
Identification of Cyclic Sulfonamides with an N-Arylacetamide Group as α-Glucosidase and α-Amylase Inhibitors: Biological Evaluation and Molecular Modeling

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
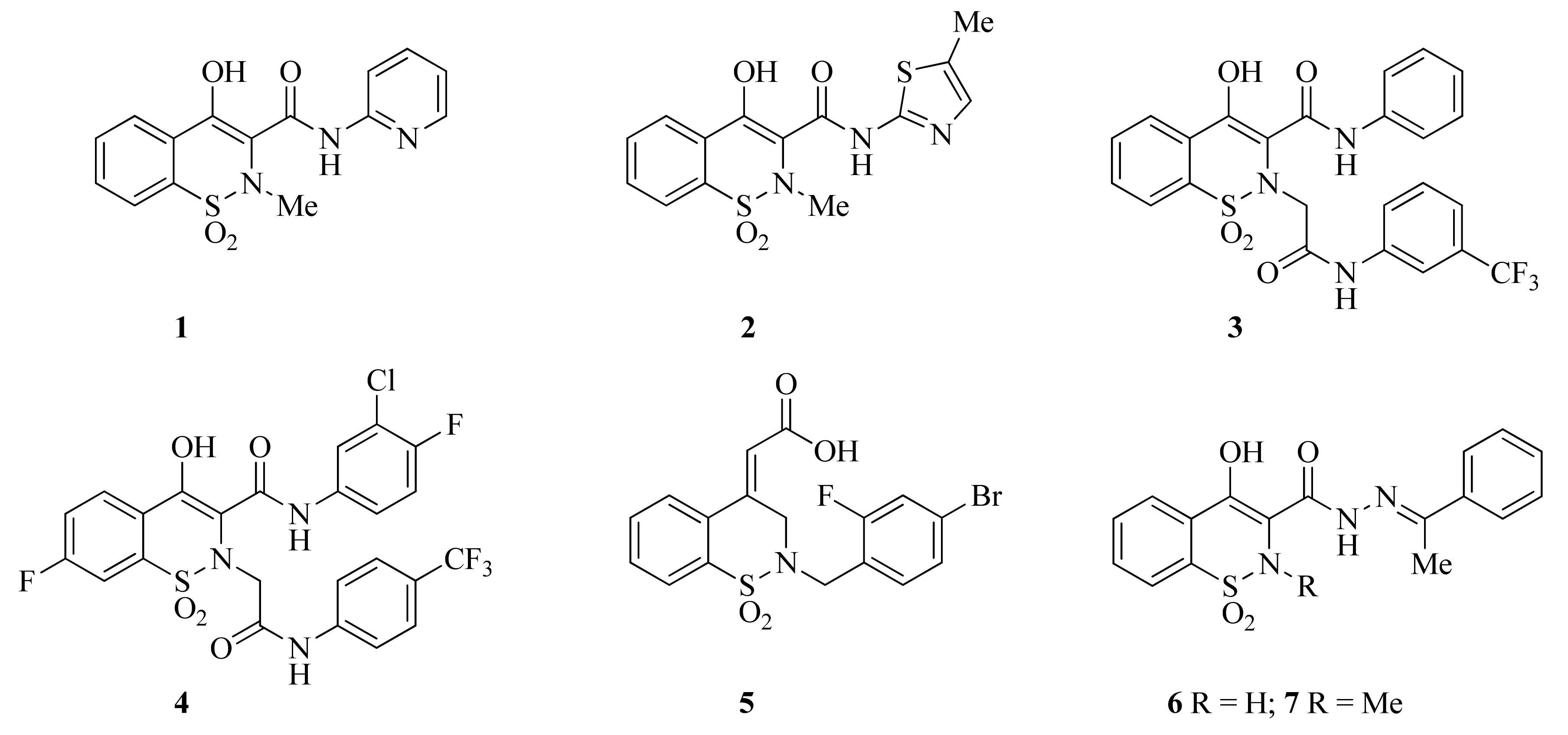
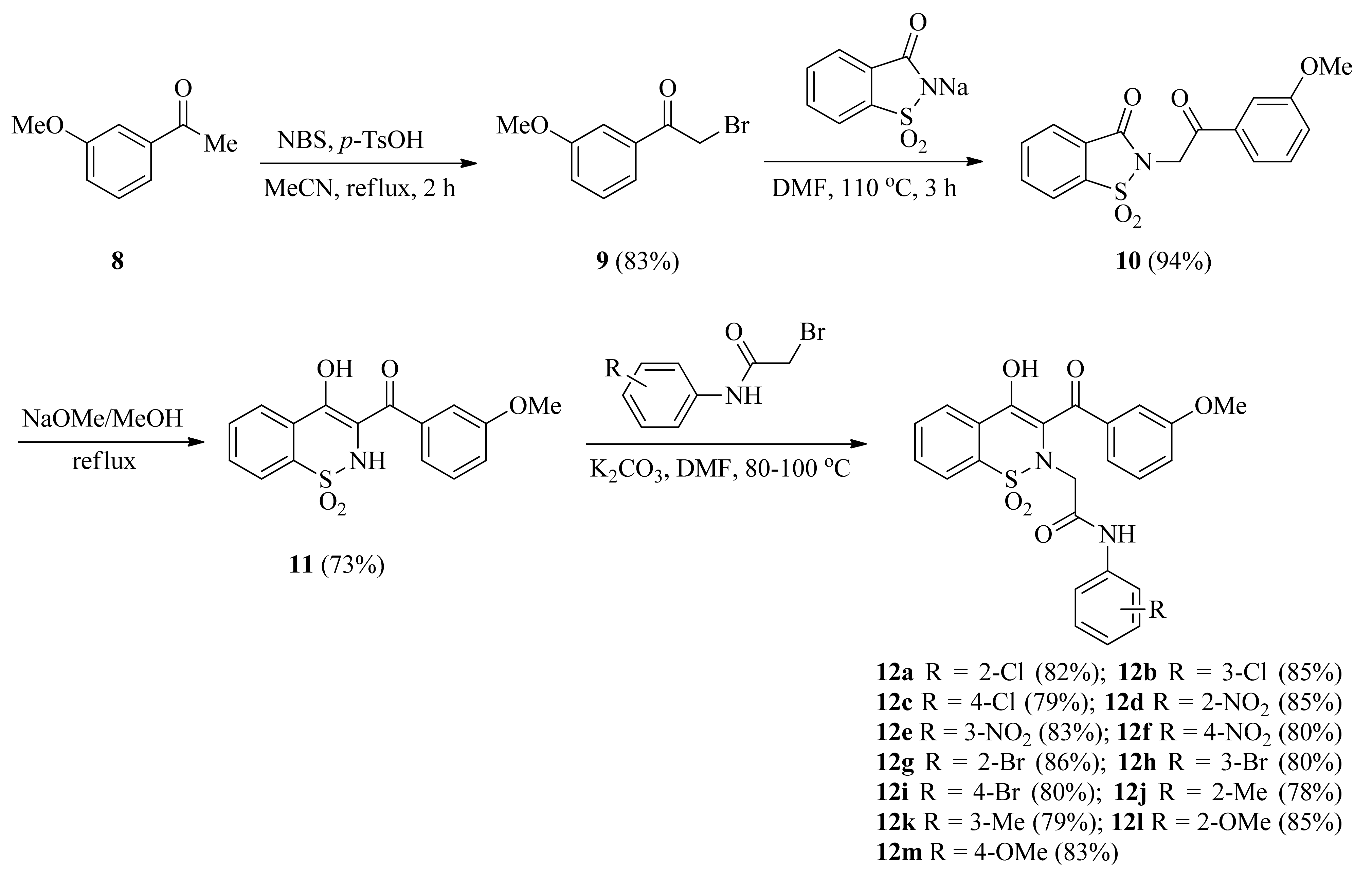
2.1. Synthetic Chemistry
2.2. Spectroscopic Characterization
2.3. In Silico Analysis
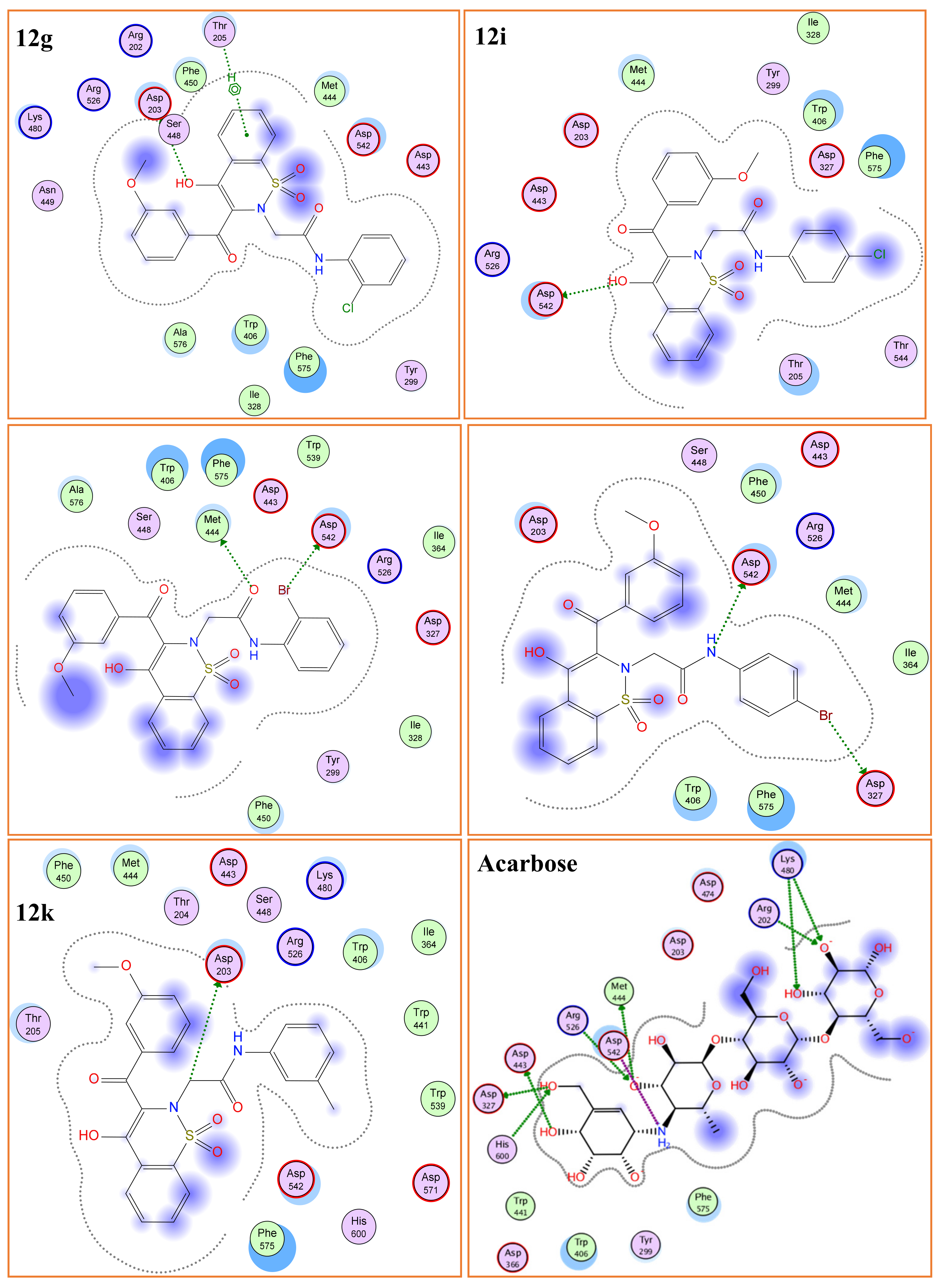
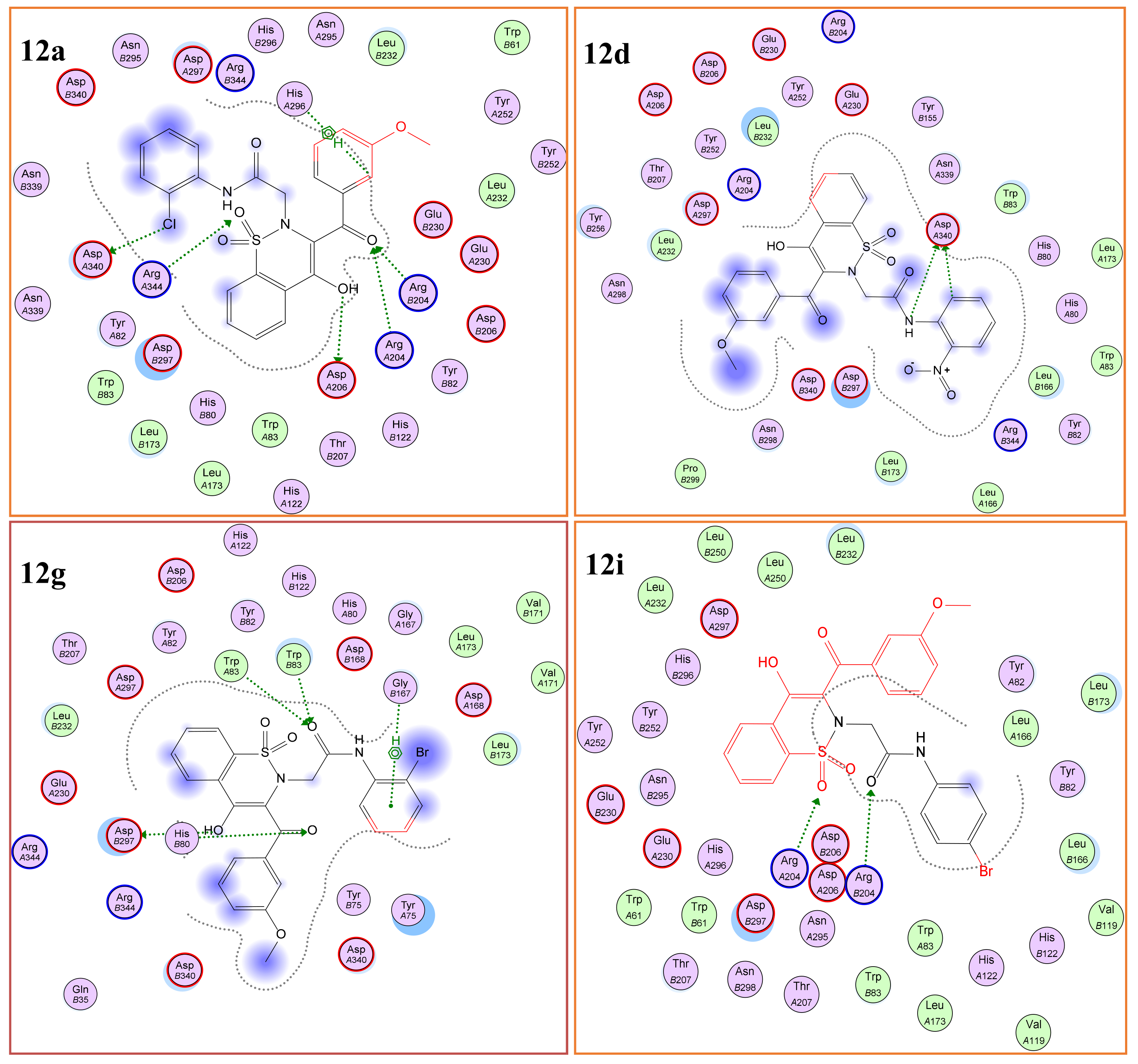
2.3.1. Molecular Docking
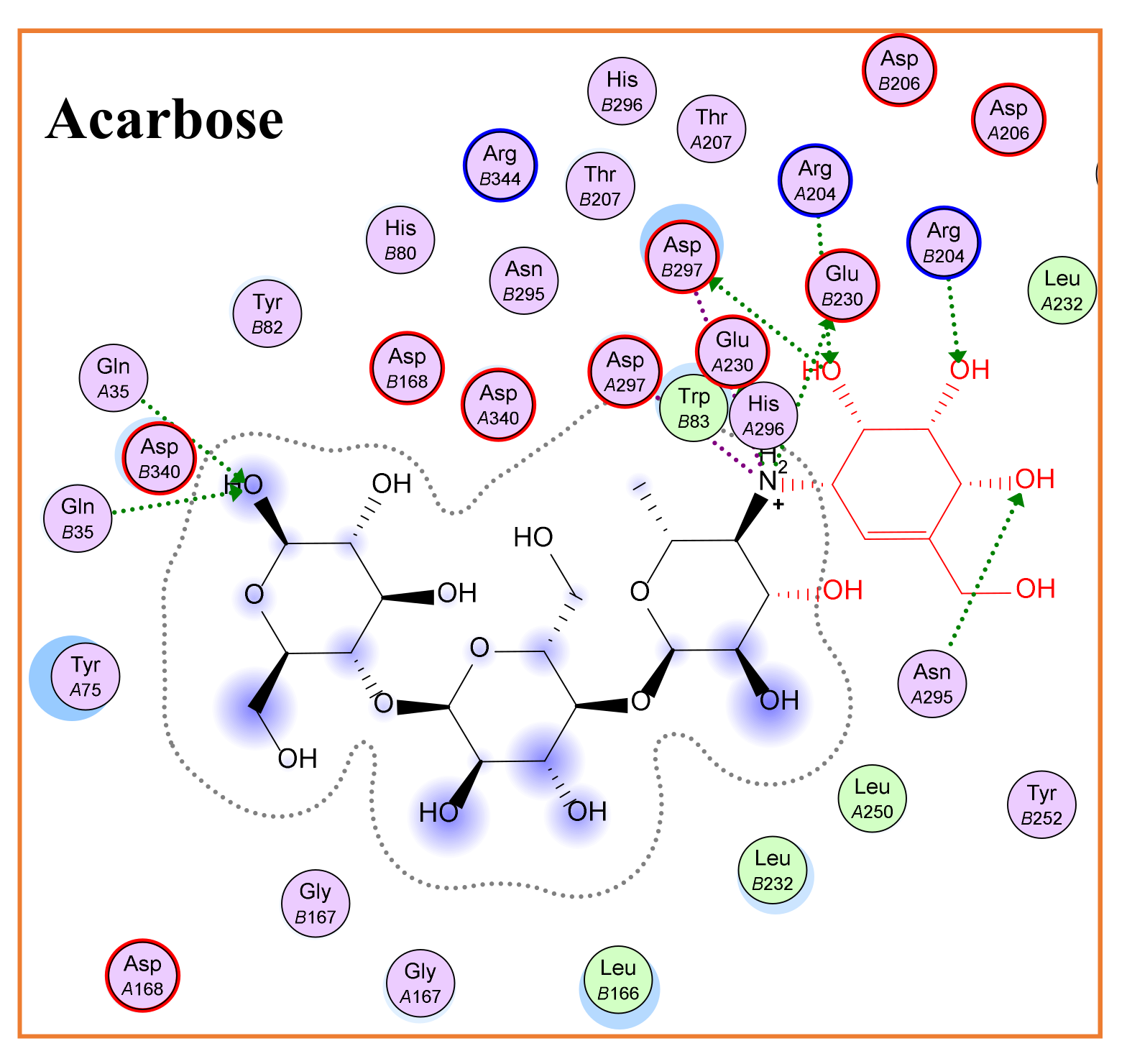
α-Glucosidase (Molecular Docking)
α-Amylase (Molecular Docking)
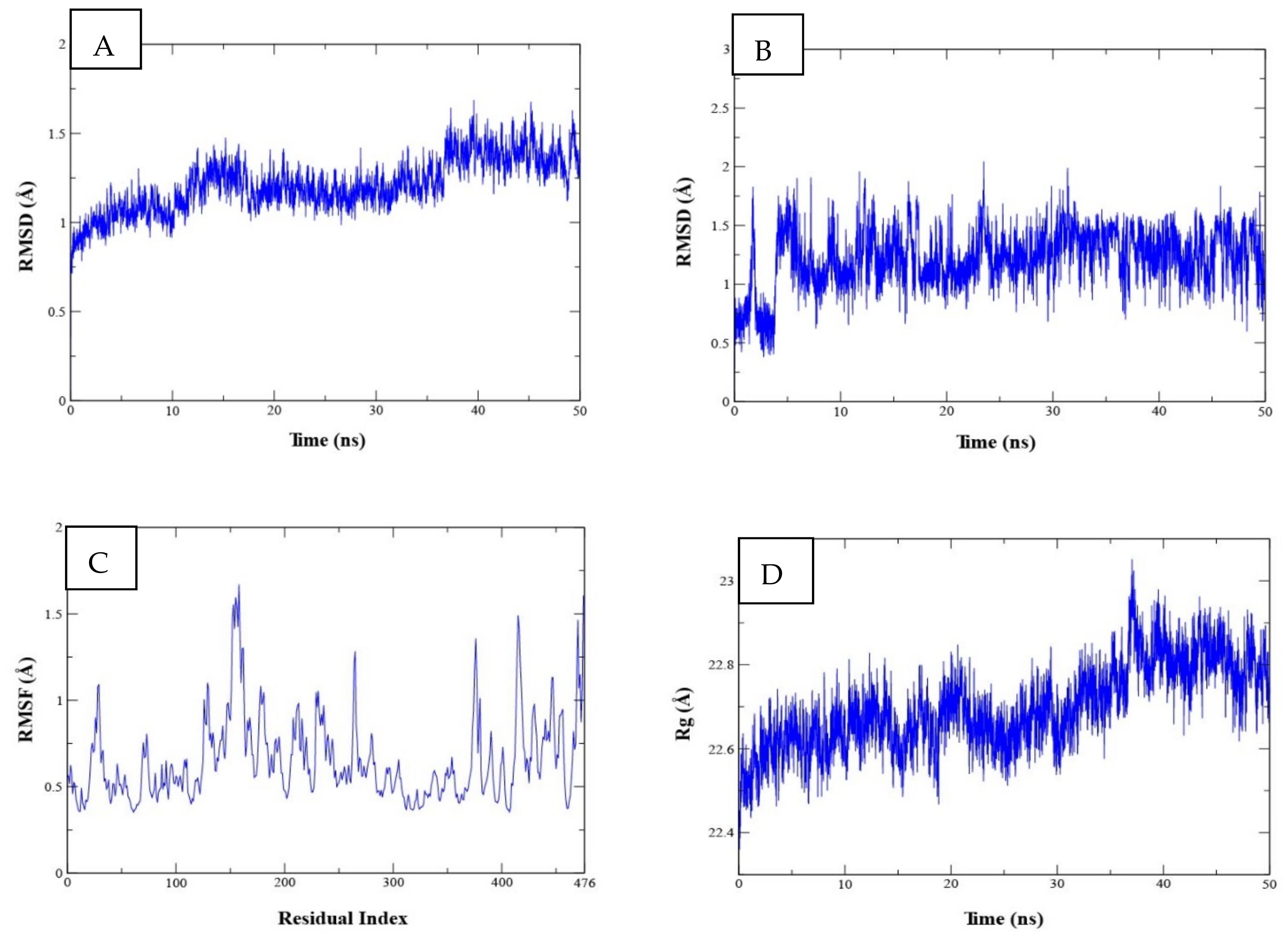
2.3.2. Molecular Dynamics (MD) Studies for Complex Stability Analysis
2.3.3. Binding Free Energy Calculations
2.4. Biological Activity
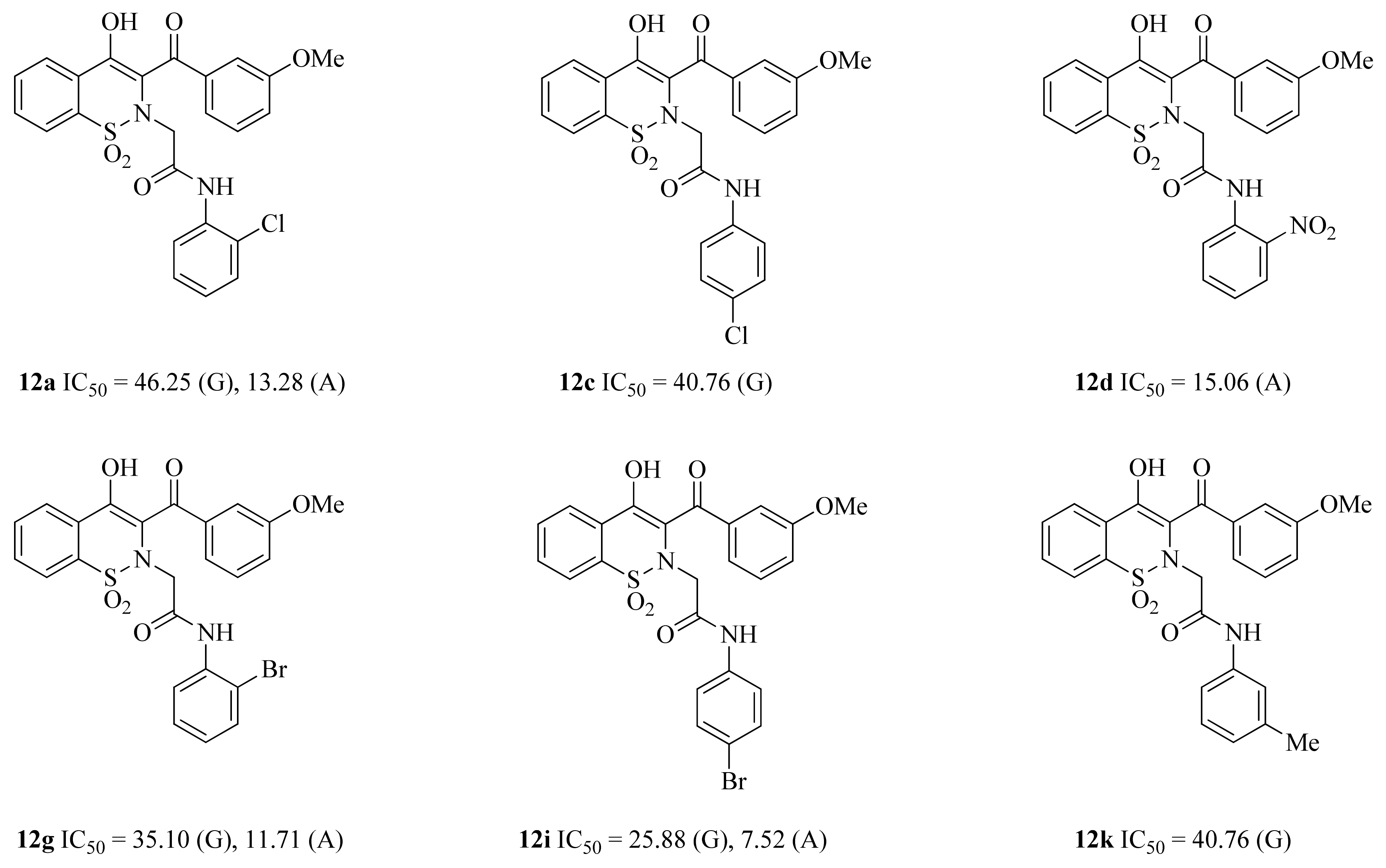
2.4.1. α-Glucosidase Inhibition
2.4.2. α-Amylase Inhibition
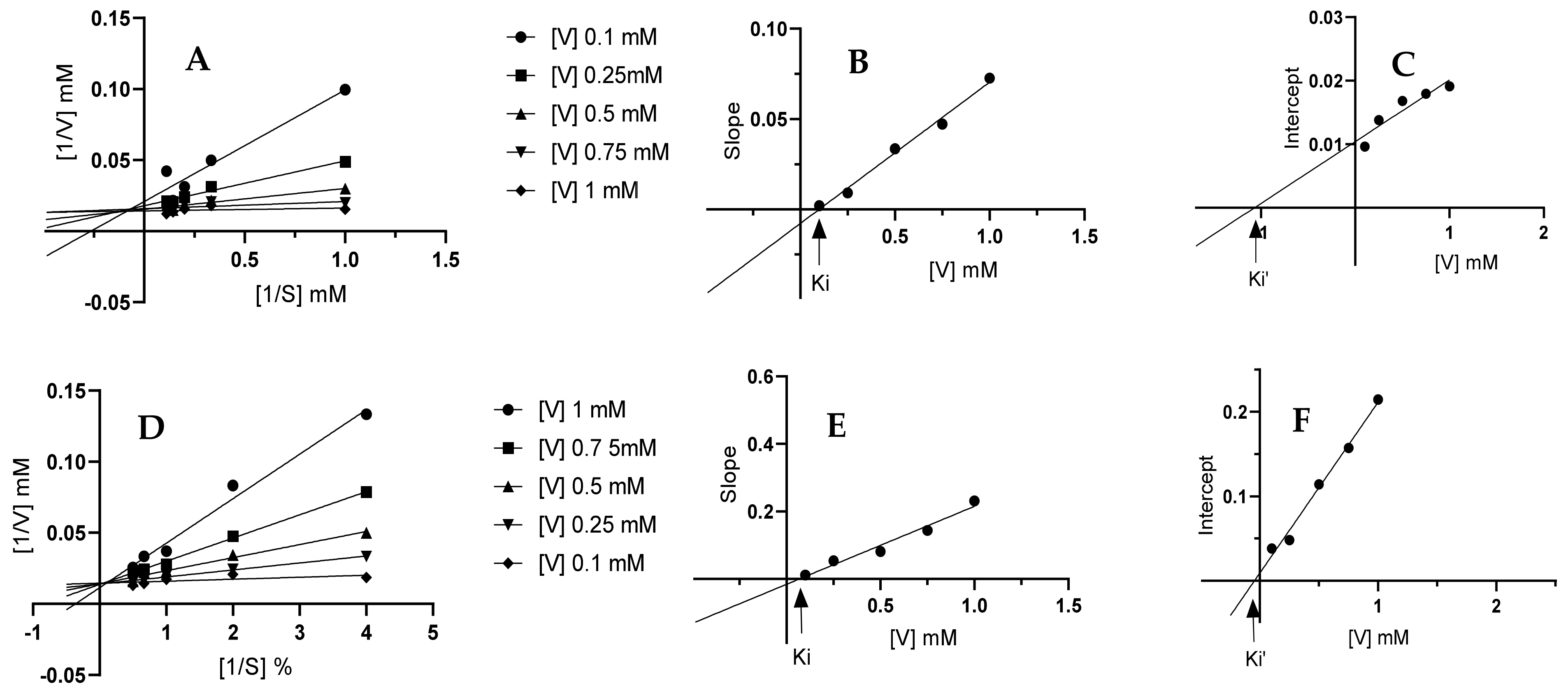
2.4.3. Enzyme Inhibition Kinetics
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of 2-(3-(3-methoxybenzoyl)-4-hydroxy-1,1-dioxido-2H-benzo[e][1,2] thiazin-2-yl)-N-arylacetamides (12a–m)
3.3. In Silico Analysis
3.3.1. Molecular Docking
3.3.2. MD Simulation
3.3.3. Binding Free Energy Calculations
3.4. Biological Activity
3.4.1. α-Glucosidase Inhibition
3.4.2. α-Amylase Inhibition
3.4.3. Enzyme Inhibition Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ogurtsova, K.; Fernandes, J.D.D.R.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hassan, N. Definition of diabetes mellitus. Br. J. Gen. Pract. 2003, 53, 567–568. [Google Scholar] [PubMed]
- Cade, W.T. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys. Ther. 2008, 88, 1322–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Care, D. Classification and diagnosis of diabetes. Diabetes Care 2015, 38, 8–16. [Google Scholar]
- Musselman, D.L.; Betan, E.; Larsen, H.; Phillips, L.S. Relationship of depression to diabetes types 1 and 2: Epidemiology, biology, and treatment. Biol. Psychiatry 2003, 54, 317–329. [Google Scholar] [CrossRef]
- Knol, M.J.; Twisk, J.W.; Beekman, A.T.; Heine, R.J.; Snoek, F.J.; Pouwer, F. Depression as a risk factor for the onset of type 2 diabetes mellitus. A meta-analysis. Diabetologia 2006, 49, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Song, L.; Wang, H.; Huang, D. A high-through put assay for quantification of starch hydrolase inhibition based on turbidity measurement. J. Agric. Food Chem. 2011, 59, 9756–9762. [Google Scholar] [CrossRef]
- Sarikaya, E.; Higassa, T.; Adachi, M.; Mikami, B. Comparison of degradation abilities of α- and β-amylases on raw starch granules. Proc. Biochem. 2000, 35, 711–715. [Google Scholar] [CrossRef]
- Ogawa, S.; Nako, K.; Okamura, M.; Sakamoto, T.; Ito, S. Stabilization of postprandialblood glucose fluctuations by addition of glucagon like polypeptide analog administration to intensive insulin therapy. J. Diabetes Investig. 2015, 6, 436–442. [Google Scholar] [CrossRef]
- Ghani, U. Re-exploring promising α-glucosidase inhibitors for potential development into oral anti-diabetic drugs: Finding needle in the haystack. Eur. J. Med. Chem. 2015, 103, 133–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Bao, L.; Ma, K.; Zhang, L.; Chen, B.; Han, J.; Ren, J.; Luo, H.; Liu, H. A novel class of α-glucosidase and HMG-CoA reductase inhibitors from Ganoderma leucocontextum and the anti-diabetic properties of ganomycin 1 in KK-Ay mice. Eur. J. Med. Chem. 2017, 127, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Bao, L.; Zhou, N.; Zhang, J.; Liao, M.; Zheng, Z.; Wang, Y.; Liu, C.; Wang, L.; Wang, W.; et al. Structural modification of natural product ganomycin 1 leading to discovery of a α-glucosidase and HMG-CoA reductase dual inhibitor improving obesity and metabolic dysfunction in vivo. J. Med. Chem. 2018, 61, 3609–3625. [Google Scholar] [CrossRef]
- Rocha, S.; Ribeiro, D.; Fernandes, E.; Freitas, M. A systematic review on anti-diabetic properties of chalcones. Curr. Med. Chem. 2020, 27, 2257–2321. [Google Scholar] [CrossRef]
- Ceriello, A. Postprandial hyperglycemia and diabetes complications: Is it time to treat? Diabetes 2005, 54, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Palanisamy, S.; Yien, E.L.H.; Shi, L.W.; Si, L.Y.; Qi, S.H.; Ling, L.S.C.; Lun, T.W.; Chen, Y.N. Systematic review of efficacy and safety of newer anti-diabetic drugs approved from 2013 to 2017 in controlling HbA1c in Diabetes Patients. Pharmacy 2018, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Etsassala, N.G.E.R.; Badmus, J.A.; Waryo, T.; Marnewick, J.L.; Cupido, C.N.; Hussein, A.A.; Iwuoha, E.I. Alpha-glucosidase and alpha-amylase inhibitory activities of novel abietane diterpenes from Salvia africana-lutea. Antioxidants 2019, 8, 421. [Google Scholar] [CrossRef] [Green Version]
- Murai, A.; Iwamura, K.; Takada, M.; Ogawa, K.; Usui, T.; Okumura, J. Control of postprandial hyperglycaemia by galactosyl maltobionolactone and its novel anti-amylase effect in mice. Life Sci. 2002, 71, 1405–1415. [Google Scholar] [CrossRef]
- Amarowicz, R.; Troszyńska, A.; Shahidi, F. Antioxidant activity of almond seed extract and its fractions. J. Food Lipids 2005, 12, 344–358. [Google Scholar] [CrossRef]
- Fujisawa, T.; Ikegami, H.; Inoue, K.; Kawabata, Y.; Ogihara, T. Effect of two alpha-glucosidase inhibitors, voglibose and acarbose, on postprandial hyperglycemia correlates with subjective abdominal symptoms. Metabolism 2005, 54, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Shobana, S.; Sreerama, Y.N.; Malleshi, N.G. Composition and enzyme inhibitory properties of finger millet (Eleusine coracana L.) seed coat phenolics: Mode of inhibition of α-glucosidase and pancreatic amylase. Food Chem. 2009, 115, 1268–1273. [Google Scholar] [CrossRef]
- Bedekar, A.; Shah, K.; Koffas, M. Natural products for type II diabetes treatment. In Advances in Appliedmicrobiology; Laskin, A.I., Sariaslani, S., Gadd, G.M., Eds.; Elsevier Inc.: San Diego, CA, USA, 2010; Volume 71, pp. 21–73. [Google Scholar]
- Lombardino, J.G.; Wiseman, E.H.; Mclamore, W. Synthesis and antiinflammatory activity of some 3-carboxamides of 2-alkyl-4-hydroxy-2H-1, 2-benzothiazine 1, 1-dioxide. J. Med. Chem. 1971, 14, 1171–1175. [Google Scholar] [CrossRef]
- Engelhardt, G.; Homma, D.; Schlegel, K.; Utzlnann, R.; Schnitzler, C. Anti-inflammatory, analgesic, antipyretic and related properties of meloxicam, a new non-steroidal anti-inflammatory agent with favorable gastrointestinal tolerance. Inflamm. Res. 1995, 44, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Churchill, L.; Graham, A.G.; Shih, C.K.; Pauletti, D.; Farina, P.R.; Grob, P.M. Selective inhibition of human cyclo-oxygenase-2 by meloxicam. Inflammopharmacology 1996, 4, 125–135. [Google Scholar] [CrossRef]
- Dequeker, J.; Hawkey, C.; Kahan, A.O.; Steinbrück, K.; Alegre, C.; Baumelou, E.; Begaud, B.; Isomäki, H.; Littlejohn, G.; Mau, J.; et al. Improvement in gastrointestinal tolerability of the selective cyclooxygenase (COX)-2 inhibitor, meloxicam, compared with piroxicam: Results of the safety and efficacy large-scale evaluation of COX-inhibiting therapies (SELECT) trial in osteoarthritis. Brit. J. Rheumatol. 1998, 37, 946–951. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kwon, S.W.; Chu, S.Y.; Lee, J.H.; Narsaiah, B.; Kim, C.H.; Kang, S.K.; Kang, N.S.; Rhee, S.D.; Bae, M.A.; et al. Identification of cyclicsulfonamide derivatives with an acetamide group as 11β-hydroxysteroid dehydrogenase 1 inhibitors. Chem. Pharm. Bull. 2011, 59, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.S.; Lee, J.Y.; Noh, S.; Kwak, Y.; Jeon, S.; Kwon, S.; Jin, Y.H.; Jang, M.S.; Kim, S.; Song, J.H.; et al. Discovery of cyclic sulfonamide derivatives as potent inhibitors of SARS-CoV-2. Bioorg. Med. Chem. Lett. 2021, 31, 127667. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, S.; Yang, Y.; Hussain, S.; He, M.; Gui, D.; Ma, B.; Jing, C.; Qiao, Z.; Zhu, C.; et al. 1,2-Benzothiazine 1,1-dioxide carboxylate derivatives as novel potent inhibitors of aldose reductase. Bioorg. Med. Chem. 2011, 19, 7262–7269. [Google Scholar] [CrossRef] [PubMed]
- Saddique, F.A.; Ahmad, M.; Ashfaq, U.A.; Aslam, A.; Khan, S.G. Alpha-glucosidase inhibition and molecular docking studies of 4-hydroxy-N’-[benzylidene/1-phenylethylidene]-2H-1,2-benzothiazine-3-carbohydrazide 1,1-dioxides. Chiang Mai J. Sci. 2021, 48, 460–469. [Google Scholar]
- Saddique, F.A.; Ahmad, M.; Ashfaq, U.A.; Ahmad, M.N.; Anjum, M.N.; Mohsin, N.U.A.; Aslam, S. Alpha-glucosidase inhibition and molecular docking studies of 1,2-benzothiazine 1,1-dioxide based carbohydrazides. Pak. J. Pharm. Sci. 2019, 32, 2829–2834. [Google Scholar] [PubMed]
- Saddique, F.A.; Zaib, S.; Jalil, S.; Aslam, S.; Ahmad, M.; Sultan, S.; Naz, H.; Iqbal, M.; Iqbal, J. Synthesis, monoamine oxidase inhibition activity and molecular docking studies of novel 4-hydroxy-N’-[benzylidene or 1-phenylethylidene]-2-H/methyl/benzyl-1,2-benzothiazine-3-carbohydrazide 1,1-dioxides. Eur. J. Med. Chem. 2018, 143, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Saddique, F.A.; Aslam, S.; Ahmad, M.; Ashfaq, U.A.; Muddassar, M.; Sultan, S.; Taj, S.; Hussain, M.; Lee, D.S.; Zaki, M.E.A. Synthesis and α-Glucosidase Inhibition Activity of 2-[3-(Benzoyl/4-bromobenzoyl)-4-hydroxy-1,1-dioxido-2H-benzo[e][1,2]thiazin-2-yl]-N-arylacetamides: An In silico and Biochemical Approach. Molecules 2021, 26, 3043. [Google Scholar] [CrossRef]
- Walayat, K.; Ahmad, M.; Ashfaq, U.A.; Khan, Z.A.; Sultan, S. Synthesis and α-glucosidase inhibition studies of norfloxacin-acetanilide hybrids. Pak. J. Pharm. Sci. 2021, 34, 1909–1915. [Google Scholar] [PubMed]
- Shabbir, A.; Shahzad, M.; Ali, A.; Zia-ur-Rehman, M. Discovery of new benzothiazine derivative as modulator of pro- and anti-inflammatory cytokines in rheumatoid arthritis. Inflammation 2016, 39, 1918–1929. [Google Scholar] [CrossRef]
- Ahmad, M.; Aslam, S.; Bukhari, M.H.; Montero, C.; Detorio, M.; Parvez, M.; Schinazi, R.F. Synthesis of novel pyrazolobenzothiazine 5,5-dioxide derivatives as potent anti-HIV-1 agents. Med. Chem. Res. 2014, 23, 1309–1319. [Google Scholar] [CrossRef]
- Ahmad, M.; Aslam, S.; Rizvi, S.U.F.; Muddassar, M.; Ashfaq, U.A.; Montero, C.; Ollinger, O.; Detorio, M.; Gardiner, J.M.; Schinazi, R.F. Molecular docking and anti-viral screening of N-substituted benzyl/phenyl-2-(3,4-dimethyl-5,5-dioxidopyrazolo[4,3-c][1,2]benzothiazin-2(4H)-yl) acetamides. Bioorg. Med. Chem. Lett. 2015, 25, 1348–1351. [Google Scholar] [CrossRef]
- Aslam, S.; Ahmad, M.; Athar, M.M.; Ashfaq, U.A.; Gardiner, J.M.; Montero, C.; Detorio, M.; Parvez, M.; Schinazi, R.F. Synthesis, molecular docking and antiviral screening of novel N’-substitutedbenzylidene-2-(4-methyl-5,5-dioxido-3-phenylbenzo[e]pyrazolo[4,3-c][1,2]thiazin-1(4H)-yl) acetohydrazides. Med. Chem. Res. 2014, 6, 2930–2946. [Google Scholar] [CrossRef]
- Khalid, Z.; Aslam, S.; Ahmad, M.; Munawar, M.A.; Montero, C.; Detorio, M.; Parvez, M.; Schinazi, R.F. Anti-HIV Activity of New Pyrazolobenzothiazine 5,5-dioxide Based Acetohydrazides. Med. Chem. Res. 2015, 24, 3671–3680. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Tang, S.; Aslam, S.; Ahmad, M.; To, K.K.W.; Wang, F.; Huang, Z.; Cai, J.; Fu, L. UMMS-4 enhanced sensitivity of chemotherapeutic agents to ABCB1-overexpressing cells via inhibiting function of ABCB1 transporter. Am. J. Cancer Res. 2014, 4, 148–160. [Google Scholar] [PubMed]
- Aslam, S.; Xi, Y.; Ahmad, M.; Mansha, A.; Farooq, T.; Zheng, Z.; Saddique, F.A.; Wang, F.; Fu, L. Anticancer activity of structural hybrids of various 5/6-memberedheterocycles with pyrazolobenzothiazine 5,5-dioxide. Pak. J. Pharm. Sci. 2020, 33, 1239–1243. [Google Scholar]
- Rai, A.; Raj, V.; Singh, A.K.; Keshari, A.K.; Kumar, U.; Kumar, D.; Saha, S. Design and synthesis of 1,4-benzothiazine derivatives with promising effects against colorectal cancer cells. Cogent. Chem. 2017, 3, 1303909. [Google Scholar] [CrossRef]
- Aslam, S.; Zaib, S.; Ahmad, M.; Gardiner, J.M.; Ahmad, A.; Hameed, A.; Furtmann, N.; Gutschow, M.; Bajorath, J.; Iqbal, J. Novel structural hybrids of pyrazolobenzothiazines with benzimidazoles as cholinesterase inhibitors. Eur. J. Med. Chem. 2014, 78, 106–117. [Google Scholar] [CrossRef]
- Sajid, Z.; Ahmad, M.; Aslam, S.; Ashfaq, U.A.; Zahoor, A.F.; Saddique, F.A.; Parvez, M.; Hameed, A.; Sultan, S.; Zgou, H.; et al. Novel armed pyrazolobenzothiazine derivatives: Synthesis, X-ray crystal structure and POM analyses of biological activity against drug resistant clinical isolate of staphylococus aureus. Pharm. Chem. J. 2016, 50, 172–180. [Google Scholar] [CrossRef]
- Seo, W.D.; Kim, J.H.; Kang, J.E.; Ryu, H.W.; Curtis-Long, M.J.; Lee, H.S.; Yang, M.S.; Park, K.H. Sulfonamide chalcone as a new class of a-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 5514–5516. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Irshad, M.; Imran, S.; Chigurupati, S.; Selvaraj, M.; Rahim, F.; Ismail, N.H.; Nawaz, F.; Khan, K.M. Synthesis of piperazine sulfonamide analogs as diabetic-II inhibitors and their molecular docking study. Eur. J. Med. Chem. 2017, 141, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Gul, S.; Siddiqui, H.L.; Ahmad, M.; Parvez, M. (4-Hydroxy-1, 1-dioxo-2H-1, 2-benzothiazin-3-yl)(3-methoxyphenyl) methanone. Acta Crystallogr. E 2010, 66, o1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, K.; Hua, X.W.; Tao, Y.Y.; Liu, Y.; Liu, N.; Ma, Y.; Li, Y.H.; Xu, X.H.; Kong, C.H. Discovery of (2-benzoylethen-1-ol)-containing 1, 2-benzothiazine derivatives as novel 4-hydroxyphenylpyruvate dioxygenase (HPPD) inhibiting-based herbicide lead compounds. Bioorg. Med. Chem. 2016, 24, 92–103. [Google Scholar] [CrossRef]
- Lee, J.C.; Bae, Y.H.; Chang, S.K. Efficient α-halogenation of carbonyl compounds by N-bromosuccinimide and N-chlorosuccinimde. Bull. Korean Chem. Soc. 2003, 24, 407–408. [Google Scholar] [CrossRef]
- Dudek-Wicher, R.K.; Szczęśniak-Sięga, B.M.; Wiglusz, R.J.; Janczak, J.; Bartoszewicz, M.; Junka, A.F. Evaluation of 1,2-benzothiazine 1,1-dioxide derivatives in vitro activity towards clinical-relevant microorganisms and fibroblasts. Molecules 2020, 25, 3503. [Google Scholar] [CrossRef]
- Henderson, B.J.; Carper, D.J.; Gonzalez-Cestari, T.F.; Yi, B.; Mahasenan, V.; Pavlovicz, R.E.; Dalefield, M.L.; Coleman, R.S.; Li, C.; McKay, D.B. Structure–activity relationship studies of sulfonylpiperazine analogues as novel negative allosteric modulators of human neuronal nicotinic receptors. J. Med. Chem. 2011, 54, 8681–8692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zia-ur-Rehman, M.; Choudary, J.A.; Elsegood, M.R.J.; Siddiqui, H.L.; Khan, K.M. A facile synthesis of novel biologically active 4-hydroxy-N’-(benzylidene)-2H-benzo[e][1,2]thiazine-3-carbohydrazide 1,1-dioxides. Eur. J. Med. Chem. 2009, 44, 1311–1316. [Google Scholar] [CrossRef]
- Ahmad, N.; Zia-ur-Rehman, M.; Siddiqui, H.L.; Ullah, M.F.; Parvez, M. Microwave assisted synthesis and structure-activity relationship of 4-hydroxy-N′-[1-phenylethylidene]-2H/2-methyl-1,2-benzothiazine-3-carbohydrazide 1,1-dioxides as anti-microbial agents. Eur. J. Med. Chem. 2011, 46, 2368–2377. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Haroon, R.A.; Bardaweel, S.K.; Hajjo, R.; Sweidan, K. N-phenyl-6-chloro-4-hydroxy-2-quinolone-3-carboxamides: Molecular Docking, Synthesis, and Biological Investigation as Anticancer Agents. Molecules 2021, 26, 73. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Alshamrani, F.J.; Rahim, F.; Hayat, S.; Ullah, H.; Zaman, K.; Imran, S.; Khan, K.M.; Naz, F. Synthesis of novel triazinoindole-based thiourea hybrid: A study on α-glucosidase inhibitors and their molecular docking. Molecules 2019, 24, 3819. [Google Scholar] [CrossRef] [Green Version]
- El-Azab, I.H.; El-Sheshtawy, H.S.; Bakr, R.B.; Elkanzi, N. New 1, 2, 3-Triazole-Containing Hybrids as Antitumor Candidates: Design, Click Reaction Synthesis, DFT Calculations, and Molecular Docking Study. Molecules 2021, 26, 708. [Google Scholar] [CrossRef]
- Munir, R.; Zia-ur-Rehman, M.; Murtaza, S.; Zaib, S.; Javid, N.; Awan, S.J.; Iftikhar, K.; Athar, M.M.; Khan, I. Microwave-Assisted Synthesis of (Piperidin-1-yl) quinolin-3-yl) methylene) hydrazinecarbothioamides as Potent Inhibitors of Cholinesterases: A Biochemical and In silico Approach. Molecules 2021, 26, 656. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Giovanni, C.D. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef]
- Ruyck, J.D.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Liebeschuetz, J.W.; Cole, J.C.; Korb, O. Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput. Aided Mol. Des. 2012, 26, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Gani, R.S.; Kudva, A.K.; Timanagouda, K.; Mujawar, S.B.H.; Joshi, S.D.; Raghu, S.V. Synthesis of novel 5-(2, 5-bis (2, 2, 2-trifluoroethoxy) phenyl)-1, 3, 4-oxadiazole-2-thiol derivatives as potential glucosidase inhibitors. Bioorg. Chem. 2021, 114, 105046. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Takahira, K.; Tanabe, G.; Morikawa, T.; Sakano, M.; Ninomiya, K.; Yoshikawa, M.; Muraoka, O.; Nakanishi, I. Docking and SAR studies of salacinol derivatives as α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 4420–4423. [Google Scholar] [CrossRef]
- Promyos, N.; Temviriyanukul, P.; Suttisansanee, U. Investigation of anthocyanidins and anthocyanins for targeting α-glucosidase in diabetes mellitus. Prev. Nutr. Food Sci. 2020, 25, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Nursamsiar, N.; Mangande, M.M.; Awaluddin, A.; Nur, S.; Asnawi, A. In Silico study of aglycon curculigoside A and its derivatives as α-amylase inhibitors. Indones. J. Pharm. Sci. Technol. 2020, 7, 29–37. [Google Scholar] [CrossRef]
- Saidi, I.; Manachou, M.; Znati, M.; Bouajila, J.; Jannet, H.B. Synthesis of new halogenated flavonoid-based isoxazoles: In vitro and in silico evaluation of a-amylase inhibitory potential, a SAR analysis and DFT studies. J. Mol. Struct. 2022, 1247, 131379. [Google Scholar] [CrossRef]
- Duhan, M.; Sindhu, J.; Kumar, P.; Devi, M.; Singh, R.; Kumar, R.; Lal, S.; Kumar, A.; Kumar, S.; Hussain, K. Quantitative structure activity relationship studies of novel hydrazone derivatives as α-amylase inhibitors with index of ideality of correlation. J. Biomol. Struct. Dyn. 2020, 38, 1–22. [Google Scholar] [CrossRef]
- Duhan, M.; Singh, R.; Devi, M.; Sindhu, J.; Bhatia, R.; Kumar, A.; Kumar, P. Synthesis, molecular docking and QSAR study of thiazole clubbed pyrazole hybrid as α-amylase inhibitor. J. Biomol. Struct. Dyn. 2021, 39, 91–107. [Google Scholar] [CrossRef]
- Flores-Bocanegra, L.; Pérez-Vásquez, A.; Torres-Piedra, M.; Bye, R.; Linares, E.; Mata, R. α-Glucosidase inhibitors from Vauquelinia corymbosa. Molecules 2015, 20, 15330–15342. [Google Scholar] [CrossRef] [Green Version]
- Caputo, A.T.; Alonzi, D.S.; Marti, L.; Reca, I.B.; Kiappes, J.L.; Struwe, W.B.; Cross, A.; Basu, S.; Lowe, E.D.; Darlot, B.; et al. Structures of mammalian ER α-glucosidase II capture the binding modes of broad-spectrum iminosugar antivirals. Proc. Natl. Acad. Sci. USA 2016, 113, E4630–E4638. [Google Scholar] [CrossRef] [Green Version]
- Dinparast, L.; Valizadeh, H.; Bahadori, M.B.; Soltani, S.; Asghari, B.; Rashidi, M.R. Design, synthesis, α-glucosidase inhibitory activity, molecular docking and QSAR studies of benzimidazole derivatives. J. Mol. Struct. 2016, 1114, 84–94. [Google Scholar] [CrossRef]
- Kashtoh, H.; Muhammad, M.T.; Khan, J.J.; Rasheed, S.; Khan, A.; Perveen, S.; Javaid, K.; Khan, K.M.; Choudhary, M.I. Dihydropyrano [2, 3-c] pyrazole: Novel in vitro inhibitors of yeast α-glucosidase. Bioorg. Chem. 2016, 65, 61–72. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Ahmad, R.; Anouar, E.; Azman, N.I.; Marzouk, M.; Abuelizz, H.A. 3-Benzyl (phenethyl)-2-thioxobenzo [g] quinazolines as a new class of potent α-glucosidase inhibitors: Synthesis and molecular docking study. Future Med. Chem. 2018, 16, 1889–1905. [Google Scholar] [CrossRef]
- Abbasi, M.A.; Riaz, S.; Rehman, A.U.; Siddiqui, S.Z.; Shah, S.A.; Ashraf, M.; Lodhi, M.A.; Khan, F.A. Synthesis of new 2-{2, 3-dihydro-1, 4-benzodioxin-6-yl [(4-methylphenyl) sulfonyl] amino}-N-(un/substituted-phenyl) acetamides as α-glucosidase and acetylcholinesterase inhibitors and their in silico study. Braz. J. Pharm. Sci. 2019, 55, e17032. [Google Scholar] [CrossRef] [Green Version]
- Algethami, F.K.; Saidi, I.; Abdelhamid, H.N.; Elamin, M.R.; Abdulkhair, B.Y.; Chrouda, A.; Ben Jannet, H. Trifluoromethylated Flavonoid-Based Isoxazoles as Antidiabetic and Anti-Obesity Agents: Synthesis, In Vitro α-Amylase Inhibitory Activity, Molecular Docking and Structure–Activity Relationship Analysis. Molecules 2021, 26, 5214. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, M.; Taha, M.; Qureshi, F.; Ullah, N.; Selvaraj, M.; Shahzad, S.; Chigurupati, S.; Waheed, A.; Almutairi, F.A. Structural elucidation, molecular docking, α-amylase and α-glucosidase inhibition studies of 5-amino-nicotinic acid derivatives. BMC Chem. 2020, 14, 43. [Google Scholar] [CrossRef]
- Aispuro-Pérez, A.; López-Ávalos, J.; García-Páez, F.; Montes-Avila, J.; Picos-Corrales, L.A.; Ochoa-Terán, A.; Bastidas, P.; Montaño, S.; Calderón-Zamora, L.; Osuna-Martínez, U.; et al. Synthesis and molecular docking studies of imines as α-glucosidase and α-amylase inhibitors. Bioorg. Chem. 2020, 94, 103491. [Google Scholar] [CrossRef]
- Altaff, S.M.; Rajeswari, T.R.; Subramanyam, C. Synthesis, α-amylase inhibitory activity evaluation and in silico molecular docking study of some new phosphoramidates containing heterocyclic ring. Phosphorus Sulfur Silicon Relat. Elem. 2020, 196, 389–397. [Google Scholar] [CrossRef]
- Kim, G.Y.; Chun, H.G.; Lee, S.D.; Kim, H.Y.; Jung, W.H.; Kang, N.S.; Bae, M.A.; Ahn, J.H.; Kang, S.G. Patent Publication 10-2011-0060653. KR 0060653. 2011. [Google Scholar]
- Salar, U.; Taha, M.; Khan, K.M.; Ismail, N.H.; Imran, S.; Perveen, S.; Gul, S.; Wadood, A. Syntheses of new 3-thiazolyl coumarin derivatives, in vitro α-glucosidase inhibitory activity, and molecular modeling studies. Eur. J. Med. Chem. 2016, 122, 196–204. [Google Scholar] [CrossRef]
- Taha, M.; Ismail, N.H.; Imran, S.; Wadood, A.; Rahim, F.; Saad, S.M.; Khan, K.M.; Nasir, A. Synthesis, molecular docking and α-glucosidase inhibition of 5-aryl-2-(6′-nitrobenzofuran-2′-yl)-1, 3, 4-oxadiazoles. Bioorg. Chem. 2016, 66, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Zawawi, N.K.N.A.; Taha, M.; Ahmat, N.; Wadood, A.; Ismail, N.H.; Rahim, F.; Azam, S.S.; Abdullah, N. Benzimidazole derivatives as new α-glucosidase inhibitors and in silico studies. Bioorg. Chem. 2016, 64, 29–36. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N-log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Mosihuzzman, M.; Naheed, S.; Hareem, S.; Talib, S.; Abbas, G.; Khan, S.N.; Israr, M. Studies on α-glucosidase inhibition and anti-glycation potential of Iris loczyi and Iris unguicularis. Life Sci. 2013, 92, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Salar, U.; Khan, K.M.; Chigurupati, S.; Syed, S.; Vijayabalan, S.; Wadood, A.; Riaz, M.; Ghufran, M.; Perveen, S. New hybrid scaffolds based on hydrazinylthiazole substituted coumarin; as novel leads of dual potential; in vitro α-amylase inhibitory and antioxidant (DPPH and ABTS radical scavenging) activities. Med. Chem. 2019, 15, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Baharudin, M.S.; Ismail, N.H.; Imran, S.; Khan, M.N.; Rahim, F.; Selvaraj, M.; Chigurupati, S.; Nawaz, M.; Qureshi, F.; et al. Synthesis, α-amylase inhibitory potential and molecular docking study of indole derivatives. Bioorg. Chem. 2018, 80, 36–42. [Google Scholar] [CrossRef]
- Imran, S.; Taha, M.; Selvaraj, M.; Ismail, N.H.; Chigurupati, S.; Mohammad, J.I. Synthesis and biological evaluation of indole derivatives as α-amylase inhibitor. Bioorg. Chem. 2017, 73, 121–127. [Google Scholar] [CrossRef]
- Khan, K.M.; Gollapalli, M.; Taha, M.; Hayat, U.; Nawaz, M.; Al-Muqarrabun, L.M.R.; Rahim, F.; Qureshi, F.; Mosaddik, A.; Ahmat, N. Synthesis of Bisindolylmethanesulfonohydrazides derivatives as potent α-Glucosidase inhibitors. Bioorg. Chem. 2018, 80, 112–120. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Substituent | α-Glucosidase | α-Amylase | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Code | R | Docking Score (Kcal/mol) | RMSD Value (Å) | Binding Residues | IC50 (μM) | Docking Score (Kcal/mol) | RMSD Value (Å) | Binding Residues | IC50 (μM) |
| 11 | - | - | - | ND | - | - | 147.32 | ||
| 12a | 2-Cl | −12.50 | 1.28 | Asp203, Thr205 | 46.25 | −15.24 | 1.03 | Arg204, Asp340 | 13.28 |
| 12b | 3-Cl | - | - | ND | - | - | ND | ||
| 12c | 4-Cl | −12.80 | 1.29 | Asp542 | 40.76 | - | - | 124.6 | |
| 12d | 2-NO2 | - | - | 102.33 | −14.89 | 1.22 | Asp340 | 15.06 | |
| 12e | 3-NO2 | - | - | ND | - | - | 37.5 | ||
| 12f | 4-NO2 | - | - | 97.03 | −14.60 | - | - | 28.97 | |
| 12g | 2-Br | −12.98 | 1.22 | Asp542, Met444 | 35.10 | −15.89 | 0.88 | Trp83 | 11.71 |
| 12h | 3-Br | - | - | 86.21 | - | - | 109.6 | ||
| 12i | 4-Br | −13.30 | 1.01 | Asp327, Asp542 | 25.88 | −16.13 | 0.75 | Arg204 | 7.52 |
| 12j | 2-Me | - | - | ND | - | - | ND | ||
| 12k | 3-Me | −13.22 | 1.12 | Asp203 | 30.45 | - | - | 67.23 | |
| 12l | 2-OMe | - | - | 110.23 | - | - | ND | ||
| 12m | 4-OMe | - | - | ND | - | - | 121.88 | ||
| Acarbose | - | −16.18 | 2.12 | His600, Asp542, Arg526, Asp327, Met444, Lys480 | 58.8 | −17.69 | 2.01 | Arg204, Glu230 | 17.0 |
| Binding Energy Components | Amylase–Ligand | Std. Dev. | Std. Err. of Mean |
|---|---|---|---|
| ΔGbind = (ΔGcomplex) − (ΔGprotein + ΔGligand) | |||
| MMGBSA | |||
| VDWAALS | −50.6534 | 5.6170 | 0.7944 |
| EEL | −28.0565 | 13.2835 | 1.8786 |
| EGB | 53.3866 | 10.3616 | 1.4654 |
| ESURF | −6.1459 | 0.3115 | 0.0440 |
| DELTA G gas | −78.7099 | 17.7063 | 2.5041 |
| DELTA G solv | 47.2406 | 10.1638 | 1.4374 |
| DELTA TOTAL | −31.4693 | 8.6701 | 1.2261 |
| 12i | Ki | Ki’ |
|---|---|---|
| α-glucosidase | 0.1005 | −1.067 |
| α-amylase | 0.0704 | −0.0564 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saddique, F.A.; Ahmad, M.; Ashfaq, U.A.; Muddassar, M.; Sultan, S.; Zaki, M.E.A. Identification of Cyclic Sulfonamides with an N-Arylacetamide Group as α-Glucosidase and α-Amylase Inhibitors: Biological Evaluation and Molecular Modeling. Pharmaceuticals 2022, 15, 106. https://doi.org/10.3390/ph15010106
Saddique FA, Ahmad M, Ashfaq UA, Muddassar M, Sultan S, Zaki MEA. Identification of Cyclic Sulfonamides with an N-Arylacetamide Group as α-Glucosidase and α-Amylase Inhibitors: Biological Evaluation and Molecular Modeling. Pharmaceuticals. 2022; 15(1):106. https://doi.org/10.3390/ph15010106
Chicago/Turabian StyleSaddique, Furqan Ahmad, Matloob Ahmad, Usman Ali Ashfaq, Muhammad Muddassar, Sadia Sultan, and Magdi E. A. Zaki. 2022. "Identification of Cyclic Sulfonamides with an N-Arylacetamide Group as α-Glucosidase and α-Amylase Inhibitors: Biological Evaluation and Molecular Modeling" Pharmaceuticals 15, no. 1: 106. https://doi.org/10.3390/ph15010106