
Stability Study, Quantification Method and Pharmacokinetics Investigation of a Coumarin–Monoterpene Conjugate Possessing Antiviral Properties against Respiratory Syncytial Virus

 ,
,  , ,
, ,
Abstract
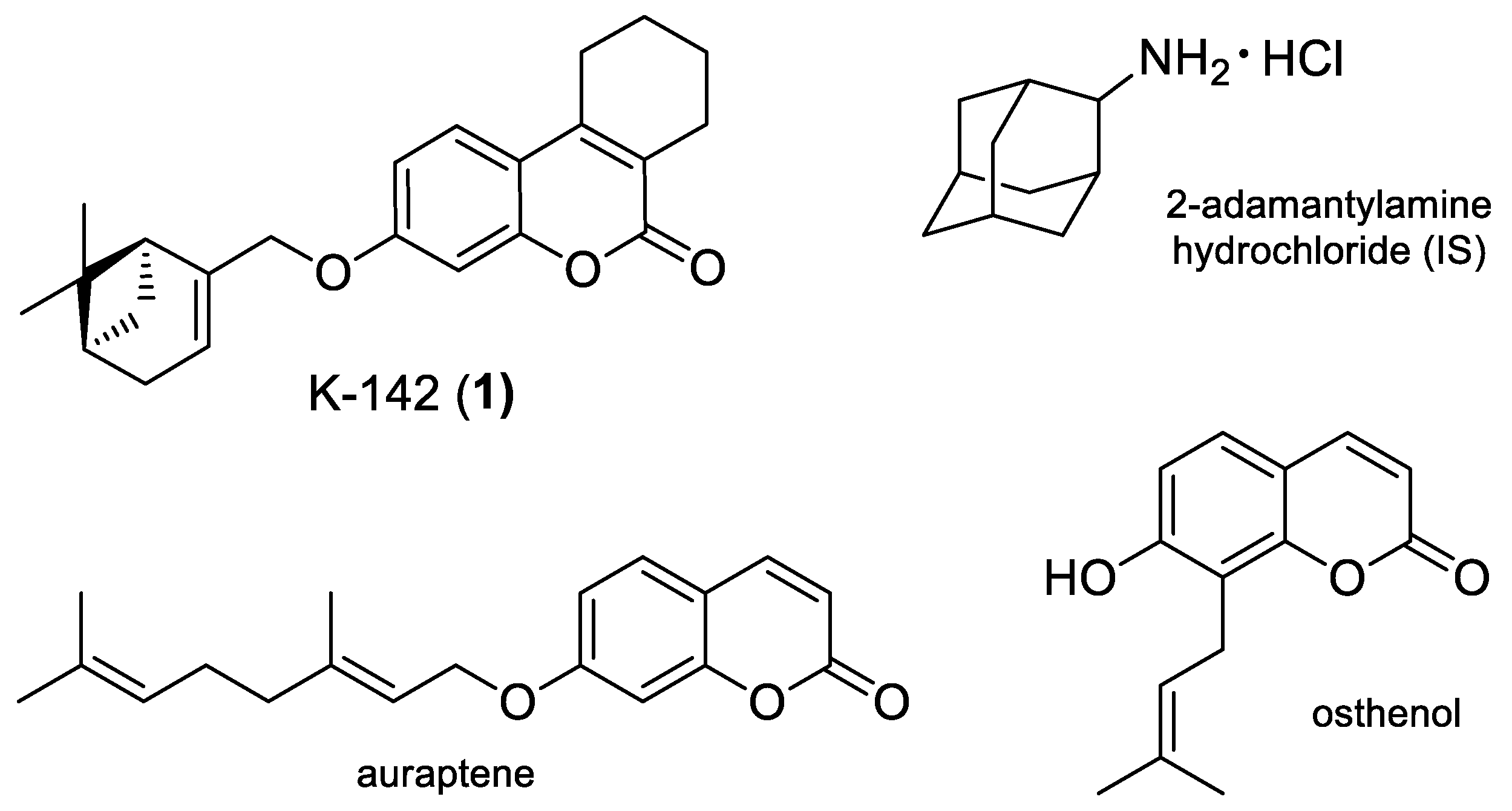
:1. Introduction
2. Results and Discussion
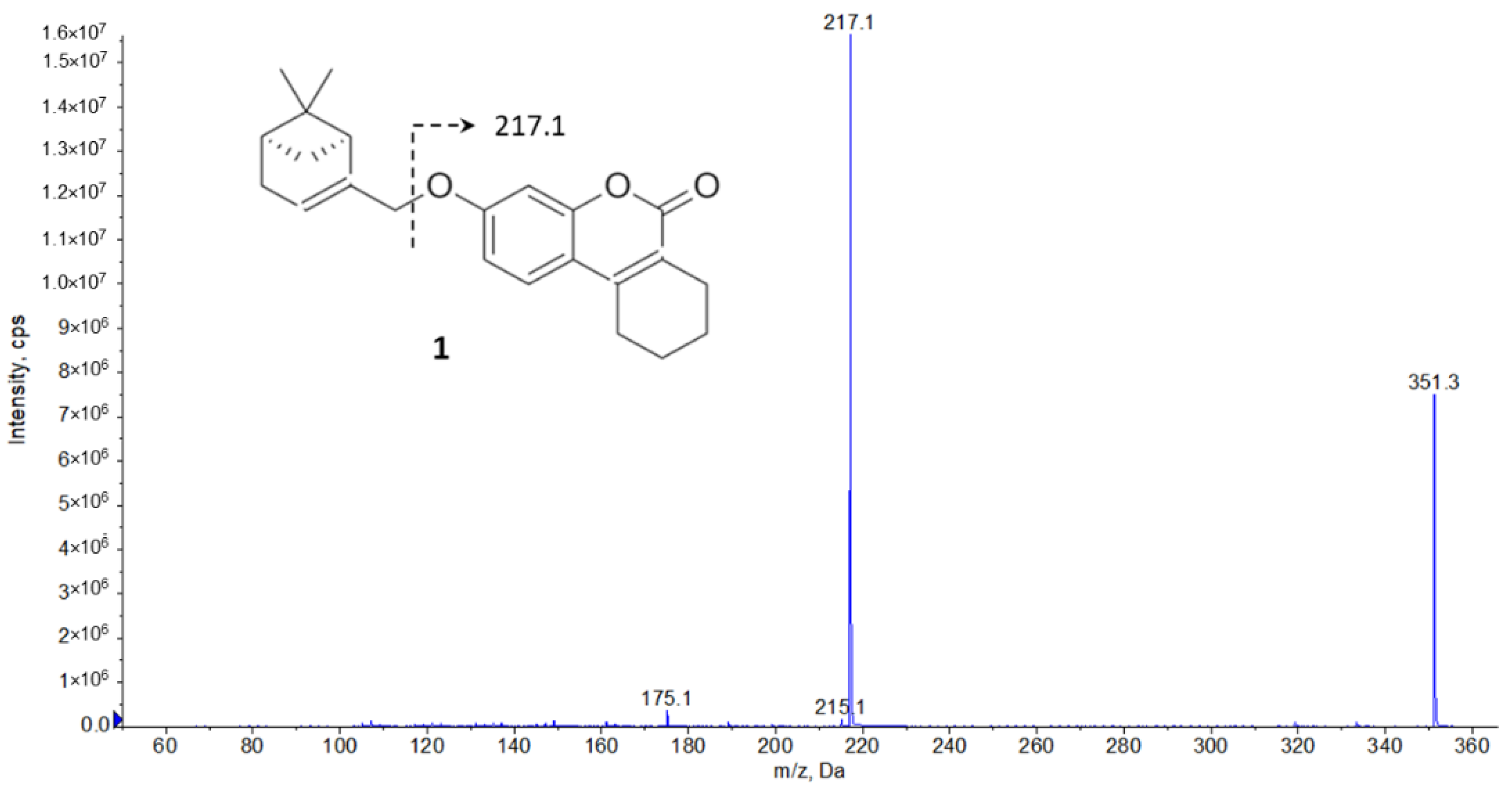
2.1. LC-MS/MS Method Development
2.2. Stability Study of the Agent K-142 in Whole Mice Blood
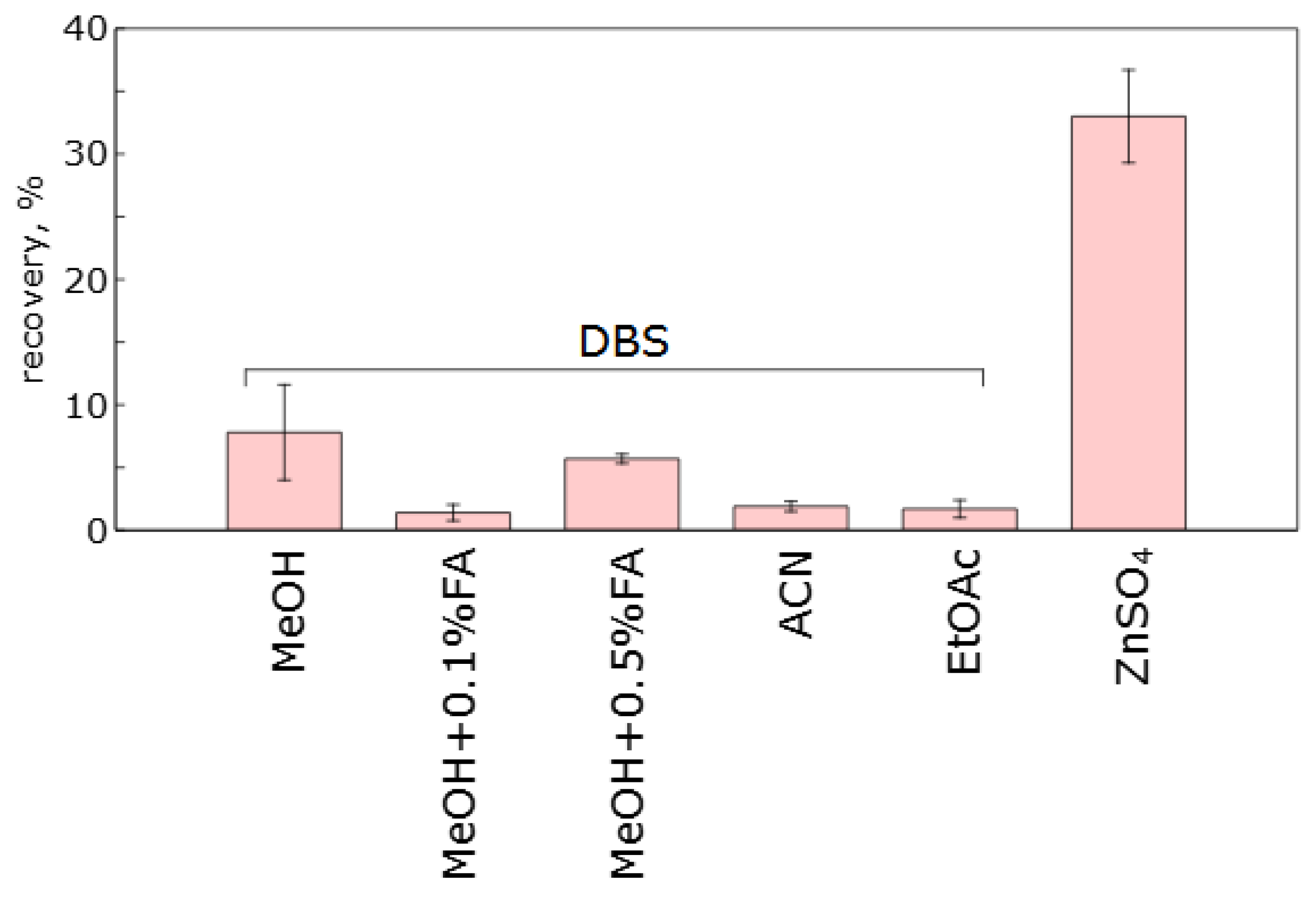
2.3. Selection of the Method for Whole Blood Samples Preparation
2.4. Method Validation
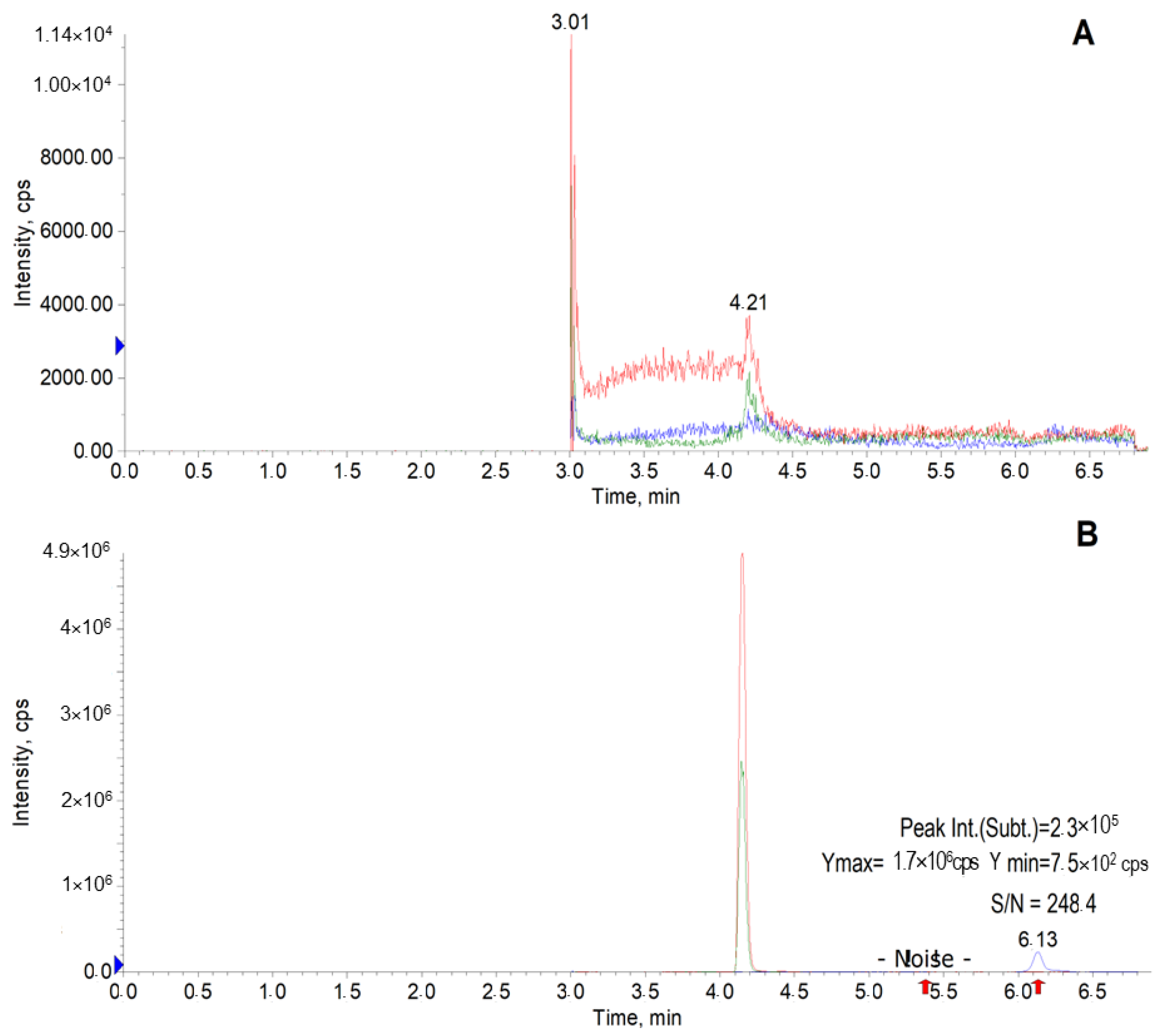
2.4.1. Selectivity and LLOQ
2.4.2. Calibration Curve
2.4.3. Accuracy and Precision
2.4.4. Extraction Recovery and Matrix Factor
2.4.5. Carry-Over
2.5. Pharmacokinetics Study of K-142 after Oral Administration to Mice
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Stock Solutions, Calibrators and Quality Control Samples
3.3. Sample Preparation
3.3.1. Dried Blood Spots Sampling and Preparation
3.3.2. Whole Blood Precipitation
3.4. Apparatus and LC-MS/MS Conditions
3.5. Method Validation
3.5.1. Selectivity and LLOQ
3.5.2. Linearity of Calibration Curves and Lower Limit of Quantification (LLOQ)
3.5.3. Accuracy and Precision
3.5.4. Extraction Recovery, Matrix Factor and Prepared Samples Stability
3.5.5. Carry-Over
3.6. Pharmacokinetics Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Karron, R.A. Preventing respiratory syncytial virus (RSV) disease in children. Science 2021, 372, 686–687. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Wainwright, C. Acute viral bronchiolitis in children—A very common condition with few therapeutic options. Paediatr. Respir. Rev. 2010, 11, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Cockerill, G.S.; Good, J.; Mathews, N. State of the Art in Respiratory Syncytial Virus Drug Discovery and Development. J. Med. Chem. 2019, 62, 3206–3227. [Google Scholar] [CrossRef] [PubMed]
- Curini, M.; Cravotto, G.; Epifano, F.; Giannone, G. Chemistry and biological activity of natural and synthetic prenyloxycoumarins. Curr. Med. Chem. 2006, 13, 199–222. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, A.; Iranshahy, M.; Iranshahi, M. Biological properties and molecular targets of umbelliprenin—A mini-review. J. Asian Nat. Prod. Res. 2014, 16, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Preziuso, F.; Genovese, S.; Marchetti, L.; Sharifi-Rad, M.; Palumbo, L.; Epifano, F.; Fiorito, S. 7-Isopentenyloxycoumarin: What Is New across the Last Decade. Molecules 2020, 25, 5923. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, S.; Preziuso, F.; Sharifi-Rad, M.; Marchetti, L.; Epifano, F.; Genovese, S. Auraptene and umbelliprenin: A review on their latest literature acquisitions. Phytochem. Rev. 2022, 21, 317–326. [Google Scholar] [CrossRef]
- Tayarani-Najaran, Z.; Tayarani-Najaran, N.; Eghbali, S. A Review of Auraptene as an Anticancer Agent. Front. Pharmacol. 2021, 12, 698352. [Google Scholar] [CrossRef]
- Ito, C.; Itoigawa, M.; Ju-ichi, M.; Sakamoto, N.; Tokuda, H.; Nishino, H.; Furukawa, H. Antitumor-promoting activity of coumarins from citrus plants. Planta Med. 2005, 71, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Khomenko, T.M.; Shtro, A.A.; Galochkina, A.V.; Nikolaeva, Y.V.; Petukhova, G.D.; Borisevich, S.S.; Korchagina, D.V.; Volcho, K.P.; Salakhutdinov, N.F. Monoterpene-Containing Substituted Coumarins as Inhibitors of Respiratory Syncytial Virus (RSV) Replication. Molecules 2021, 26, 7493. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G. Animal models of respiratory syncytial virus infection. Vaccine 2017, 35, 469–480. [Google Scholar] [CrossRef]
- Neganova, M.; Aleksandrova, Y.; Suslov, E.; Mozhaitsev, E.; Munkuev, A.; Tsypyshev, D.; Chicheva, M.; Rogachev, A.; Sukocheva, O.; Volcho, K.; et al. Novel Multitarget Hydroxamic Acids with a Natural Origin CAP Group against Alzheimer’s Disease: Synthesis, Docking and Biological Evaluation. Pharmaceutics 2021, 13, 1893. [Google Scholar] [CrossRef]
- Bhuket, P.R.N.; Niwattisaiwong, N.; Limpikirati, P.; Khemawoot, P.; Towiwat, P.; Ongpipattanakul, B.; Rojsitthisak, P. Simultaneous determination of curcumin diethyl disuccinate and its active metabolite curcumin in rat plasma by LC-MS/MS: Application of esterase inhibitors in the stabilization of an ester-containing prodrug. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1033, 301–310. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, A.; Locatelli, M.; Tartaglia, A.; Molteni, L.; D’Ovidio, C.; Savini, F.; Rudge, J.; de Grazia, U. Therapeutic Drug Monitoring of Antiseizure Medications Using Volumetric Absorptive Microsampling: Where Are We? Pharmaceuticals 2021, 14, 627. [Google Scholar] [CrossRef]
- Alampanos, V.; Kabir, A.; Furton, K.G.; Samanidou, V. Rapid exposure monitoring of six bisphenols and diethylstilbestrol in human urine using fabric phase sorptive extraction followed by high performance liquid chromatography—Photodiode array analysis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2021, 1177, 122760. [Google Scholar] [CrossRef] [PubMed]
- Okhina, A.A.; Rogachev, A.D.; Yarovaya, O.I.; Pokrovsky, A.G.; Salakhutdinov, N.F. Stability study of the antiviral agent camphecene in dried blood spots at different temperatures. Drug Test. Anal. 2021, 13, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Göen, T. Human metabolism of α-pinene and metabolite kinetics after oral administration. Arch. Toxicol. 2017, 91, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.G. Coumarin metabolism, toxicity and carcinogenicity: Relevance for human risk assessment. Food Chem. Toxicol. 1999, 37, 423–453. [Google Scholar] [CrossRef]
- Egan, D.; O’Kennedy, R.; Moran, E.; Cox, D.; Prosser, E.; Thornes, R.B. The pharmacology, metabolism, analysis, and applications of coumarin and coumarin-related compounds. Drug Metab. Rev. 1990, 22, 503–529. [Google Scholar] [CrossRef] [PubMed]
- Cho, P.; Choi, S.M.; Kim, Y.; Lee, D.H.; Noh, Y.; Kim, S.; Kim, J.H.; Lee, T.; Lee, S. Characterization of osthenol metabolism in vivo and its pharmacokinetics. Xenobiotica 2020, 50, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.D.; Ouyang, H.; Zhong, L.Y.; Li, T.E.; Rao, X.Y.; Feng, Y.L.; Yang, W.L. Rapid and sensitive LC-MS/MS method for the determination of auraptene in rat plasma and its application in a pharmacokinetic and bioavailability study in rats. Genet. Mol. Res. 2016, 15, gmr.8786. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Guidance for Industry: Bioanalytical Method Validation; US Food and Drug Administration: Silver Spring, MD, USA, 2018.
- European Medicines Agency Guideline on Bioanalytical Method Validation; European Medicines Agency: London, UK, 2015.
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | K-142 |
|---|---|
| Cmax, ng/mL | 63.4 ± 19.1 |
| T1/2, min | 27.7 ± 5.4 |
| AUC0–∞, ng/mL×min | (1.57 ± 0.27) × 103 |
| MRT0–∞, min | 40.5 ± 2.9 |
| CL, (mg/kg)/(ng/mL)/min | (1.3 ± 0.2) × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemolochnova, A.G.; Rogachev, A.D.; Salnikova, O.P.; Khomenko, T.M.; Volcho, K.P.; Yarovaya, O.I.; Fatianova, A.V.; Pokrovsky, A.G.; Salakhutdinov, N.F. Stability Study, Quantification Method and Pharmacokinetics Investigation of a Coumarin–Monoterpene Conjugate Possessing Antiviral Properties against Respiratory Syncytial Virus. Pharmaceuticals 2022, 15, 1158. https://doi.org/10.3390/ph15091158
Nemolochnova AG, Rogachev AD, Salnikova OP, Khomenko TM, Volcho KP, Yarovaya OI, Fatianova AV, Pokrovsky AG, Salakhutdinov NF. Stability Study, Quantification Method and Pharmacokinetics Investigation of a Coumarin–Monoterpene Conjugate Possessing Antiviral Properties against Respiratory Syncytial Virus. Pharmaceuticals. 2022; 15(9):1158. https://doi.org/10.3390/ph15091158
Chicago/Turabian StyleNemolochnova, Arina G., Artem D. Rogachev, Olga P. Salnikova, Tatyana M. Khomenko, Konstantin P. Volcho, Olga I. Yarovaya, Alina V. Fatianova, Andrey G. Pokrovsky, and Nariman F. Salakhutdinov. 2022. "Stability Study, Quantification Method and Pharmacokinetics Investigation of a Coumarin–Monoterpene Conjugate Possessing Antiviral Properties against Respiratory Syncytial Virus" Pharmaceuticals 15, no. 9: 1158. https://doi.org/10.3390/ph15091158