reduced insulin within higher body weight” [14] | |
| “the occurrence of low blood sugar after RYGB” [15] | “moderate hypoglycemia impairs neurological function; severe hypoglycemia leads to the death of neurons” [16] “hypoglycemia may cause a proarrhythmic effect” [17] | |
| “calcium-sensitive kinase, calcium/calmodulin dependent-protein kinase II (CaMKK), plays a critical role in glucose homeostasis” [18,19,20] | “CaMKK regulates insulin signaling and glucose homeostasis throughout the body and in lipolysis and adipocyte inflammation.” [21] | |
| Smoking | “nicotine determinate aging of β cells of the pancreas” [22] | “impaired glucose metabolism, higher glucose variability and worse course of previously diagnosed T2DM” [22] |
| “exposure to nicotine changed the expression of aging proteins such as p16, p19, p21 when exposed to higher values” [22] | ||
| “nicotine exposition might lead to hdl accumulation in pancreatic islets cells” | ||
| “gene TCF7L2 was densely expressed in the mHb region of the rodent brain and regulated the function of nicotinic acetylcholine receptors” [23] | “nicotine-induced increase in blood glucose may affect the response of mHb neurons and stop local NACHR function causing habitual tobacco smoking” [23] | |
| “increased circulating glucose levels can modulate mHb function” [23] | ||
| Physical activity | “the elevation of the plasma levels of extracellular HSP70 is connected to obesity and diabetes” [24] | “reduced extracellular HSP70 concentration can inhibit inflammation, mitochondrial fatty acid oxidation, and increase activation of SREBP-1c, which is a gene transcription factor in ER stress” [25] |
| “increased HSP70 expression in brain cells can increase insulin sensitivity, normalize blood glucose level” [24] | ||
| “long-term exercises increased the production of intracellular HSP70 in the muscle, the liver, kidneys, and heart” [26] | ||
| “HSP72 has exercise-induced expression” [27] | “reduce the pro-inflammatory cytokines, increase insulin sensitivity” [27] | |
| “intracellular HSP-72 is anti-inflammatory by blocking the activity of the jnk and nk-kb pathways” [27] | ||
| “physical activity in connection to an increase in tissue temperature that occurs during exercise can increase HSP72 concentration” [27] | ||
| “in human skeletal muscles is noted two proteins of the orphan nuclear receptor family, Nur77 and NOR1” [28] | “induce insulin response genes and glucose and fat metabolism” [29] | |
| “aerobic exercise strongly increased Nur77 and NOR1 in a healthy population” [30] | ||
| “diabetes can lead to increased levels of tau protein and Aβ” [31] | “HIIT significantly reduced blood glucose and tau and Aβ protein levels.” [32] | |
| Diet | “there is no difference in HbA1c decrease in the Mediterranean diet compared a low-fat diet. there was a significant decrease in fasting glucose concentration in the Mediterranean group compared to the low-fat diet” [33] | “it can be concluded that the fluctuations in both hyperglycemia that make up the mean HbA1c score are reduced in the Mediterranean diet” [33] |
| “human islet amyloid polypeptide—hIAPP is probably responsible for the loss of β cell function, and death” [34] | “the polyphenols found in virgin olive oil have antioxidant properties and the ability to inhibit the growth of human islet amyloid polypeptide—hIAPP” [35] | |
| “lipotoxicity can disrupt Cx36 gap junctions couplings within the islets in diet-induced obesity” [36] | “decrease in insulin secretion and variability in glycemic homeostasis [36] | |
| “the mechanism of action of HFD contributing to the disorders of homeostasis is multidirectional.” [37] | “aggregation of Aβ and hyperphosphorylated Tau protein” [37] “chronic overconsumption of HFDs leads to impaired glucose homeostasis, insulin resistance and T2DM” [38] |
2.1. Body Mass
2.2. Smoking
2.3. Physical Activity
2.4. Diet
3. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- World Health Organisation. Diabetes-Key Facts. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 28 May 2022).
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polskie Towarzystwo Diabetologiczne. Guidelines on the Managment of Diabetic Patients. 2020. Available online: https://cukrzyca.info.pl/zalecenia_kliniczne/2020_guidelines_on_the_management_of_diabetic_patients (accessed on 28 May 2022).
- Association, A.D. 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes-2021. Diabetes Care 2021, 44, S15–S33. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, R.A.; Guthrie, D.W. Pathophysiology of Diabetes Mellitus. Crit. Care Nurs. Q. 2004, 27, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Pasquel, F.J.; Lansang, M.C.; Dhatariya, K.; Umpierrez, G.E. Management of Diabetes and Hyperglycaemia in the Hospital. Lancet Diabetes Endocrinol. 2021, 9, 174–188. [Google Scholar] [CrossRef]
- Lauffenburger, J.C.; Lewey, J.; Jan, S.; Lee, J.; Ghazinouri, R.; Choudhry, N.K. Association of Potentially Modifiable Diabetes Care Factors with Glycemic Control in Patients with Insulin-Treated Type 2 Diabetes. JAMA Netw. Open 2020, 3, e1919645. [Google Scholar] [CrossRef]
- Khattab, M.; Khader, Y.S.; Al-Khawaldeh, A.; Ajlouni, K. Factors Associated with Poor Glycemic Control among Patients with Type 2 Diabetes. J. Diabetes Complicat. 2010, 24, 84–89. [Google Scholar] [CrossRef]
- Cisińska, G.; Moczulski, D. Analiza Czynników Ryzyka Cukrzycy Na Podstawie Ankiety FINDRISC Analysis of Risk Factors of Diabetes Based on FINDRISC Survey. Hygeia Public Health 2013, 48, 200–204. [Google Scholar]
- Aghili, R.; Polonsky, W.H.; Valojerdi, A.E.; Malek, M.; Keshtkar, A.A.; Esteghamati, A.; Heyman, M.; Khamseh, M.E. Type 2 Diabetes: Model of Factors Associated with Glycemic Control. Can. J. diabetes 2016, 40, 424–430. [Google Scholar] [CrossRef]
- Böni-Schnetzler, M.; Meier, D.T. Islet inflammation in type 2 diabetes. Semin. Immunopathol. 2019, 41, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; Seo, J.B.; Yang, B.H.; Wollam, J.; Riopel, M.; et al. Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting β Cell Proliferation and Function in Obesity. Cell Metab. 2019, 29, 457–474.e5. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Gordillo, R.; Shao, M.; Onodera, T.; Chen, Z.; Chen, S.; Lin, X.; SoRelle, J.A.; Li, X.; Tang, M.; et al. Intracellular lipid metabolism impairs β cell compensation during diet-induced obesity. J. Clin. Investig. 2018, 128, 1178–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, M.; Esguerra, J.L.S.; Asai, A.; Ofori, J.K.; Edlund, A.; Wendt, A.; Sugihara, H.; Wollheim, C.B.; Oikawa, S.; Eliasson, L. Potential protection against type 2 diabetes in obesity through lower CD36 expression and improved exocytosis in β-cells. Diabetes 2020, 69, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Salehi, M.; Vella, A.; McLaughlin, T.; Patti, M.E. Hypoglycemia After Gastric Bypass Surgery: Current Concepts and Controversies. J. Clin. Endocrinol. Metab. 2018, 103, 2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaev, N.K.; Stel’Mashuk, E.V.; Zorov, D.B. Cellular Mechanisms of Brain Hypoglycemia. Biochemistry 2007, 72, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Languren, G.; Montiel, T.; Julio-Amilpas, A.; Massieu, L. Neuronal Damage and Cognitive Impairment Associated with Hypoglycemia: An Integrated View. Neurochem. Int. 2013, 63, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Wong, C.C.L.; Li, G.; Xu, T.; Pajvani, U.; Park, S.K.R.; Wronska, A.; Chen, B.X.; Marks, A.R.; Fukamizu, A.; et al. Calcium Signaling through CaMKII Regulates Hepatic Glucose Production in Fasting and Obesity. Cell Metab. 2012, 15, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yan, S.; Xiao, B.; Zuo, S.; Zhang, Q.; Chen, G.; Yu, Y.; Chen, D.; Liu, Q.; Liu, Y.; et al. Prostaglandin F2α Facilitates Hepatic Glucose Production Through CaMKIIγ/P38/FOXO1 Signaling Pathway in Fasting and Obesity. Diabetes 2018, 67, 1748–1760. [Google Scholar] [CrossRef] [Green Version]
- Perry, R.J.; Zhang, D.; Guerra, M.T.; Brill, A.L.; Goedeke, L.; Nasiri, A.R.; Rabin-Court, A.; Wang, Y.; Peng, L.; Dufour, S.; et al. Glucagon Stimulates Gluconeogenesis by INSP3R1-Mediated Hepatic Lipolysis. Nature 2020, 579, 279–283. [Google Scholar] [CrossRef]
- Dai, W.; Choubey, M.; Patel, S.; Singer, H.A.; Ozcan, L. Adipocyte CAMK2 Deficiency Improves Obesity-Associated Glucose Intolerance. Mol. Metab. 2021, 53, 101300. [Google Scholar] [CrossRef]
- Sun, L.; Wang, X.; Gu, T.; Hu, B.; Luo, J.; Qin, Y.; Wan, C. Nicotine triggers islet β cell senescence to facilitate the progression of type 2 diabetes. Toxicology 2020, 441, 152502. [Google Scholar] [CrossRef]
- Duncan, A.; Heyer, M.P.; Ishikawa, M.; Caligiuri, S.P.B.; Liu, X.-A.; Chen, Z.; Di Bonaventura, M.V.M.; Elayouby, K.S.; Ables, J.L.; Howe, W.M.; et al. Habenular TCF7L2 links nicotine addiction to diabetes. Nature 2019, 574, 372–377. [Google Scholar] [CrossRef]
- Krause, M.; Bock, P.M.; Takahashi, H.K.; De Bittencourt, P.I.H.; Newsholme, P. The regulatory roles of NADPH oxidase, intra- and extra-cellular HSP70 in pancreatic islet function, dysfunction and diabetes. Clin. Sci. 2015, 128, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar]
- Noble, E.G.; Ho, R.; Dzialoszynski, T. Exercise is the primary factor associated with Hsp70 induction in muscle of treadmill running rats. Acta Physiol. 2006, 187, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, T.; Kobayashi, H.; Yoshihara, T.; Kakigi, R.; Ichinoseki-Sekine, N.; Naito, H. Attenuation of exercise-induced heat shock protein 72 expression blunts improvements in whole-body insulin resistance in rats with type 2 diabetes. Cell Stress Chaperones 2017, 22, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Wang, J.; Cui, X.; Maianu, L.; Rhees, B.; Rosinski, J.; So, W.V.; Willi, S.M.; Osier, M.V.; Hill, H.S.; et al. The effect of insulin on expression of genes and biochemical pathways in human skeletal muscle. Endocrine 2007, 31, 5–17. [Google Scholar] [CrossRef]
- Chao, L.C.; Wroblewski, K.; Ilkayeva, O.R.; Stevens, R.D.; Bain, J.; Meyer, G.A.; Schenk, S.; Martinez, L.; Vergnes, L.; Narkar, V.A.; et al. Skeletal muscle Nur77 expression enhances oxidative metabolism and substrate utilization. J. Lipid Res. 2012, 53, 2610–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mey, J.T.; Solomon, T.P.J.; Kirwan, J.P.; Haus, J.M. Skeletal muscle Nur77 and NOR1 insulin responsiveness is blunted in obesity and type 2 diabetes but improved after exercise training. Physiol. Rep. 2019, 7, e14042. [Google Scholar] [CrossRef]
- Castellani, R.J.; Rolston, R.K.; Smith, M.A. Alzheimer Disease. Dis. Mon. 2010, 56, 484. [Google Scholar] [CrossRef]
- Orumiyehei, A.; Khoramipour, K.; Rezaei, M.H.; Madadizadeh, E.; Meymandi, M.S.; Mohammadi, F.; Chamanara, M.; Bashiri, H.; Suzuki, K. High-Intensity Interval Training-Induced Hippocampal Molecular Changes Associated with Improvement in Anxiety-like Behavior but Not Cognitive Function in Rats with Type 2 Diabetes. Brain Sci. 2022, 12, 1280. [Google Scholar] [CrossRef]
- Shai, I.; Schwarzfuchs, D.; Henkin, Y.; Shahar, D.R.; Witkow, S.; Greenberg, I.; Golan, R.; Fraser, D.; Bolotin, A.; Vardi, H.; et al. Weight Loss with a Low-Carbohydrate, Mediterranean, or Low-Fat Diet. N. Engl. J. Med. 2008, 359, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Leri, M.; Natalello, A.; Bruzzone, E.; Stefani, M.; Bucciantini, M. Oleuropeinaglycone and hydroxytyrosol interfere differently with toxic Aβ 1-42 aggregation. Food Chem. Toxicol. 2019, 129, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M.; Rigacci, S. Protein Folding and Aggregation into Amyloid: The Interference by Natural Phenolic Compounds. Int. J. Mol. Sci. 2013, 14, 12411–12457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- do Amaral, M.E.C.; Kravets, V.; Dwulet, J.A.M.; Farnsworth, N.L.; Piscopio, R.; Schleicher, W.E.; Miranda, J.G.; Benninger, R.K.P. Caloric restriction recovers impaired b-cell-b-cell gap junction coupling, calcium oscillation coordination, and insulin secretion in prediabetic mice. Am. J. Physiol.-Endocrinol. Metab. 2020, 319, E709–E720. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Milanski, M.; Coope, A.; Torsoni, A.S.; Ropelle, E.; Carvalho, D.P.; Carvalheira, J.B.; Velloso, L.A. Low-Grade Hypothalamic Inflammation Leads to Defective Thermogenesis, Insulin Resistance, and Impaired Insulin Secretion. Endocrinology 2011, 152, 1314–1326. [Google Scholar] [CrossRef] [Green Version]
- Johnston-Cox, H.; Koupenova, M.; Yang, D.; Corkey, B.; Gokce, N.; Farb, M.G.; LeBrasseur, N.; Ravid, K. The A2b Adenosine Receptor Modulates Glucose Homeostasis and Obesity. PLoS ONE 2012, 7, e40584. [Google Scholar] [CrossRef] [Green Version]
- Krishan, P.; Bedi, O.; Rani, M. Impact of diet restriction in the management of diabetes: Evidences from preclinical studies. Naunyn. Schmiedebergs. Arch. Pharmacol. 2018, 391, 235–245. [Google Scholar] [CrossRef]
- Lee, Y.S.; Wollam, J.; Olefsky, J.M. An Integrated View of Immunometabolism. Cell 2018, 172, 22–40. [Google Scholar] [CrossRef] [Green Version]
- Butcher, M.J.; Hallinger, D.; Garcia, E.; Machida, Y.; Chakrabarti, S.; Nadler, J.; Galkina, E.V.; Imai, Y. Association of proinflammatory cytokines and islet resident leucocytes with islet dysfunction in type 2 diabetes. Diabetologia 2014, 57, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Wang, M.; Wang, Q.A.; Scherer, P.E. Adiponectin-mediated antilipotoxic effects in regenerating pancreatic islets. Endocrinology 2015, 156, 2019–2028. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Fu, W.; Lee, Y.S.; Olefsky, J.M. The role of macrophages in obesity-associated islet inflammation and β-cell abnormalities. Nat. Rev. Endocrinol. 2020, 16, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Rosengren, A.H.; Braun, M.; Mahdi, T.; Andersson, S.A.; Travers, M.E.; Shigeto, M.; Zhang, E.; Almgren, P.; Ladenvall, C.; Axelsson, A.S.; et al. Reduced insulin exocytosis in human pancreatic β-cells with gene variants linked to type 2 diabetes. Diabetes 2012, 61, 1726–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noushmehr, H.; D’Amico, E.; Farilla, L.; Hui, H.; Wawrowsky, K.A.; Mlynarski, W.; Doria, A.; Abumrad, N.A.; Perfetti, R. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic β-cells and mediates fatty acid effects on insulin secretion. Diabetes 2005, 54, 472–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, C.; Borg, L.A.H.; Welsh, N. Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology 1999, 140, 3422–3428. [Google Scholar] [CrossRef]
- Dixon, J.B.; Blazeby, J.M. Quality of life after bariatric surgery. Lancet Diabetes Endocrinol. 2014, 2, 100–102. [Google Scholar] [CrossRef]
- Zuo, D.; Xiao, X.; Yang, S.; Gao, Y.; Wang, G.; Ning, G. Effects of bariatric surgery in Chinese with obesity and type 2 diabetes mellitus: A 3-year follow-up. Medicine 2020, 99, e21673. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.J.; Moon, J.H.; Choi, S.H.; Lim, S.; Park, K.S.; Cho, N.H.; Jang, H.C. Body-Weight Fluctuation and Incident Diabetes Mellitus, Cardiovascular Disease, and Mortality: A 16-Year Prospective Cohort Study. J. Clin. Endocrinol. Metab. 2019, 104, 639–646. [Google Scholar] [CrossRef]
- Yeboah, P.; Hsu, F.C.; Bertoni, A.G.; Yeboah, J. Body Mass Index, Change in Weight, Body Weight Variability and Outcomes in Type 2 Diabetes Mellitus [from the ACCORD Trial]. Am. J. Cardiol. 2019, 123, 576–581. [Google Scholar] [CrossRef]
- Salehi, M.; Gastaldelli, A.; D’Alessio, D.A. Altered Islet Function and Insulin Clearance Cause Hyperinsulinemia in Gastric Bypass Patients with Symptoms of Postprandial Hypoglycemia. J. Clin. Endocrinol. Metab. 2014, 99, 2008–2017. [Google Scholar] [CrossRef] [Green Version]
- Salehi, M.; Gastaldelli, A.; D’Alessio, D.A. Blockade of Glucagon-like Peptide 1 Receptor Corrects Postprandial Hypoglycemia after Gastric Bypass. Gastroenterology 2014, 146, 669–680.e2. [Google Scholar] [CrossRef] [Green Version]
- Salehi, M.; D’Alessio, D.A. Effects of Glucagon like Peptide-1 to Mediate Glycemic Effects of Weight Loss Surgery. Rev. Endocr. Metab. Disord. 2014, 15, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Laferrère, B.; Teixeira, J.; McGinty, J.; Tran, H.; Egger, J.R.; Colarusso, A.; Kovack, B.; Bawa, B.; Koshy, N.; Lee, H.; et al. Effect of Weight Loss by Gastric Bypass Surgery versus Hypocaloric Diet on Glucose and Incretin Levels in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 2479–2485. [Google Scholar] [CrossRef]
- Sang, W.S.; Hamby, A.M.; Swanson, R.A. Hypoglycemia, Brain Energetics, and Hypoglycemic Neuronal Death. Glia 2007, 55, 1280–1286. [Google Scholar] [CrossRef]
- Nordin, C. The Proarrhythmic Effect of Hypoglycemia: Evidence for Increased Risk from Ischemia and Bradycardia. Acta Diabetol. 2014, 51, 5–14. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, H.; Wang, J.; Wang, H.; Yang, B.; Wang, Z. Impairment of Human Ether-à-Go-Go-Related Gene (HERG) K+ Channel Function by Hypoglycemia and Hyperglycemia. Similar Phenotypes but Different Mechanisms. J. Biol. Chem. 2003, 278, 10417–10426. [Google Scholar] [CrossRef] [Green Version]
- Priori, S.G.; Corr, P.B. Mechanisms Underlying Early and Delayed Afterdepolarizations Induced by Catecholamines. Am. J. Physiol. 1990, 258, H1796–H1805. [Google Scholar] [CrossRef]
- Okazaki, O.; Suda, N.; Hongo, K.; Konishi, M.; Kurihara, S. Modulation of Ca2+ Transients and Contractile Properties by Beta-Adrenoceptor Stimulation in Ferret Ventricular Muscles. J. Physiol. 1990, 423, 221–240. [Google Scholar] [CrossRef]
- Choi, B.R.; Burton, F.; Salama, G. Cytosolic Ca2+ Triggers Early Afterdepolarizations and Torsade de Pointes in Rabbit Hearts with Type 2 Long QT Syndrome. J. Physiol. 2002, 543, 615–631. [Google Scholar] [CrossRef]
- Chen, J.; Fleming, T.; Katz, S.; Dewenter, M.; Hofmann, K.; Saadatmand, A.; Kronlage, M.; Werner, M.P.; Pokrandt, B.; Schreiter, F.; et al. CaM Kinase II-δ Is Required for Diabetic Hyperglycemia and Retinopathy but Not Nephropathy. Diabetes 2021, 70, 616–626. [Google Scholar] [CrossRef]
- Segal, S.; Lloyd, S.; Sherman, N.; Sussman, K.; Draznin, B. Postprandial Changes in Cytosolic Free Calcium and Glucose Uptake in Adipocytes in Obesity and Non-Insulin-Dependent Diabetes Mellitus. Horm. Res. 1990, 34, 39–44. [Google Scholar] [CrossRef]
- Baumbach, J.; Hummel, P.; Bickmeyer, I.; Kowalczyk, K.M.; Frank, M.; Knorr, K.; Hildebrandt, A.; Riedel, D.; Jäckle, H.; Kühnlein, R.P. A Drosophila in Vivo Screen Identifies Store-Operated Calcium Entry as a Key Regulator of Adiposity. Cell Metab. 2014, 19, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, M.; Metya, S.K.; Sadaf, S.; Kumar, S.; Schwudke, D.; Hasan, G. Altered Lipid Homeostasis in Drosophila InsP3 Receptor Mutants Leads to Obesity and Hyperphagia. DMM Dis. Model. Mech. 2013, 6, 734–744. [Google Scholar] [CrossRef]
- Willi, C.; Bodenmann, P.; Ghali, W.A.; Faris, P.D.; Cornuz, J. Active smoking and the risk of type 2 diabetes: A systematic review and meta-analysis. J. Am. Med. Assoc. 2007, 298, 2654–2664. [Google Scholar] [CrossRef]
- Chang, S.A. Smoking and type 2 diabetes mellitus. Diabetes Metab. J. 2012, 36, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Akter, S.; Goto, A.; Mizoue, T. Smoking and the risk of type 2 diabetes in Japan: A systematic review and meta-analysis. J. Epidemiol. 2017, 27, 553–561. [Google Scholar] [CrossRef]
- Kim, J.H.; Seo, D.C.; Kim, B.J.; Kang, J.G.; Lee, S.J.; Lee, S.H.; Kim, B.S.; Kang, J.H. Association between cigarette smoking and new-onset diabetes mellitus in 78,212 Koreans using self-reported questionnaire and urine cotinine. Diabetes Metab. J. 2019, 43, 426. [Google Scholar] [CrossRef] [Green Version]
- Epifano, L.; Di Vincenzo, A.; Fanelli, C.; Porcellati, E.; Perriello, G.; De Feo, P.; Motolese, M.; Brunetti, P.; Bolli, G.B. Effect of cigarette smoking and of a transdermal nicotine delivery system on glucoregulation in type 2 diabetes mellitus. Eur. J. Clin. Pharmacol. 1992, 43, 257–263. [Google Scholar] [CrossRef]
- Maddatu, J.; Anderson-Baucum, E.; Evans-Molina, C. Smoking and the risk of type 2 diabetes. Transl. Res. 2017, 184, 101–107. [Google Scholar] [CrossRef]
- Wang, P.; Fiaschi-Taesch, N.M.; Vasavada, R.C.; Scott, D.K.; García-Ocaña, A.; Stewart, A.F. Diabetes mellitus-advances and challenges in human β-cell proliferation. Nat. Rev. Endocrinol. 2015, 11, 201–212. [Google Scholar] [CrossRef]
- Pan, A.; Wang, Y.; Talaei, M.; Hu, F.B.; Wu, T. Relation of active, passive, and quitting smoking with incident type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2015, 3, 958–967. [Google Scholar] [CrossRef]
- Goya Wannamethee, S.; Gerald Shaper, A.; Perry, I.J. Smoking as a modifiable risk factor for type 2 diabetes in middle-aged men. Diabetes Care 2001, 24, 1590–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, R.J.; Riggs, D.W.; Conklin, D.J.; Lorkiewicz, P.; Srivastava, S.; Bhatnagar, A.; Defilippis, A.P. Nicotine metabolism in adults with type 2 diabetes. Nicotine Tob. Res. 2018, 21, 846–849. [Google Scholar] [CrossRef] [PubMed]
- Chiolero, A.; Faeh, D.; Paccaud, F.; Cornuz, J. Consequences of smoking for body weight, body fat distribution, and insulin resistance. Am. J. Clin. Nutr. 2008, 87, 801–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeghate, E.; Schattner, P.; Dunn, E. An update on the etiology and epidemiology of diabetes mellitus. Ann. New York Acad. Sci. 2006, 1084, 1–29. [Google Scholar] [CrossRef]
- Srikanthan, P.; Karlamangla, A.S. Relative muscle mass is inversely associated with insulin resistance and prediabetes. Findings from the Third National Health and Nutrition Examination Survey. J. Clin. Endocrinol. Metab. 2011, 96, 2898–2903. [Google Scholar] [CrossRef] [Green Version]
- Damas, F.; Phillips, S.; Vechin, F.C.; Ugrinowitsch, C. A Review of Resistance Training-Induced Changes in Skeletal Muscle Protein Synthesis and Their Contribution to Hypertrophy. Sport. Med. 2015, 45, 801–807. [Google Scholar] [CrossRef]
- Peterson, J.M.; Bryner, R.W.; Alway, S.E. Satellite cell proliferation is reduced in muscles of obese Zucker rats but restored with loading. Am. J. Physiol.-Cell Physiol. 2008, 295, C521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katta, A.; Kundla, S.; Kakarla, S.K.; Wu, M.; Fannin, J.; Paturi, S.; Liu, H.; Addagarla, H.S.; Blough, E.R. Impaired overload-induced hypertrophy is associated with diminished mTOR signaling in insulin-resistant skeletal muscle of the obese Zucker rat. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2010, 299, R1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, D.L.; Li, Z.; Tuder, R.M.; Feinstein, E.; Kimball, S.R.; Dungan, C.M. Altered nutrient response of mTORC1 as a result of changes in REDD1 expression: Effect of obesity vs. REDD1 deficiency. J. Appl. Physiol. 2014, 117, 246–256. [Google Scholar] [CrossRef] [Green Version]
- Ato, S.; Kido, K.; Sato, K.; Fujita, S. Type 2 diabetes causes skeletal muscle atrophy but does not impair resistance training-mediated myonuclear accretion and muscle mass gain in rats. Exp. Physiol. 2019, 104, 1518–1531. [Google Scholar] [CrossRef]
- Lindström, J.; Ilanne-Parikka, P.; Peltonen, M.; Aunola, S.; Eriksson, J.G.; Hemiö, K.; Hämäläinen, H.; Härkönen, P.; Keinänen-Kiukaanniemi, S.; Laakso, M.; et al. Sustained reduction in the incidence of type 2 diabetes by lifestyle intervention: Follow-up of the Finnish Diabetes Prevention Study. Lancet 2006, 368, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-c.; Park, I.; Jun, T.-W.; Nam, B.-H.; Cho, S.-i.; Blair, S.N.; Kim, Y.-S. Physical Activity and Body Mass Index and Their Associations with the Development of Type 2 Diabetes in Korean Men. Am. J. Epidemiol. 2012, 176, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, P.C.; Gupte, A.A. Heat shock proteins are important mediators of skeletal muscle insulin sensitivity. Exerc. Sport Sci. Rev. 2011, 39, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Tytell, M.; Davis, A.T.; Giles, J.; Snider, L.C.; Xiao, R.; Dozier, S.G.; Presley, T.D.; Kavanagh, K. Alfalfa-derived HSP70 administered intranasally improves insulin sensitivity in mice. Cell Stress Chaperones 2018, 23, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Mulyani, W.R.W.; Sanjiwani, M.I.D.; Sandra; Prabawa, P.Y.; Lestari, A.A.W.; Wihandani, D.M.; Suastika, K.; Saraswati, M.R.; Bhargah, A.; Manuaba, I.B.A.P. Chaperone-based therapeutic target innovation: Heat shock protein 70 [HSP70] for type 2 diabetes mellitus. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 559–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.; Nguyen, A.K.; Henstridge, D.C.; Holmes, A.G.; Chan, M.H.S.; Mesa, J.L.; Lancaster, G.I.; Southgate, R.J.; Bruce, C.R.; Duffy, S.J.; et al. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 1739–1744. [Google Scholar] [CrossRef] [Green Version]
- Atkin, A.S.; Moin, A.S.M.; Nandakumar, M.; Al-Qaissi, A.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Impact of Severe Hypoglycemia on the Heat Shock and Related Protein Response. Sci. Rep. 2021, 11, 17057. [Google Scholar] [CrossRef]
- Pearen, M.A.; Muscat, G.E.O. The Nuclear Receptor Nor-1 Is a Pleiotropic Regulator of Exercise-Induced Adaptations. Exerc. Sport Sci. Rev. 2018, 46, 97–104. [Google Scholar] [CrossRef]
- Nisar, O.; Pervez, H.; Mandalia, B.; Waqas, M.; Sra, H.K. Type 3 Diabetes Mellitus: A Link Between Alzheimer’s Disease and Type 2 Diabetes Mellitus. Cureus 2020, 12, e11703. [Google Scholar] [CrossRef]
- Anderson, R.J.; Freedland, K.E.; Clouse, R.E.; Lustman, P.J. The Prevalence of Comorbid Depression in Adults with Diabetes: A Meta-Analysis. Diabetes Care 2001, 24, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Mikaliukštiene, A.; Žagminas, K.; Juozulynas, A.; Narkauskaite, L.; Salyga, J.; Jankauskiene, K.; Stukas, R.; Šurkiene, G. Prevalence and Determinants of Anxiety and Depression Symptoms in Patients with Type 2 Diabetes in Lithuania. Med. Sci. Monit. 2014, 20, 182–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimundo, A.F.; Ferreira, S.; Martins, I.C.; Menezes, R. Islet Amyloid Polypeptide: A Partner in Crime with Aβ in the Pathology of Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Huang, L.; Zheng, W.; An, J.; Zhan, Z.; Wang, L.; Chen, Z.; Liu, L. Recurrent Nonsevere Hypoglycemia Exacerbates Imbalance of Mitochondrial Homeostasis Leading to Synapse Injury and Cognitive Deficit in Diabetes. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E973–E986. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Wu, Y.; Chen, Z.; Huang, L.; Wang, L.; Liu, L. Severe Hypoglycemia Contributing to Cognitive Dysfunction in Diabetic Mice Is Associated with Pericyte and Blood–Brain Barrier Dysfunction. Front. Aging Neurosci. 2021, 13, 826. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Chiodini, P.; Maiorino, M.I.; Bellastella, G.; Panagiotakos, D.; Giugliano, D. Which diet for prevention of type 2 diabetes? A meta-analysis of prospective studies. Endocrine 2014, 47, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Forouhi, N.G.; Misra, A.; Mohan, V.; Taylor, R.; Yancy, W. Dietary and nutritional approaches for prevention and management of type 2 diabetes. BMJ 2018, 361, k2234. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Cousins, R.S.; Liu, S.; Phelps, B.M.; Promes, J.A. Connecting pancreatic islet lipid metabolism with insulin secretion and the development of type 2 diabetes. Ann. N. Y. Acad. Sci. 2019, 1461, 53–72. [Google Scholar] [CrossRef]
- Duan, Y.; Zeng, L.; Zheng, C.; Song, B.; Li, F.; Kong, X.; Xu, K. Inflammatory Links Between High Fat Diets and Diseases. Front. Immunol. 2018, 9, 2649. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Wang, Z.H.; Kang, S.S.; Liu, X.; Xia, Y.; Chan, C.B.; Ye, K. High-Fat Diet-Induced Diabetes Couples to Alzheimer’s Disease through Inflammation-Activated C/EBPβ/AEP Pathway. Mol. Psychiatry 2022, 27, 3396–3409. [Google Scholar] [CrossRef]
- Li, H.; Meng, Y.; He, S.; Tan, X.; Zhang, Y.; Zhang, X.; Wang, L.; Zheng, W. Macrophages, Chronic Inflammation, and Insulin Resistance. Cells 2022, 11, 3001. [Google Scholar] [CrossRef]
- Tersey, S.A.; Maier, B.; Nishiki, Y.; Maganti, A.V.; Nadler, J.L.; Mirmira, R.G. 12-Lipoxygenase Promotes Obesity-Induced Oxidative Stress in Pancreatic Islets. Mol. Cell. Biol. 2014, 34, 3735–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, L. Energy Metabolism in the Liver. Compr. Physiol. 2014, 4, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghazala, R.A.; El Medney, A.; Meleis, A.; Mohie El dien, P.; Samir, H. Role of anti-inflammatory interventions in high-fat-diet-induced obesity. Biomed. Chromatogr. 2020, 34, e4743. [Google Scholar] [CrossRef] [PubMed]
- Elhayany, A.; Lustman, A.; Abel, R.; Attal-Singer, J.; Vinker, S. A low carbohydrate Mediterranean diet improves cardiovascular risk factors and diabetes control among overweight patients with type 2 diabetes mellitus: A 1-year prospective randomized intervention study. Diabetes Obes. Metab. 2010, 12, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Giugliano, D. Mediterranean diet and type 2 diabetes. Diabetes Metab. Res. Rev. 2014, 30, 34–40. [Google Scholar] [CrossRef]
- Nani, A.; Murtaza, B.; Khan, A.S.; Khan, N.A.; Hichami, A. Antioxidant and Anti-Inflammatory Potential of Polyphenols Contained in Mediterranean Diet in Obesity: Molecular Mechanisms. Molecules 2021, 26, 985. [Google Scholar] [CrossRef]
- Galic, S.; Fullerton, M.D.; Schertzer, J.D.; Sikkema, S.; Marcinko, K.; Walkley, C.R.; Izon, D.; Honeyman, J.; Chen, Z.-P.; Van Denderen, B.J.; et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Investig. 2011, 121, 4903–4915. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, M.-S.; O’Brien, E.L.; Bigornia, S.; Mott, M.; Cacicedo, J.M.; Xu, X.J.; Gokce, N.; Apovian, C.; Ruderman, N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem. Biophys. Res. Commun. 2011, 404, 382–387. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Hu, P.; Feng, L.-P.; Huang, L.-L.; Wang, Y.; Yan, X.; Xiong, J.; Xia, H.-L. Protective Effects of Ferulic Acid on Metabolic Syndrome: A Comprehensive Review. Molecules 2022, 28, 281. [Google Scholar] [CrossRef]
- Huang, Y.; Zhu, X.; Chen, K.; Lang, H.; Zhang, Y.; Hou, P.; Ran, L.; Zhou, M.; Zheng, J.; Yi, L.; et al. Resveratrol prevents sarcopenic obesity by reversing mitochondrial dysfunction and oxidative stress via the PKA/LKB1/AMPK pathway. Aging 2019, 11, 2217–2240. [Google Scholar] [CrossRef]
- Obrenovich, M.; Li, Y.; Tayahi, M.; Reddy, V.P. Polyphenols and Small Phenolic Acids as Cellular Metabolic Regulators. Curr. Issues Mol. Biol. 2022, 44, 4152–4166. [Google Scholar] [CrossRef] [PubMed]
- Chaari, A. Inhibition of human islet amyloid polypeptide aggregation and cellular toxicity by oleuropein and derivatives from olive oil. Int. J. Biol. Macromol. 2020, 162, 284–300. [Google Scholar] [CrossRef] [PubMed]
- von Hanstein, A.-S.; Lenzen, S.; Plötz, T. Toxicity of fatty acid profiles of popular edible oils in human EndoC-βH1 beta-cells. Nutr. Diabetes 2020, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwingshackl, L.; Lampousi, A.M.; Portillo, M.P.; Romaguera, D.; Hoffmann, G.; Boeing, H. Olive oil in the prevention and management of type 2 diabetes mellitus: A systematic review and meta-analysis of cohort studies and intervention trials. Nutr. Diabetes 2017, 7, e262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosser, R.E.; Maulis, M.F.; Moullé, V.S.; Dunn, J.C.; Carboneau, B.A.; Arasi, K.; Pappan, K.; Poitout, V.; Gannon, M. High-fat diet-induced β-cell proliferation occurs prior to insulin resistance in C57Bl/6J male mice. Am. J. Physiol.-Endocrinol. Metab. 2015, 308, E573–E582. [Google Scholar] [CrossRef] [Green Version]
- Stamateris, R.E.; Sharma, R.B.; Kong, Y.; Ebrahimpour, P.; Panday, D.; Ranganath, P.; Zou, B.; Levitt, H.; Parambil, N.A.; O’Donnell, C.P.; et al. Glucose Induces mouse β-cell proliferation via IRS2, MTOR, and cyclin D2 but Not the insulin receptor. Diabetes 2016, 65, 981–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Her, T.K.; Lagakos, W.S.; Brown, M.R.; LeBrasseur, N.K.; Rakshit, K.; Matveyenko, A.V. Dietary carbohydrates modulate metabolic and β-cell adaptation to high-fat diet-induced obesity. Am. J. Physiol.-Endocrinol. Metab. 2020, 318, E856–E865. [Google Scholar] [CrossRef] [PubMed]

| Author | Years | Factors Affecting Glucose Level in the Context of Molecular Discoveries | Influence on Glucose Level ↑ Increase ↓ Decrease |
|---|---|---|---|
| Body weight | |||
| Ying, W. et al. [12] | 2019 |
| ↑ |
| ↑ | ||
| ↓ | ||
| Nagao, M. et al. [14] | 2020 |
| ↑ |
| ↓ | ||
| Zuo, D et al. [48] | 2020 |
| ↑ |
| ↓ | ||
| ↓ | ||
| ↑ | ||
| Salehi, M et al. [15] | 2018 |
| ↓ |
| Dai, W et al. [21] | 2021 |
| ↓ |
| Smoking | |||
| Kim, J.H et al. [68] | 2019 |
| ↑ |
| Sun, L et al. [22] | 2020 |
| ↑ |
| ↑ | ||
| ↓ | ||
| Duncan, A., et al. [23] | 2019 |
| ↑ |
| ↑ | ||
| ↑ | ||
| ↑ | ||
| ↓ | ||
| ↑ | ||
| ↓ | ||
| Physical activity | |||
| Ato, S et al. [82] | 2019 |
| ↓ |
| ↓ | ||
| Krause, M et al. [24] | 2015 |
| ↑ |
| ↓ | ||
| ↑ | ||
| ↓ | ||
| ↓ | ||
| ↓ | ||
| Tsuzuki, T. et al. [27] | 2017 |
| ↓ |
| ↓ | ||
| ↑ | ||
| ↓ | ||
| Pearen, M.A et al. [90] | 2018 |
| ↓ |
| ↓ | ||
| Mey, J.T. et al. [30] | 2019 |
| ↓ |
| ↓ | ||
| ↓ | ||
| Orumiyehei, A. et al. [32] | 2022 |
| ↓ |
| Diet | |||
| do Amaral, M.E.C. et al. [36] | 2020 |
| ↑ |
| ↓ | ||
| ↓ | ||
| Liu, P. et al. [101] | 2022 |
| - |
| Arruda, A.P. et al. [37] | 2011 |
| ↑ |
| Tersey, S.A. et al. [103] | 2014 |
| ↑ |
| Johnston-Cox, H. et al. [38] | 2012 |
| ↑ |
| Leri, M. et al. [34] | 2019 |
| ↓ |
| von Hanstein, A.-S et al. [115] | 2020 |
| ↑ |
| - | ||
| ↓ | ||
| ↓ | ||
| Her, T.K. et al. [119] | 2020 |
| ↓ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczechla, M.; Balewska, A.; Naskręt, D.; Zozulińska-Ziółkiewicz, D.; Uruska, A. Molecular Changes in Cells of Patients with Type 2 Diabetes Mellitus Depending on Changes in Glycemia Level in the Context of Lifestyle—An Overview of the Latest Scientific Discoveries. Curr. Issues Mol. Biol. 2023, 45, 1961-1981. https://doi.org/10.3390/cimb45030126
Szczechla M, Balewska A, Naskręt D, Zozulińska-Ziółkiewicz D, Uruska A. Molecular Changes in Cells of Patients with Type 2 Diabetes Mellitus Depending on Changes in Glycemia Level in the Context of Lifestyle—An Overview of the Latest Scientific Discoveries. Current Issues in Molecular Biology. 2023; 45(3):1961-1981. https://doi.org/10.3390/cimb45030126
Chicago/Turabian StyleSzczechla, Magdalena, Anita Balewska, Dariusz Naskręt, Dorota Zozulińska-Ziółkiewicz, and Aleksandra Uruska. 2023. "Molecular Changes in Cells of Patients with Type 2 Diabetes Mellitus Depending on Changes in Glycemia Level in the Context of Lifestyle—An Overview of the Latest Scientific Discoveries" Current Issues in Molecular Biology 45, no. 3: 1961-1981. https://doi.org/10.3390/cimb45030126