
Synthesis and Anti-Hepatoma Activities of U12 Derivatives Arresting G0/G1 Phase and Inducing Apoptosis by PI3K/AKT/mTOR Pathway

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
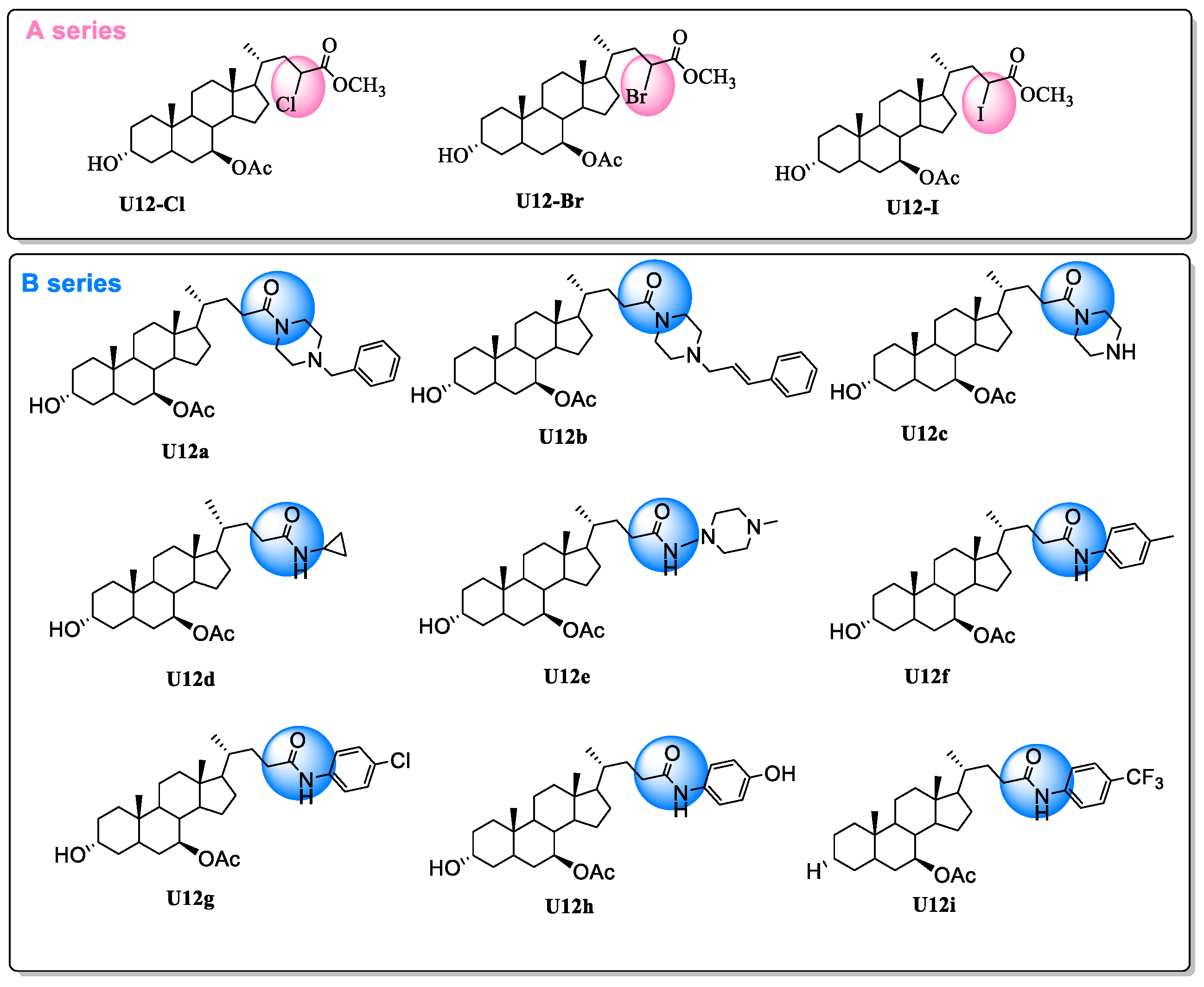
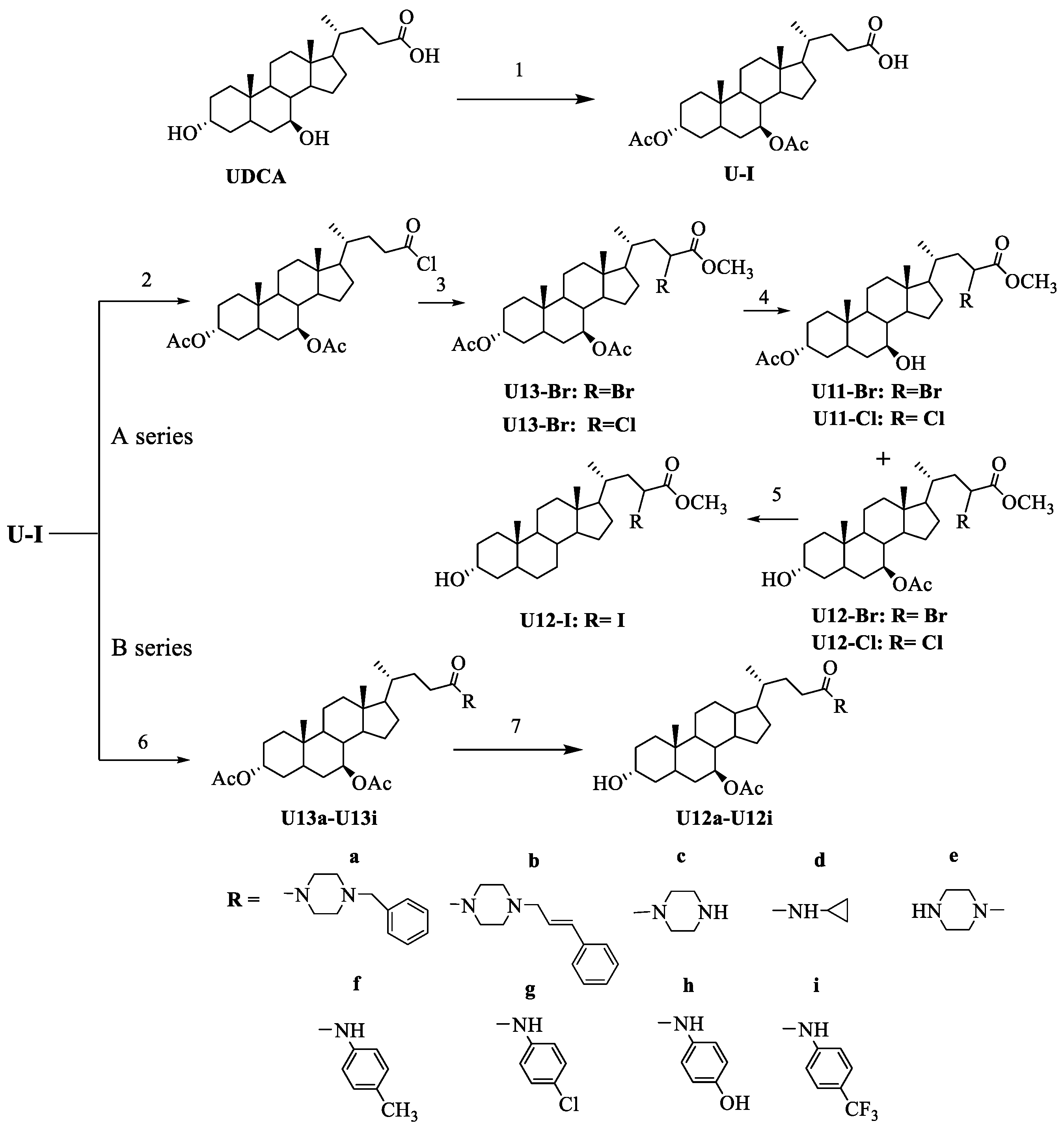
2.1. Chemistry
2.2. Biological Activities
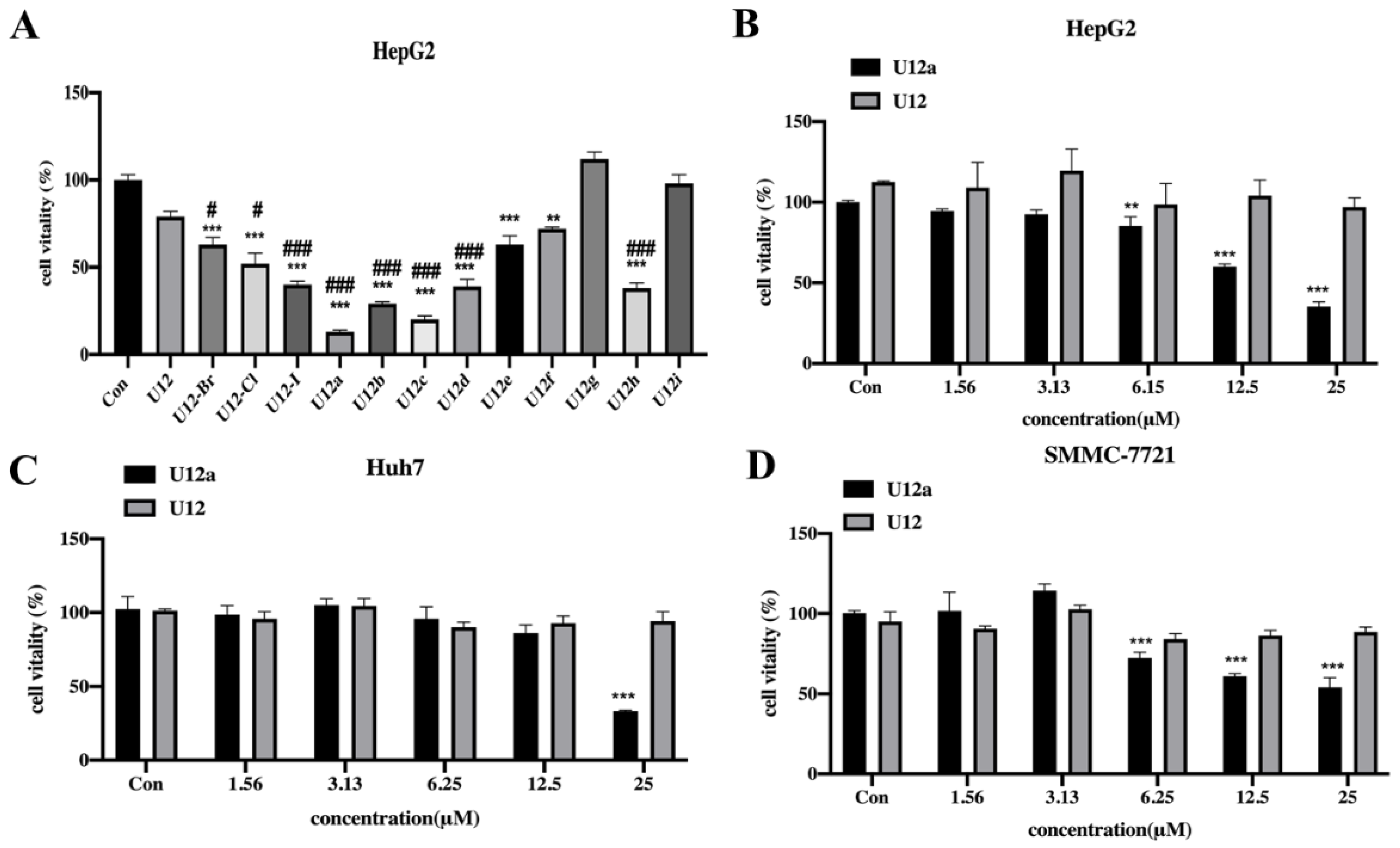
2.2.1. Cytotoxic Effects of the U12 Derivatives on HepG2 Cells
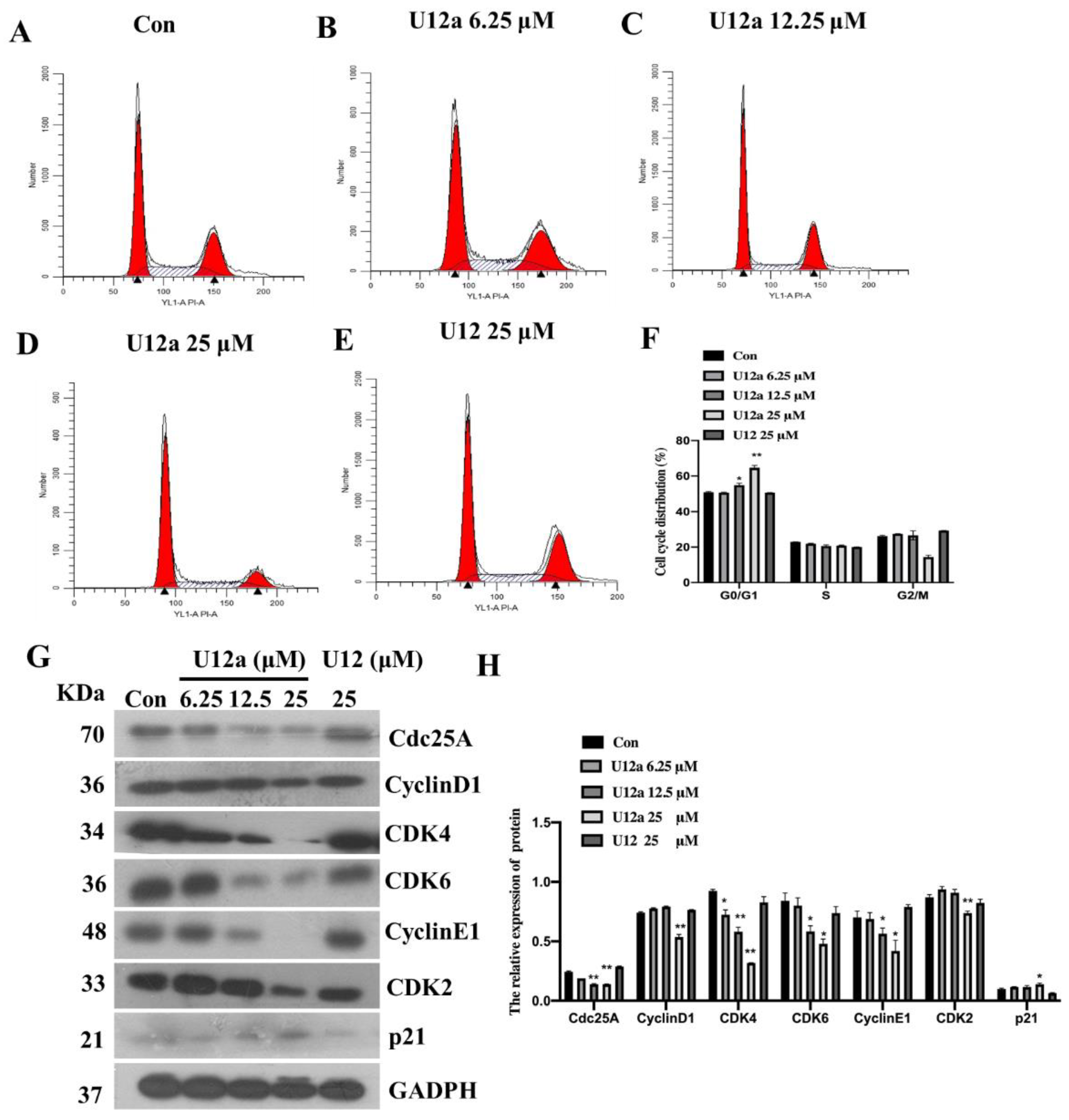
2.2.2. U12a Inhibits HepG2 Cell Proliferation by Arresting G0/G1 Phase
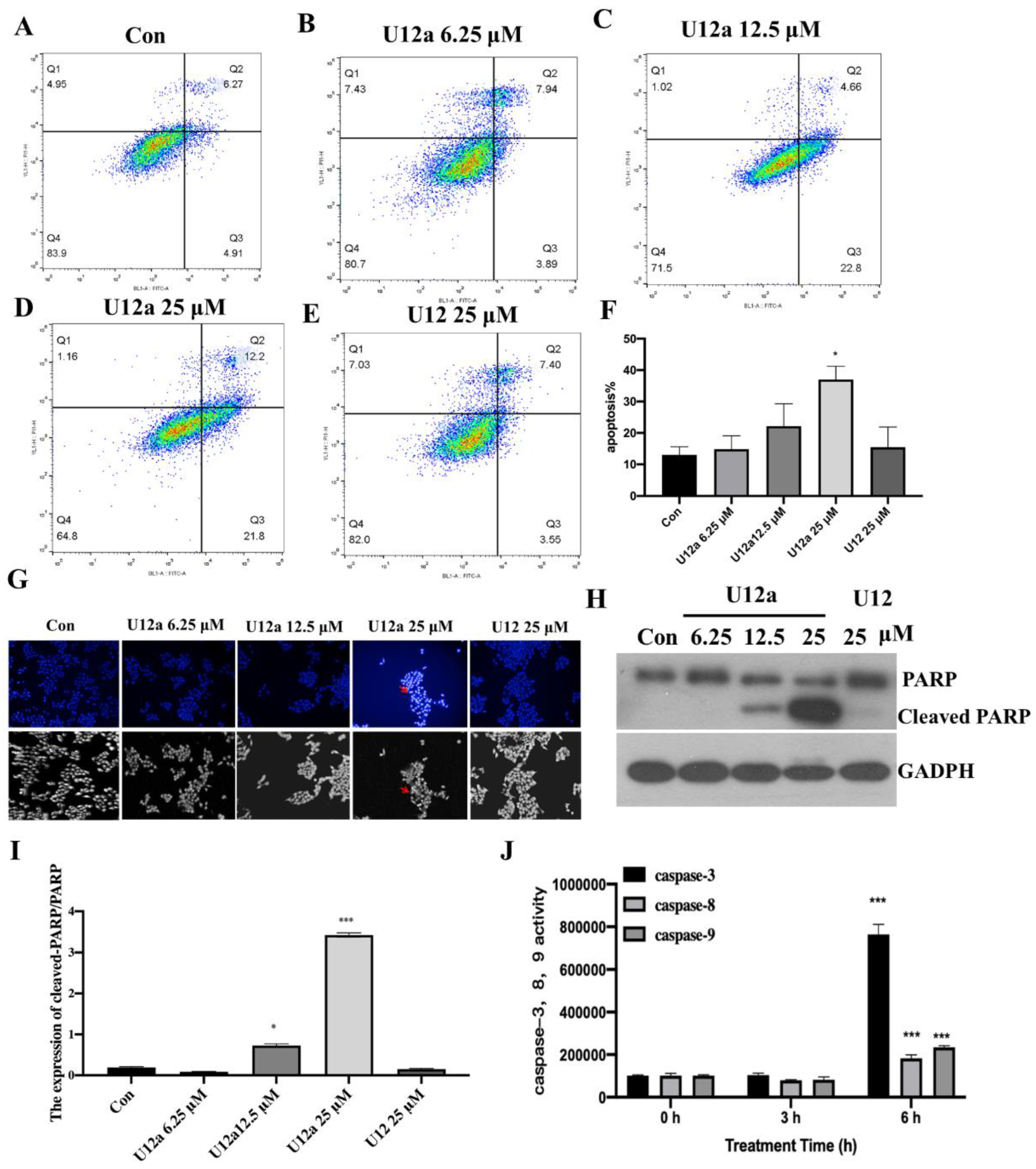
2.2.3. The Pro-Apoptotic Effect of U12a on HepG2 Cells
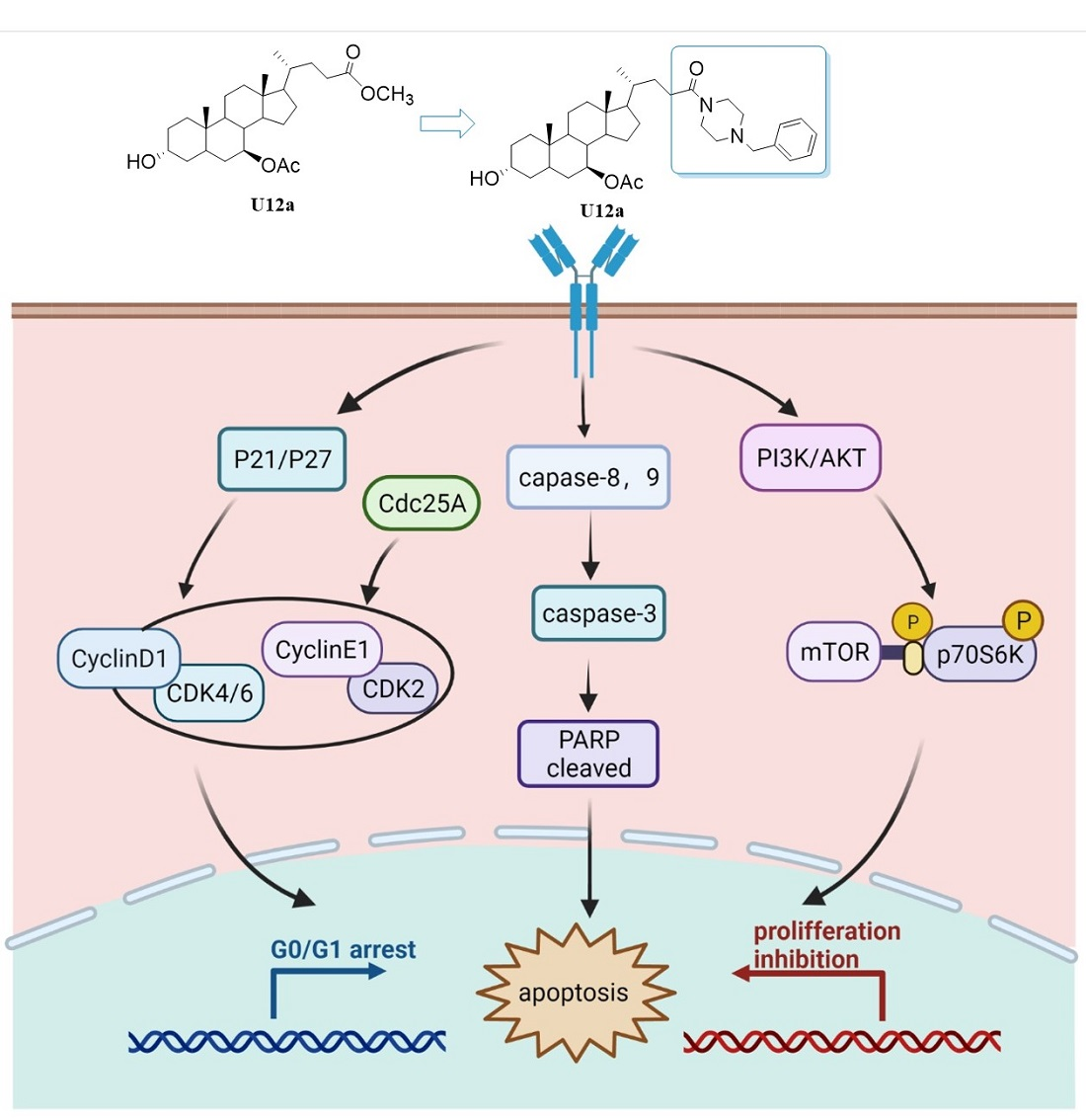
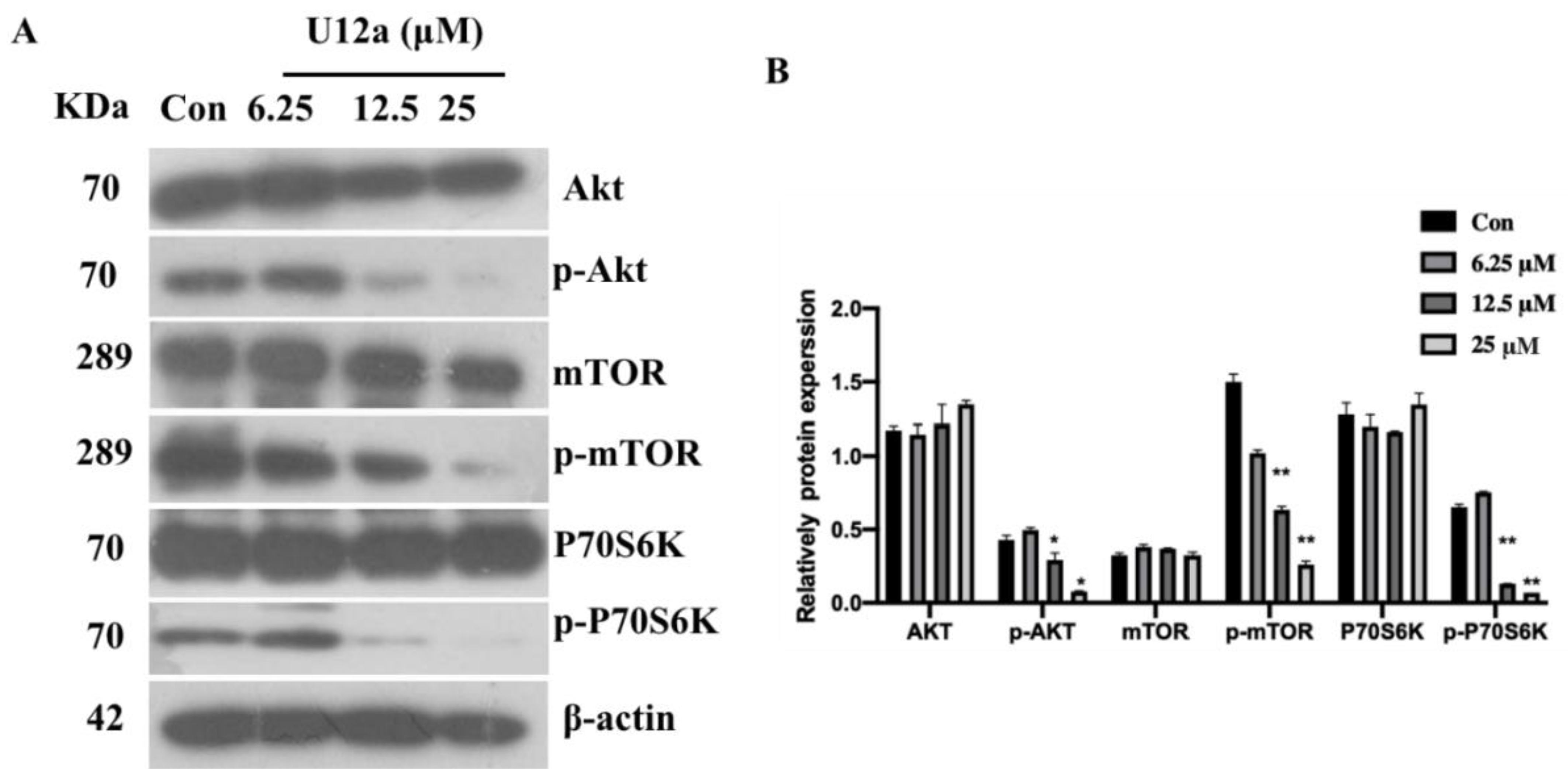
2.2.4. U12a Suppressed the Activation of the PI3K/AKT/mTOR Pathway
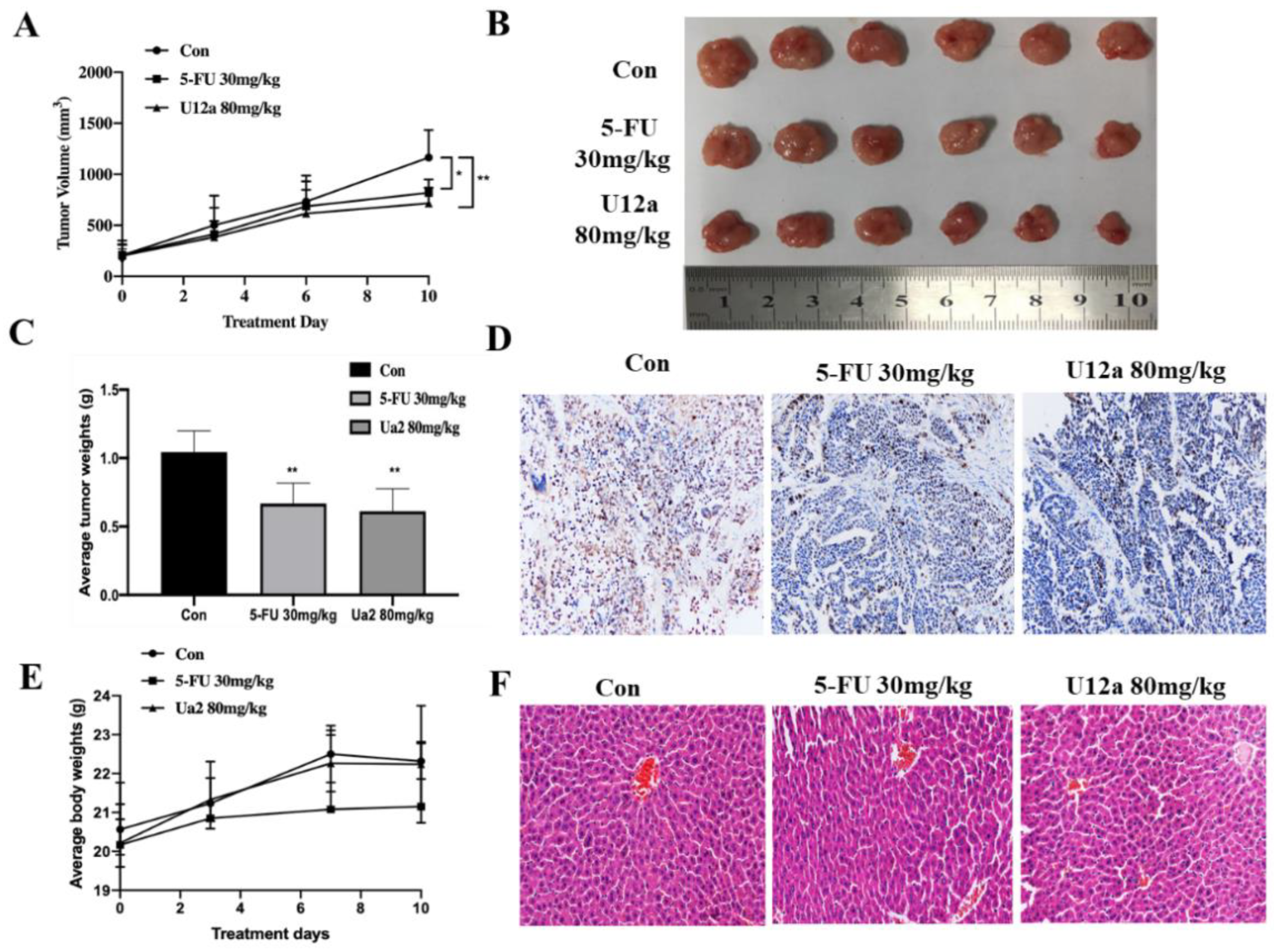
2.2.5. U12a Inhibited Tumor Growth in HepG2 Xenograft Model
3. Materials and Methods
3.1. Chemistry
General
3.2. Synthesis
3.2.1. General Procedure for 3α, 7 β-Diacetoxyursodeoxycholic Acid (U-I)
3.2.2. General Procedure for U12-Cl, U12-Br and U12-I
3.2.3. General Procedure for U12a-U12i
3.2.4. Analytical Data
3.3. Cell Viability Assay
3.4. Flow Cytometry Assay for Cell Cycle Detection
3.5. Annexin V-FITC/PI Double Staining Assay for Cellular Apoptosis Detection
3.6. Hochest 33528 Staining
3.7. Western Blot Assay
3.8. In vivo Anti-tumor Activity Detected by HepG2 Xenograft Model
3.9. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, W.; Xue, W.; Xu, P.; Deng, Z.; Zhang, D.; Zheng, S.; Qiu, X. Long noncoding RNA AURKAPS1 potentiates malignant hepatocellular carcinoma progression by regulating miR-142, miR-155 and miR-182. Sci. Rep. 2019, 9, 19645. [Google Scholar] [CrossRef] [Green Version]
- Llopiz, D.; Ruiz, M.; Villanueva, L.; Iglesias, T.; Silva, L.; Egea, J.; Lasarte, J.J.; Pivette, P.; Trochon-Joseph, V.; Vasseur, B.; et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 2019, 3, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.M.; Choi, J.; Lee, D.; Shim, J.H.; Lim, Y.S.; Lee, H.C.; Kim, K.M.; Chung, Y.-H.; Lee, Y.S.; Park, S.R.; et al. Regorafenib Versus Nivolumab After Sorafenib Failure: Real-World Data in Patients With Hepatocellular Carcinoma. Hepatol. Commun. 2020, 4, 1073–1086. [Google Scholar] [CrossRef]
- Lindor, K.D.; Gershwin, M.E.; Poupon, R.; Kaplan, M.; Bergasa, N.V.; Heathcote, E.J.; American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology 2009, 50, 291–308. [Google Scholar] [CrossRef]
- Beuers, U.; Boyer, J.L.; Paumgartner, G. Ursodeoxycholic acid in cholestasis: Potential mechani- sms of action and therapeutic applications. Hepatology 2003, 28, 1449–1453. [Google Scholar] [CrossRef]
- Park, I.H.; Kim, M.K.; Kim, S.U. Ursodeoxycholic acid prevents apoptosis of mouse sensory neurons induced by cisplatin by reducing P53 accumulation. Biochem. Biophys. Res. Commun. 2008, 377, 1025–1030. [Google Scholar] [CrossRef]
- Khare, S.; Mustafi, R.; Cerda, S.; Yuan, W.; Jagadeeswaran, S.; Dougherty, U.; Tretiakova, M.; Samarel, A.; Cohen, G.; Wang, J.; et al. Ursodeoxycho -lic acid suppresses Cox-2 expression in colon cancer: Roles of Ras, p38, and CCAAT/enhancer-binding protein. Nutr. Cancer 2008, 60, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Alberts, D.S.; Martínez, M.E.; Hess, L.M.; Einspahr, J.G.; Green, S.B.; Bhattacharyya, A.K.; Guillen, J.; Krutzsch, M.; Batta, A.K.; Salen, G.; et al. Phoenix and Tucson Gastroenterologist Networks. Phase III trial of ursodeoxycholic acid to prevent colorectal adenoma recurrence. J. Natl. Cancer Inst. 2005, 97, 846–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centuori, S.M.; Martinez, J.D. Differential regulation of EGFR-MAPK signaling by deoxycholic acid (DCA) and ursodeoxy- cholic acid (UDCA) in colon cancer. Dig. Dis. Sci. 2014, 59, 2367–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsumura, T.; Sato, H.; Yamamoto, K.; Ueyama, T. Ursodeoxycholic acid prevents gastrointestinal disorders caused by anticancer drugs. Jpn. J. Surg. 1981, 11, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Halilbasic, E.; Steinacher, D.; Trauner, M. Nor-Ursodeoxycholic Acid as a Novel Therapeutic Approach for Cholestatic and Metabolic Liver Diseases. Dig. Dis. 2017, 35, 288–292. [Google Scholar] [CrossRef]
- Sombetzki, M.; Fuchs, C.D.; Fickert, P.; Österreicher, C.H.; Mueller, M.; Claudel, T.; Trauner, M.; Loebermann, M.; Engelmann, R.; Langner, C.; et al. 24-nor-ursodeoxycholic acid ameliorates inflammatory response and liver fibrosis in a murine model of hepatic schistosomiasis. J. Hepatol. 2015, 62, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Fickert, P.; Wagner, M.; Marschall, H.U.; Fuchsbichler, A.; Zollner, G.; Tsybrovskyy, O.; Zatloukal, K.; Liu, J.; Waalkes, M.P.; Cover, C.; et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 2006, 130, 465–481. [Google Scholar] [CrossRef]
- Berzigotti, A.; Bellot, P.; De Gottardi, A.; Garcia-Pagan, J.C.; Gagnon, C.; Spénard, J.; Bosch, J. NCX-1000, a nitric oxide-releasing derivative of UDCA, does not decrease portal pressure in patients with cirrhosis: Results of a randomized, double-blind, dose-escalating study. Am. J. Gastroenterol. 2010, 105, 1094–1101. [Google Scholar] [CrossRef]
- Fiorucci, S.; Antonelli, E.; Brancaleone, V.; Sanpaolo, L.; Orlandi, S.; Distrutti, E.; Morelli, A.; Acuto, G.; Clerici, C.; Baldoni, M.; et al. NCX-1000, a nitric oxide-releasing derivative of ursodeoxycholic acid, ameliorates portal hypertension and lowers norepinephrine-induced intrahepatic resistance in the isolated and perfused rat liver. J. Hepatol. 2003, 39, 932–939. [Google Scholar] [CrossRef]
- Im, E.; Choi, S.H.; Suh, H.; Choi, Y.H.; Yoo, Y.H.; Kim, N.D. Synthetic bile acid derivatives induce apoptosis through a c-Jun N-terminal kinase and NF-kappaB-dependent process in human cervical carcinoma cells. Cancer Lett. 2005, 229, 49–57. [Google Scholar] [CrossRef]
- Choi, Y.H.; Im, E.O.; Suh, H.; Jin, Y.; Yoo, Y.H.; Kim, N.D. Apoptosis and modulation of cell cycle control by synthetic derivatives of ursodeoxycholic acid and chenodeoxycholic acid in human prostate cancer cells. Cancer Lett. 2003, 199, 157–167. [Google Scholar] [CrossRef]
- Im, E.O.; Choi, Y.H.; Paik, K.J.; Suh, H.; Jin, Y.; Kim, K.W.; Yoo, Y.H.; Kima, N.D. Novel bile acid derivatives induce apoptosis via a p53-independent pathway in human breast carcinoma cells. Cancer Lett. 2001, 163, 83–93. [Google Scholar] [CrossRef]
- Brossard, D.; Lechevrel, M.; El Kihel, L.; Quesnelle, C.; Khalid, M.; Moslemi, S.; Reimund, J.M. Synthesis and biological evaluation of bile carboxamide derivatives with pro-apoptotic effect on human colon adenocarcinoma cell lines. Eur. J. Med. Chem. 2014, 30, 279–290. [Google Scholar] [CrossRef]
- Brossard, D.; El Kihel, L.; Clément, M.; Sebbahi, W.; Khalid, M.; Roussakis, C.; Rault, S. Synthesis of bile acid derivatives and in vitro cytotoxic activity with pro-apoptotic process on multiple myeloma (KMS-11), glioblastoma multiforme (GBM), and colonic carcinoma(HCT-116) human cell lines. Eur. J. Med. Chem. 2010, 45, 2912–2918. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Luo, Q.; Lin, T.; Zeng, Z.; Wang, G.; Zeng, D.; Ding, R.; Sun, C.; Zhang, X.; Chen, H. U12, a UDCA derivative, acts as an anti-hepatoma drug lead and inhibits the mTOR/S6K1 and cyclin/CDK complex pathways. PLoS ONE 2014, 9, e113479. [Google Scholar] [CrossRef] [PubMed]
- Marcelo, Z.H.; Suellen, M.T.C.; Diogo, R.M.M.; Walter, F.D.A.J.; Ana, C.L.L. Halogen Atoms in the Modern Medicinal Chemistry: Hints for the Drug Design. Curr. Drug Targets 2010, 11, 303–314. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Mou, Z.; Wang, Y.; Li, Y. Brazilein induces apoptosis and G1/G0 phase cell cycle arrest by up-regulation of miR-133a in human vestibular schwannoma cells. Exp. Mol. Pathol. 2019, 107, 95–101. [Google Scholar] [CrossRef]
- Adhami, V.M.; Aziz, M.H.; Reagan-Shaw, S.R.; Nihal, M.; Mukhtar, H.; Ahmad, N. Sanguinarine causes cell cycle blockade and apoptosis of human prostate carcinoma cells via modulation of cyclin kinase inhibitor-cyclin-cyclin-dependent kinase machinery. Mol. Cancer Ther. 2004, 3, 933–940. [Google Scholar]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Zhu, P.; Zhao, Y.; Zhang, J.; Li, H.; Zhao, H.; Qin, J. Effect of PI3K/AKT/mTOR Signaling Pathway on Regulating and Controlling the Anti-Invasion and Metastasis of Hepatoma Cells by Bufalin. Recent Pat. Anticancer Drug Discov. 2021, 16, 54–65. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, R.; Du, C.; Cao, T.; Wang, G.; Jiang, X.; Gao, J.; Lin, T.; Sun, C.; Ding, R.; Tian, W.; et al. Synthesis and Anti-Hepatoma Activities of U12 Derivatives Arresting G0/G1 Phase and Inducing Apoptosis by PI3K/AKT/mTOR Pathway. Pharmaceuticals 2022, 15, 107. https://doi.org/10.3390/ph15010107
Yang R, Du C, Cao T, Wang G, Jiang X, Gao J, Lin T, Sun C, Ding R, Tian W, et al. Synthesis and Anti-Hepatoma Activities of U12 Derivatives Arresting G0/G1 Phase and Inducing Apoptosis by PI3K/AKT/mTOR Pathway. Pharmaceuticals. 2022; 15(1):107. https://doi.org/10.3390/ph15010107
Chicago/Turabian StyleYang, Renjing, Chunchun Du, Ting Cao, Guanghui Wang, Xin Jiang, Jun Gao, Ting Lin, Cuiling Sun, Rong Ding, Wenjing Tian, and et al. 2022. "Synthesis and Anti-Hepatoma Activities of U12 Derivatives Arresting G0/G1 Phase and Inducing Apoptosis by PI3K/AKT/mTOR Pathway" Pharmaceuticals 15, no. 1: 107. https://doi.org/10.3390/ph15010107