
Mini-Tablets: A Valid Strategy to Combine Efficacy and Safety in Pediatrics

 ,
,  ,
,  ,
,
Abstract
:
1. Introduction
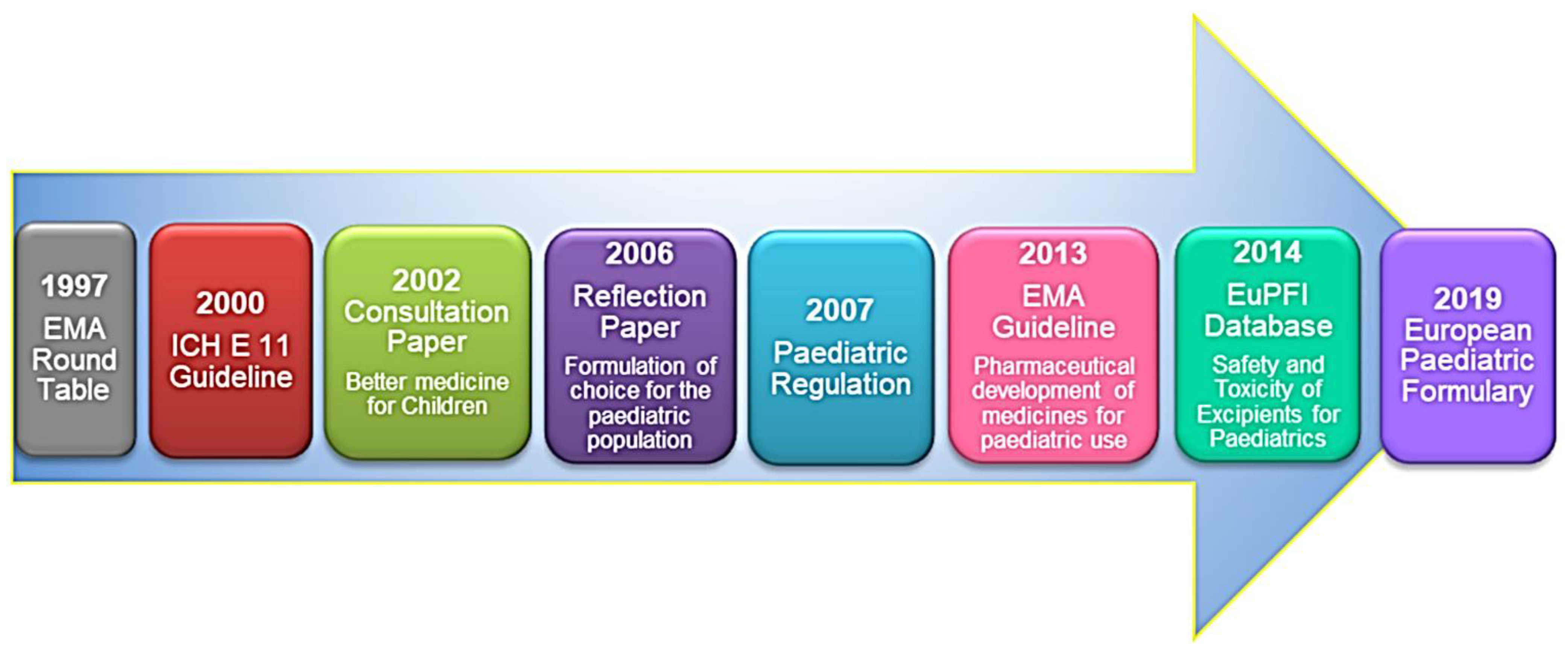
2. European Regulatory Aspects
Enalapril: From an Off-Label to a Licensed Use via Oro-Dispersible Mini-Tablet Preparation
3. Appropriate Excipients for Pediatrics
- CHMP scientific opinions
- Already authorized in pediatric medicines with known quantitative composition
- Included in the European Food Legislation or Included in EFSA opinions
- Other sources such as the expert committee on food additives (JECFA), indexed
- Literature, or in-house scientific evidence
Focus on Flavors for Paediatrics: How to Select the Right One
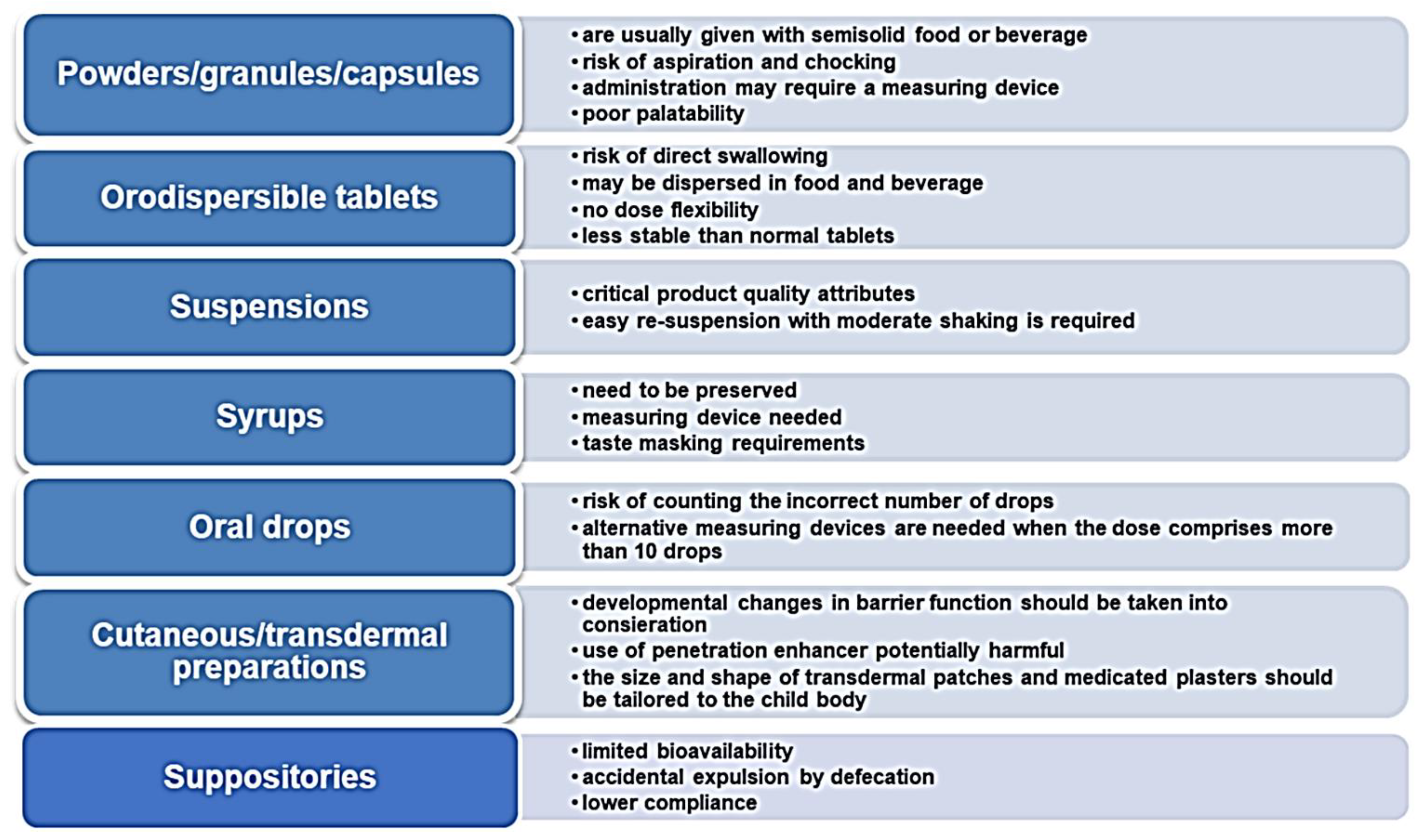
4. Appropriate Medications for Pediatrics: The Paradigm Shift towards Oral Solid Dosage Forms
4.1. Mini-Tablets
4.1.1. Mini-Tablet Manufacturing Techniques: Compression Methods
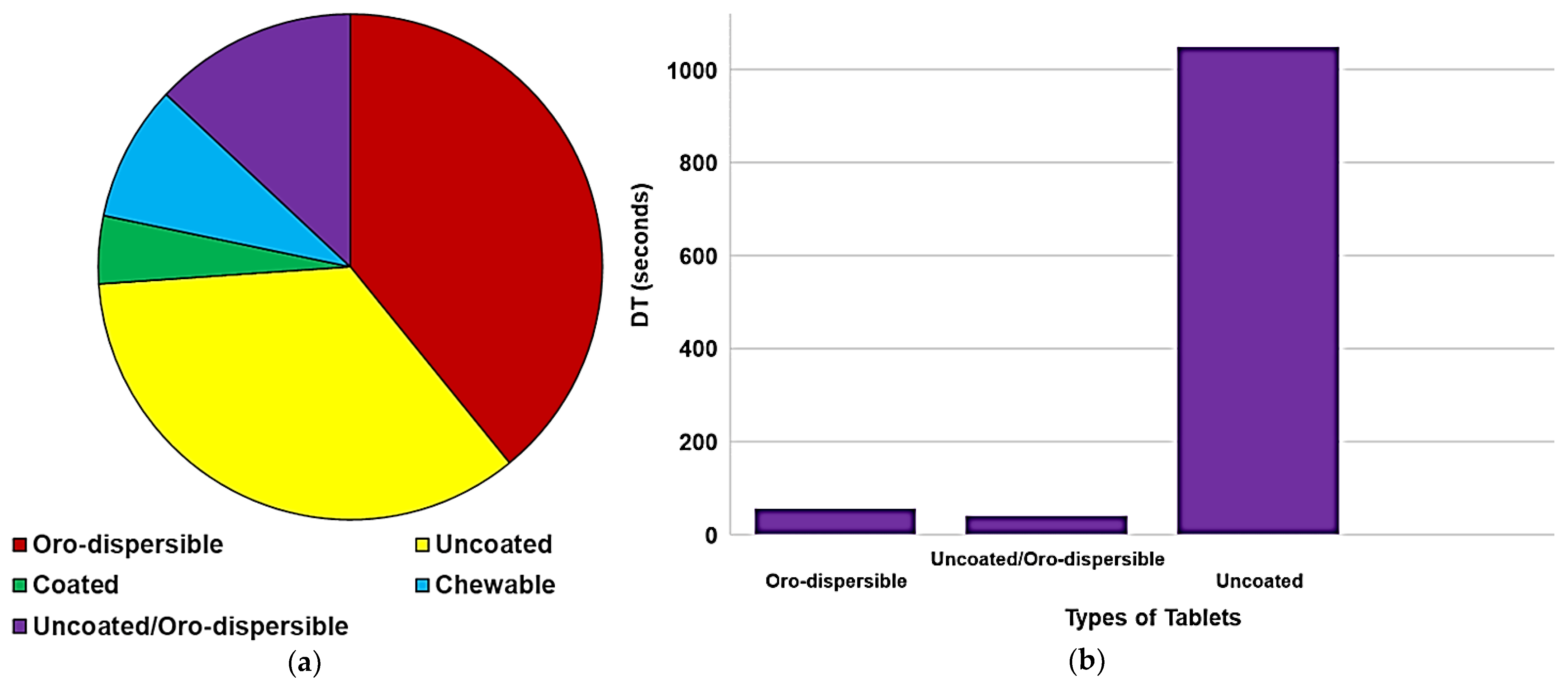
Case Studies on Oro-Dispersible MTs Obtained by Compression Methods
4.1.2. Hot Melt Extrusion Technique
4.1.3. Electrospinning Technique
4.1.4. 3D Printing Technique
Case Studies on MTs Obtained by 3DP
4.1.5. 3D Printing for Production of Friendly-Shape Medicines
4.2. Low-Dose Micro-Tablets
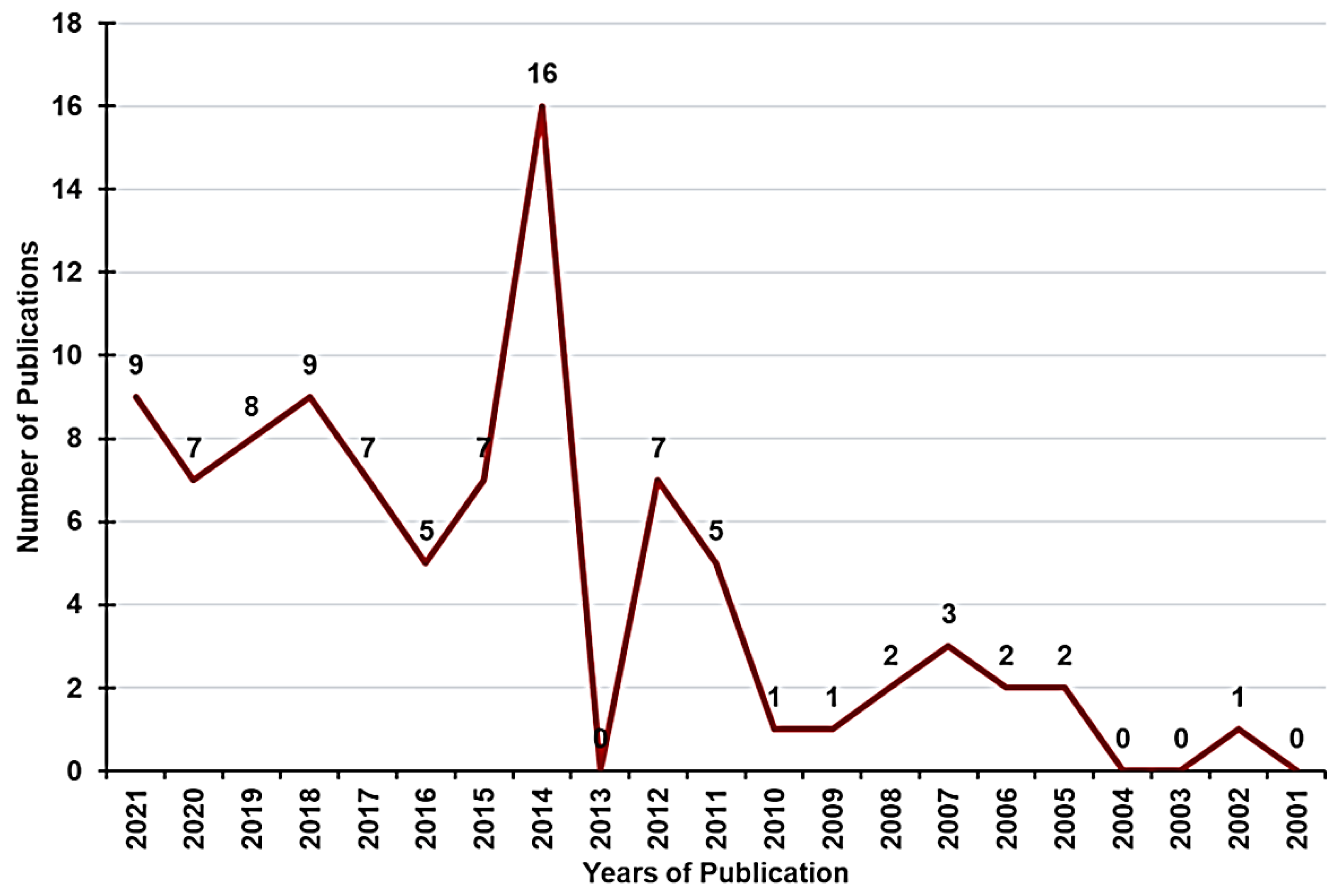
5. Clinical Trials
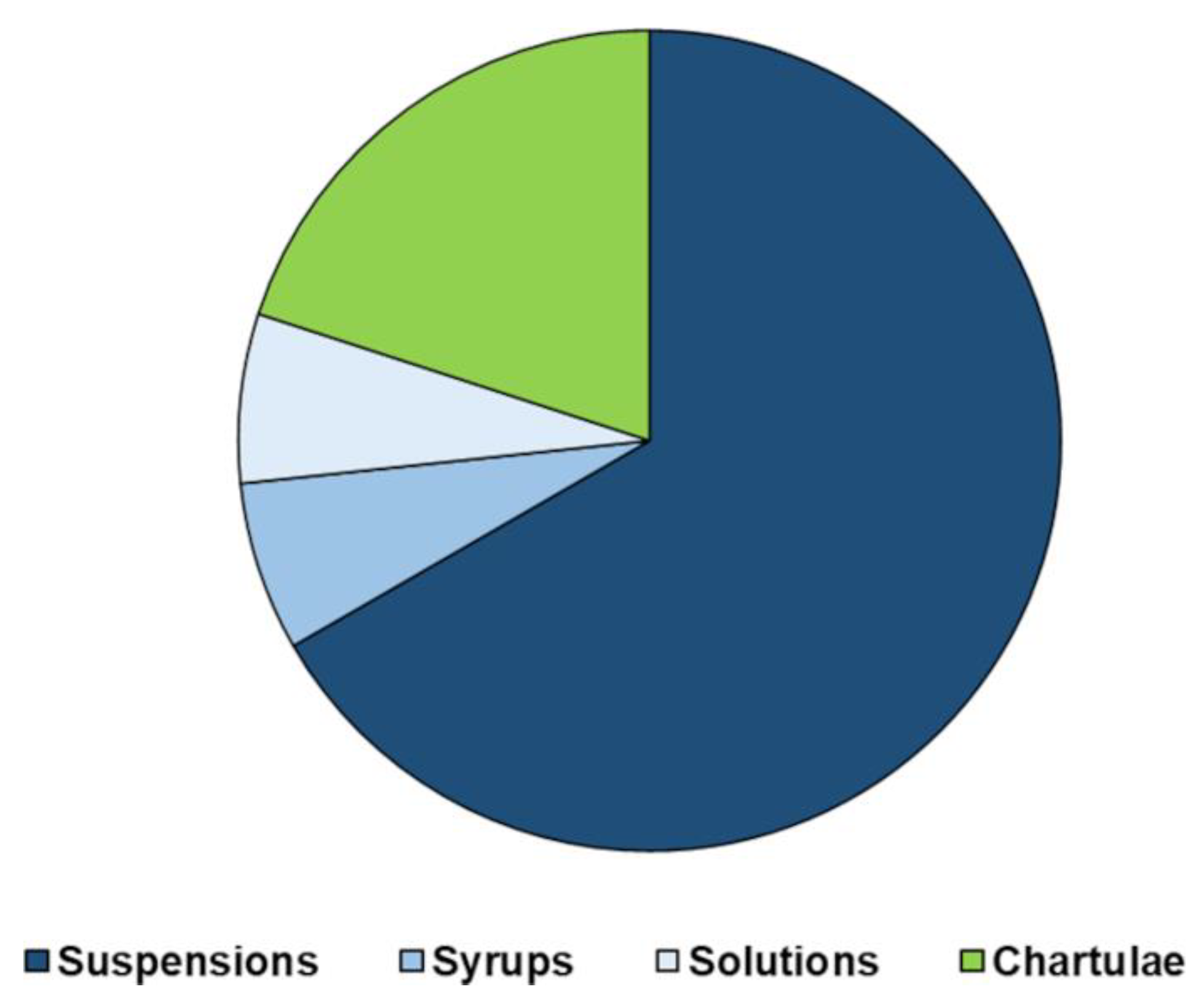
6. Manipulation of Pediatric Medicinal Products: The G. Gaslini Children’s Hospital Experience
- Dose adjustment according to patient’s age, weight, creatinine clearance and symptoms;
- change in the dosage form due to inability in swallowing capsules or tablets in patients such as neonates, dysphagic patients and patients fed through PEG (percutaneous endoscopic gastrostomy);
- two drug association non commercially available.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| API | Available Dosage Form | Compounding Process | Dispensed Preparation |
|---|---|---|---|
| Omeprazole | 20 mg capsules | Opened and dispersed in a liquid vehicle A proportion of the liquid given | Suspension 2 mg/mL |
| Mycophenolate mofetil | 250 mg capsules | Syrup 100 mg/mL | |
| Tracolimus | 5 mg Capsules (Adoport) | Suspension 0.5 mg/mL | |
| Gabapentin | 300 mg capsules | Suspension 100 mg/mL | |
| Hydrochlorthiazide | 25 mg Capsules (Esidrex) | Suspension 5 mg/mL | |
| Hydrocortisone | 10 mg Tablets (Roussel) | Triturated and dispersed in a liquid vehicle A proportion of the liquid given | Suspension 2 mg/mL |
| Flecainide | 100 mg tablets | Suspension 2 or 20 mg/mL | |
| Amlodipine | 10 mg tablets (Norvasc) | Suspension 1 mg/mL | |
| Levodopa/carbidopa | 100/25 mg tablets (Sinemet) | Suspension 5/1.25 mg/mL | |
| Captopril | Powder Pharm. Eur. | Magistral preparation | Solution 1 mg/mL |
| Ursodeoxycholic acid | Powder Pharm. Eur. | Suspension 20 mg/mL | |
| Riboflavin | Vitamin B2 Powder Pharm. Eur. | Suspension 10 mg/mL | |
| Phenytoin sodium | 100 mg dintoina tablets | Triturated and a portion of the powder given | Chartulae |
| Bosentan | 32 or 125 mg tablets | Chartulae | |
| Indomethacin | 50 or 25 mg capsules (Indoxen) | Opened and a portion of the powder given | Chartulae |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rocchi, F.; Tomasi, P. The development of medicines for children: Part of a series on Pediatric Pharmacology, guest edited by Gianvincenzo Zuccotti, Emilio Clementi, and Massimo Molteni. Pharmacol. Res. 2011, 64, 169–175. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use (CHMP), European Medicines Agency. Reflection Paper: Formulations of Choice for the Paediatric Population (EMEA/CHMP/PEG/194810/2005). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003782.pdf/ (accessed on 20 December 2021).
- Carome, M.M.D. For Some Drugs, Crushing Tablets or Opening Capsules Can Yield Fatal Consequences. Health Letter. 2019. Available online: https://www.citizen.org/article/for-some-drugs-crushing-tablets-or-opening-capsules-can-yield-fatal-cosequences/ (accessed on 20 December 2021).
- Report on the Experts Round Table on the Difficulties Related to the Use of New Medicinal Products in Children’ (EMEA/27164/98 Revision 1). Available online: https://www.ema.europa.eu/en/documents/report/report-experts-round-table-difficulties-related-use-new-medicinal-products-children-held-18-december_en.pdf (accessed on 20 December 2021).
- European Medicines Agency. ICH Topic E11 Clinical Investigation of Medicinal Products in the Paediatric Population, CPMP/ICH/2711/99. 2001. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-1.pdf (accessed on 20 December 2021).
- European Commission. Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the approximation of the laws, regulations, and administrative provisions of Member States relating to the implementation of good clinical practice in the conduct of clinical trials for human use. Off. J. Eur. Communities 2001, L281, 31–50. [Google Scholar]
- European Commission. Better Medicines for Children—Proposed Regulatory Actions on Paediatric Medicinal Products. Consultation Document. Available online: http://ec.europa.eu/health/files/pharmacos/docs/doc2002/feb/cd_pediatrics_en.pdf/ (accessed on 20 December 2021).
- European Union. Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for paediatric use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004. Official Journal of the European Union L378/1. 2006. Available online: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=OJ%3AL%3A2006%3A378%3ATOC (accessed on 20 December 2021).
- European Medicines Agency Committee on Medicinal Products for Human Use (CHMP) & Paediatric Commitee (PDCO). Guideline on the Pharmaceutical Development of Medicines for Paediatric Use. 2013. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/07/WC500147002.pdf (accessed on 20 December 2021).
- Council of Europe. European Paediatric Formulary—EDQM. Available online: https://paedform.edqm.eu/home (accessed on 20 December 2021).
- European Medicines Agency. ICH Topic E11: Note for Guidance on Clinical Investigation of Medicinal Products in the Paediatric Population (CPMP/ICH/2711/99). Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002926.pdf/ (accessed on 20 December 2021).
- ICH. Final Concept Paper Pediatric Extrapolation. 3 October 2017. Available online: https://database.ich.org/sites/default/files/E11A_EWG_Concept_Paper.pdf (accessed on 20 December 2021).
- European Medicines Agency (EMA). 10-Year Report to the European Commission General: Report on the Experience Acquired as a Result of the Application of the Paediatric Regulation. Available online: http://EMA/231225/2015.ec.europa.eu/health/sites/health/files/files/paediatrics/2016_pc_report_2017/ema_10_year_report_for_consultation.pdf (accessed on 20 December 2021).
- Bajcetic, M.; de Wildt, S.N.; Dalinghaus, M.; Breitkreutz, J.; Klingmann, I.; Lagler, F.B.; Keatley-Clarke, A.; Breur, J.M.P.J.; Male, C.; Jovanovic, I.; et al. Orodispersible Minitablets of Enalapril for Use in Children with Heart Failure (LENA): Rationale and Protocol for a Multicentre Pharmacokinetic Bridging Study and Follow-up Safety Study. Contemp. Clin. Trials Commun. 2019, 15, 100393. [Google Scholar] [CrossRef] [PubMed]
- Thabet, Y.; Walsh, J.; Breitkreutz, J. Flexible and Precise Dosing of Enalapril Maleate for All Paediatric Age Groups Utilizing Orodispersible Minitablets. Int. J. Pharm. 2018, 541, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Lena Final Report Grant Agreement NO: 602295 ©LENA Consortium Public Page 4/26. Available online: https://cordis.europa.eu/docs/results/602/602295/final1-lena-602295-final-report-publishable-summary-v1-0.pdf (accessed on 7 January 2022).
- Turner, M.A.; Duncan, J.C.; Shah, U.; Metsvaht, T.; Varendi, H.; Nellis, G.; Lutsar, I.; Yakkundi, S.; McElnay, J.C.; Pandya, H.; et al. Risk assessment of neonatal excipient exposure: Lessons from food safety and other areas. Adv. Drug Deliv. Rev. 2014, 73, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Nellis, G.; Metsvaht, T.; Varendi, H.; Toompere, K.; Lass, J.; Mesek, I.; Nunn, A.J.; Turner, M.A.; Lutsar, I. Potentially harmful excipients in neonatal medicines: A pan-European observational study. Arch. Dis. Child. 2015, 100, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, L. Neonates exposed to excipients: Concern about safety. J. Pediatric Neonatal Individ. Med. 2018, 7, e070112. [Google Scholar]
- Zuccotti, G.V.; Fabiano, V. Safety issues with ethanol as an excipient in drugs intended for pediatric use. Expert Opin. Drug Saf. 2011, 10, 10499–10502. [Google Scholar] [CrossRef]
- De Cock, R.F.; Allegaert, K.; Vanhaesebrouck, S.; de Hoon, J.; Verbesselt, R.D.; PharmD, M.; Knibbe, C.A.J. Low but inducible contribution of renal elimination to clearance of propyleneglycol in preterm and term neonates. Ther. Drug Monit. 2014, 36, 278–287. [Google Scholar] [CrossRef]
- Kraft, W.K.; Adeniyi-Jones, S.C.; Chervoneva, I.; Greenspan, J.S.; Abatemarco, D.; Kaltenbach, K.; Ehrlich, M.E. Buprenorphine for the treatment of the neonatal abstinence syndrome. N. Engl. J. Med. 2017, 376, 2341–2348. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use (CHMP), EMA. Questions and Answers on Ethanol in the Context of the Revision of the Guideline on ‘Excipients in the Label and Package Leaflet of Medical Products for Human Use’. EMA/CHMP/507988/2013. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/questions-answers-ethanol-context-revision-guideline-excipients-label-package-leaflet-medicinal_en.pdf (accessed on 20 December 2021).
- Bove, K.E.; Kosmetatos, N.; Wedig, K.E.; Frank, D.J.; Whitlatch, S.; Saldivar, V.; Haas, J.; Bodenstein, C.; Balistreri, W.F. Vasculopathic hepatotoxicity associated with E-Ferol syndrome in low-birth-weight infants. JAMA 1985, 254, 2422–2430. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.G.; Fletcher, A.B.; Johnson, E.L.; Boeckx, R.L.; Getson, P.R.; Miller, M.K. The potential toxicity to neonates of multivitamin preparations used in parenteral nutrition. JPEN J. Parenter. Enter. Nutr. 1987, 11, 169–171. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use; Paediatric Committee. Benzyl Alcohol. Adopted Questions and Answers. European Medicines Agency, 2017. Available online: https://www.ema.europa.eu/documents/scientific-guideline/questions-answers-benzyl-alcohol-used-excipient-medicinal-products-human-use_en.pdf (accessed on 20 December 2021).
- Valeur, K.S.; Hertel, S.A.; Lundstrøm, K.E.; Holst, H. Safe excipient exposure in neonates and small children—protocol for the SEEN project. Dan. Med. J. 2017, 64, A5324. [Google Scholar] [PubMed]
- Valeur, K.S.; Holst, H.; Allegaert, K. Excipients in Neonatal Medicinal Products: Never Prescribed, Commonly Administered. Pharm. Med. 2018, 32, 251–258. [Google Scholar] [CrossRef] [Green Version]
- STEP Data Base. Available online: www.eupfi.org/step-database-info/ (accessed on 4 December 2021).
- EMA. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-excipients-dossier-application-marketing-authorisation-medicinal-product-revision-2_en.pdf (accessed on 20 December 2021).
- Kogermann, K.; Lass, J.; Nellis, G.; Metsvaht, T.; Lutsar, I. Age-Appropriate Formulations Including Pharmaceutical Excipients in Neonatal Medicines. CPD 2018, 23, 5779–5789. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Regulation (EC) No 1334/2008 of the European Parliament and of the Council of 16 December 2008 on Flavourings and Certain Food Ingredients with Flavouring Properties for Use in and on Foods and Amending Council Regulation (EEC) No 1601/91, Regulations (EC) No 2232/96 and (EC) No 110/2008 and Directive 2000/13/EC. 2008. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2008:354:0034:0050:en:PDF (accessed on 20 December 2021).
- Commission Implementing Regulation (EU) No. 872/2012 Adopting the List of Flavouring Substances Provided for by Regulation (EC) No. 2232/96 of the European Parliament and of the Council, Introducing It in Annex I to Regulation (EC) No. 1334/2008 of the European Parliament and of the Council and Repealing Commission Regulation (EC) No. 1565/2000 and Commission Decision 1999/217/EC. Available online: https://eur-lex.europa.eu/legal-content/IT/TXT/PDF/?uri=CELEX:32012R0872&from=en (accessed on 20 December 2021).
- Commission Regulation (EC) No 1881/2006 of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs (Text with EEA Relevance) Text with EEA Relevance. Available online: http://data.europa.eu/eli/reg/2006/1881/2021-09-19 (accessed on 20 December 2021).
- Regulation (EC) No 396/2005 of the European Parliament and of the Council of 23 February 2005 on Maximum Residue Levels of Pesticides in or on Food and Feed of Plant and Animal Origin and Amending Council Directive 91/414/EEC Text with EEA Relevance. Available online: http://data.europa.eu/eli/reg/2005/396/oj (accessed on 3 December 2021).
- Regulation (EU) No 1169/2011 of the European Parliament and of the Council of 25 October 2011 on the Provision of Food Information to Consumers, Amending Regulations (EC) No 1924/2006 and (EC) No 1925/2006 of the European Parliament and of the Council, and Repealing Commission Directive 87/250/EEC, Council Directive 90/496/EEC, Commission Directive 1999/10/EC, Directive 2000/13/EC of the European Parliament and of the Council, Commission Directives 2002/67/EC and 2008/5/EC and Commission Regulation (EC) No 608/2004 Text with EEA Relevance. Available online: http://data.europa.eu/eli/reg/2011/1169/oj (accessed on 20 December 2021).
- Tanner, S.; Wells, M.; Scarbecz, M.; McCann, B.W. Parents’ Understanding of and Accuracy in Using Measuring Devices to Administer Liquid Oral Pain Medication. J. Am. Dent. Assoc. 2014, 145, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, P.; Christopherson, J.; Ambrose, P.J.; Corelli, R.L. Accuracy of Oral Liquid Measuring Devices: Comparison of Dosing Cup and Oral Dosing Syringe. Ann. Pharm. 2008, 42, 46–52. [Google Scholar] [CrossRef]
- Lopez, F.L.; Ernest, T.B.; Tuleu, C.; Gul, M.O. Formulation Approaches to Pediatric Oral Drug Delivery: Benefits and Limitations of Current Platforms. Expert Opin. Drug Deliv. 2015, 12, 1727–1740. [Google Scholar] [CrossRef] [PubMed]
- Mfoafo, K.A.; Omidian, M.; Bertol, C.D.; Omidi, Y.; Omidian, H. Neonatal and Pediatric Oral Drug Delivery: Hopes and Hurdles. Int. J. Pharm. 2021, 597, 120296. [Google Scholar] [CrossRef]
- Alessandrini, E.; Brako, F.; Scarpa, M.; Lupo, M.; Bonifazi, D.; Pignataro, V.; Cavallo, M.; Cullufe, O.; Enache, C.; Nafria, B.; et al. Children’s Preferences for Oral Dosage Forms and Their Involvement in Formulation Research via EPTRI (European Paediatric Translational Research Infrastructure). Pharmaceutics 2021, 13, 730. [Google Scholar] [CrossRef]
- Krampe, R.; Visser, J.C.; Frijlink, H.W.; Breitkreutz, J.; Woerdenbag, H.J.; Preis, M. Oromucosal Film Preparations: Points to Consider for Patient Centricity and Manufacturing Processes. Expert Opin. Drug Deliv. 2016, 13, 493–506. [Google Scholar] [CrossRef]
- Klingmann, V.; Spomer, N.; Lerch, C.; Stoltenberg, I.; Frömke, C.; Bosse, H.M.; Breitkreutz, J.; Meissner, T. Favorable Acceptance of Mini-Tablets Compared with Syrup: A Randomized Controlled Trial in Infants and Preschool Children. J. Pediatr. 2013, 163, 1728–1732.e1. [Google Scholar] [CrossRef]
- Klingmann, V.; Seitz, A.; Meissner, T.; Breitkreutz, J.; Moeltner, A.; Bosse, H.M. Acceptability of Uncoated Mini-Tablets in Neonates—A Randomized Controlled Trial. J. Pediatr. 2015, 167, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Klingmann, V.; Linderskamp, H.; Meissner, T.; Mayatepek, E.; Moeltner, A.; Breitkreutz, J.; Bosse, H.M. Acceptability of Multiple Uncoated Minitablets in Infants and Toddlers: A Randomized Controlled Trial. J. Pediatr. 2018, 201, 202–207. [Google Scholar] [CrossRef] [PubMed]
- van Riet-Nales, D.A.; de Neef, B.J.; Schobben, A.F.A.M.; Ferreira, J.A.; Egberts, T.C.G.; Rademaker, C.M.A. Acceptability of Different Oral Formulations in Infants and Preschool Children. Arch. Dis. Child. 2013, 98, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilhan, E.; Ugurulu, T.; Kerimoglu, O. Mini Tablets: A Short Review-Revision. Open J. Chem. 2017, 11, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Keerthi, M.L.; Kiran, R.S.; Rao, V.U.M.; Sannapu, A.; Dutt, A.G.; Krishna, K.S. Pharmaceutical Mini-Tablets, its Advantages, Formulation Possibilities and General Evaluation Aspects: A Review. Int. J. Pharm. Sci. Rev. Res. 2014, 28, 214–221. [Google Scholar]
- Flemming, J.; Mielck, J.B. Requirements for the production of microtablets: Suitability of direct-compression excipients estimated from powder characteristics and flow rates. Drug Dev. Ind. Pharm. 1995, 21, 2239–2251. [Google Scholar] [CrossRef]
- Zhao, J.; Yin, D.; Rowe, J.; Badawy, S.; Nikfar, F.; Pandey, P. Understanding the Factors That Control the Quality of Mini-Tablet Compression: Flow, Particle Size, and Tooling Dimension. J. Pharm. Sci. 2018, 107, 1204–1208. [Google Scholar] [CrossRef]
- Shah, B.A.; Patel, A.S.; Patel, B.; Patel, D.J.; Qu, A. Mini-Tablet Drug Delivery System for Pediatric Dosage Form (PDF): A Review of Manufacturing Perspectives. Int. J. Drug Dev. Res. 2018, 10, 47–52. [Google Scholar]
- Stoltenberg, I.; Breitkreutz, J. Orally Disintegrating Mini-Tablets (ODMTs)—A Novel Solid Oral Dosage Form for Paediatric Use. Eur. J. Pharm. Biopharm. 2011, 78, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Lura, A.; Luhn, O.; Suarez Gonzales, J.; Breitkreutz, J. New Orodispersible Mini-Tablets for Paediatric Use—A Comparison of Isomalt with a Mannitol Based Co-Processed Excipient. Int. J. Pharm. 2019, 572, 118804. [Google Scholar] [CrossRef]
- Ortega, C.A.; Favier, L.S.; Cianchino, V.A.; Cifuente, D.A. New Orodispersible Mini Tablets of Enalapril Maleate by Direct Compression for Pediatric Patients. CDD 2020, 17, 505–510. [Google Scholar] [CrossRef]
- El-Say, K.M.; Ahmed, T.A.; Abdelbary, M.F.; Ali, B.E.; Aljaeid, B.M.; Zidan, A.S. Risperidone Oral Disintegrating Mini-Tablets: A Robust-Product for Pediatrics. Acta Pharm. 2015, 65, 365–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madathilethu, J.; Roberts, M.; Peak, M.; Blair, J.; Prescott, R.; Ford, J.L. Content Uniformity of Quartered Hydrocortisone Tablets in Comparison with Mini-Tablets for Paediatric Dosing. Bmjpo 2018, 2, e000198. [Google Scholar] [CrossRef]
- EMA. EMA/CHMP/261937/2018 Committee for Medicinal Products for Human Use (CHMP). 22 March 2018. Available online: https://www.ema.europa.eu/en/documents/assessment-report/alkindi-epar-public-assessment-report_en.pdf (accessed on 20 December 2021).
- Yeleken, G.; Kotłowska, H.; Sznitowska, M.; Golenia, E.; Ustenova, G. Development of direct compressed loratadine minitablets. J. Pharm. Sci. Res. 2017, 9, 401–406. [Google Scholar]
- Freerks, L.; Sommerfeldt, J.; Löper, P.C.; Klein, S. Safe, Swallowable and Palatable Paediatric Mini-Tablet Formulations for a WHO Model List of Essential Medicines for Children Compound—A Promising Starting Point for Future PUMA Applications. Eur. J. Pharm. Biopharm. 2020, 156, 11–19. [Google Scholar] [CrossRef]
- Chen, H.; Wang, C.; Liu, S.; Sun, C.C. Development of Piroxicam Mini-Tablets Enabled by Spherical Cocrystallization. Int. J. Pharm. 2020, 590, 119953. [Google Scholar] [CrossRef] [PubMed]
- Lavan, M.; Wang, X.; McCain, R.; Jannasch, A.; Cooper, B.; Hostetler, S.; Byrn, S.; Knipp, G. Development of a Pediatric Mini-Tablet Formulation for Expedited Preclinical Studies. AAPS PharmSciTech 2021, 22, 40. [Google Scholar] [CrossRef]
- Nellan, A.; Rota, C.; Majzner, R.; Lester-McCully, C.M.; Griesinger, A.M.; Mulcahy Levy, J.M.; Foreman, N.K.; Warren, K.E.; Lee, D.W. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 2018, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshetaili, A.S.; Almutairy, B.K.; Tiwari, R.V.; Morott, J.T.; Alshehri, S.M.; Feng, X.; Alsulays, B.B.; Park, J.-B.; Zhang, F.; Repka, M.A. Preparation and Evaluation of Hot-Melt Extruded Patient-Centric Ketoprofen Mini-Tablets. CDD 2016, 13, 730–741. [Google Scholar] [CrossRef]
- Poller, B.; Strachan, C.; Broadbent, R.; Walker, G.F. A Minitablet Formulation Made from Electrospun Nanofibers. Eur. J. Pharm. Biopharm. 2017, 114, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Z.K.; Balogh, A.; Démuth, B.; Pataki, H.; Vigh, T.; Szabó, B.; Molnár, K.; Schmidt, B.T.; Horák, P.; Marosi, G.; et al. High Speed Electrospinning for Scaled-up Production of Amorphous Solid Dispersion of Itraconazole. Int. J. Pharm. 2015, 480, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, C.R.M.; Okafor-Muo, O.L.; Hassanin, H.; ElShaer, A. 3DP Printing of Oral Solid Formulations: A Systematic Review. Pharmaceutics 2021, 13, 358. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, S. Application of Hot Melt Extrusion in Pharmaceutical 3D Printing. J. Bioequiv. Bioavailab. 2018, 10, 14. [Google Scholar] [CrossRef]
- El Aita, I.; Rahman, J.; Breitkreutz, J.; Quodbach, J. 3D-Printing with Precise Layer-Wise Dose Adjustments for Paediatric Use via Pressure-Assisted Microsyringe Printing. Eur. J. Pharm. Biopharm. 2020, 157, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.; Müller, L.; Sarwinska, D.; Seidlitz, A.; Sznitowska, M.; Weitschies, W. 3D Printing of Mini Tablets for Pediatric Use. Pharmaceuticals 2021, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Palekar, S.; Nukala, P.K.; Mishra, S.M.; Kipping, T.; Patel, K. Application of 3D Printing Technology and Quality by Design Approach for Development of Age-Appropriate Pediatric Formulation of Baclofen. Int. J. Pharm. 2019, 556, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Ayyoubi, S.; Cerda, J.R.; Fernández-García, R.; Knief, P.; Lalatsa, A.; Healy, A.M.; Serrano, D.R. 3D Printed Spherical Mini-Tablets: Geometry versus Composition Effects in Controlling Dissolution from Personalised Solid Dosage Forms. Int. J. Pharm. 2021, 597, 120336. [Google Scholar] [CrossRef] [PubMed]
- van Riet-Nales, D.A.; Schobben, A.F.A.M.; Vromans, H.; Egberts, T.C.G.; Rademaker, C.M.A. Safe and Effective Pharmacotherapy in Infants and Preschool Children: Importance of Formulation Aspects. Arch. Dis. Child. 2016, 101, 662–669. [Google Scholar] [CrossRef]
- Scoutaris, N.; Ross, S.A.; Douroumis, D. 3D Printed “Starmix” Drug Loaded Dosage Forms for Paediatric Applications. Pharm. Res. 2018, 35, 34. [Google Scholar] [CrossRef]
- Rycerz, K.; Stepien, K.A.; Czapiewska, M.; Arafat, B.T.; Habashy, R.; Isreb, A.; Peak, M.; Alhnan, M.A. Embedded 3D Printing of Novel Bespoke Soft Dosage Form Concept for Pediatrics. Pharmaceutics 2019, 11, 630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soulairol, I.; Chaheen, M.; Tarlier, N.; Aubert, A.; Bataille, B.; Sharkawi, T. Evaluation of Disintegrants Functionality for Orodispersible Mini Tablets. Drug Dev. Ind. Pharm. 2017, 43, 1770–1779. [Google Scholar] [CrossRef]
- Mitra, B.; Thool, P.; Meruva, S.; Aycinena, J.A.; Li, J.; Patel, J.; Patel, K.; Agarwal, A.; Karki, S.; Bowen, W. Decoding the Small Size Challenges of Mini-Tablets for Enhanced Dose Flexibility and Micro-Dosing. Int. J. Pharm. 2020, 574, 118905. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Thool, P.; Meruva, S.; Li, J.; Patel, J.; Agrawal, A.; Karki, S.; Bowen, W.; Mitra, B. Development of Low Dose Micro-Tablets by High Shear Wet Granulation Process. Int. J. Pharm. 2020, 587, 119571. [Google Scholar] [CrossRef] [PubMed]
- Meruva, S.; Thool, P.; Gong, Y.; Agrawal, A.; Karki, S.; Bowen, W.; Mitra, B.; Kumar, S. A Novel Use of Nanocrystalline Suspensions to Develop Sub-Microgram Dose Micro-Tablets. J. Pharm. Sci. 2021, 110, 3276–3288. [Google Scholar] [CrossRef]
- Hagen, E.; Løding, F.S.; Mattsson, S.; Tho, I. Use of Interactive Mixtures to Obtain Mini-Tablets with High Dose Homogeneity for Paediatric Drug Delivery. J. Drug Deliv. Sci. Technol. 2016, 34, 51–59. [Google Scholar] [CrossRef]
- Schroder, C.M.; Malow, B.A.; Maras, A.; Melmed, R.D.; Findling, R.L.; Breddy, J.; Nir, T.; Shahmoon, S.; Zisapel, N.; Gringras, P. Pediatric Prolonged-Release Melatonin for Sleep in Children with Autism Spectrum Disorder: Impact on Child Behavior and Caregiver’s Quality of Life. J. Autism Dev. Disord 2019, 49, 3218–3230. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, P.; Narkewicz, M.R.; Yao, B.B.; Jolley, C.D.; Lobritto, S.J.; Wen, J.; Molleston, J.P.; Hsu, E.K.; Jonas, M.M.; Zha, J.; et al. Ombitasvir, Paritaprevir, Ritonavir, and Dasabuvir Mini-Tabs Plus Ribavirin for Children Aged 3–11 Years with Hepatitis C Genotype 1a. Adv. Ther. 2020, 37, 3299–3310. [Google Scholar] [CrossRef]
- World Health Organization. Model List of Essential Medicines for Children. 7th List. 2019. Available online: https://apps.who.int/iris/bitstream/handle/10665/325772/WHO-MVP-EMP-IAU-2019.07-eng.pdf?ua=1 (accessed on 20 December 2021).
- Del Moral-Sanchez, J.-M.; Gonzalez-Alvarez, I.; Gonzalez-Alvarez, M.; Navarro-Ruiz, A.; Bermejo, M. Availability of Authorizations from EMA and FDA for Age-Appropriate Medicines Contained in the WHO Essential Medicines List for Children 2019. Pharmaceutics 2020, 12, 316. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. Needs for Paediatric Medicines. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/paediatric-medicines/needs-paediatric-medicines# (accessed on 20 December 2021).
- Lajoinie, A.; Henin, E.; Nguyen, K.A.; Malik, S.; Mimouni, Y.; Sapori, J.M.; Bréant, V.; Cochat, P.; Kassai, B. Oral Drug Dosage Forms Administered to Hospitalized Children: Analysis of 117,665 Oral Administrations in a French Paediatric Hospital over a 1-Year Period. Int. J. Pharm. 2016, 500, 336–344. [Google Scholar] [CrossRef] [PubMed]







| Excipient | Function | Toxicity in Children | Limits |
|---|---|---|---|
| Acesulfame K | Sweetener | N.R. | 15 mg/kg bw for adults |
| α-Cyclodextrin | Complexing agent Solubilizing agent | N.R. | Oral toxicity [PDE 120 mg/kg/day; TH neonate—12] Ocular [safe solution <4%; TH neonate—0.1] Parenteral [PD—0.2 mg/kg/day; TH neonate—0.02] |
| Aspartame | Sweetener | Rare hypersensitivity reactions Cross-reactivity with sulphonamides | Not in homozygous autosomal recessive phenylketonuria patients In patients without dietary restrictions <5 mg/kg/day ADI ≤ 40 mg/kg bw for adults and children |
| Benzalkonium chloride | Wetting agent Penetration enhancer Antimicrobial preservative Antiseptic Disinfectant Cationic surfactant Solubilizing agent | N.R | ADI of 0.1 mg/kg bw/day ARfD of 0.1 mg/kg bw |
| Benzoic acid | Antimicrobial preservative | N.R. | 5 mg/kg bw for young children |
| Benzyl alcohol | Disinfectant Solvent Antimicrobial preservative | Gasping syndrome accumulation of metabolites in blood (metabolic acidosis) and brain (neurotoxicity) | Parenteral, Rectal TH—Zero 1 Neonates 2 Up to three years old 3 5 mg/kg/day for adults and children aged over 4 weeks |
| β-Cyclodextrin | Complexing agent Solubilizing agent | N.R. | Oral Tox [PDE 10 mg/kg/day; TH neonate—1] Nasal [safe solutio—1.5%; TH neonate—0.15] Ocular [safe solution ±1%; TH neonate—0.1] Rectal [safe amount—5 mg/kg/day or %; TH neonate—0.5] Dermal [safe amount ±0.1%; TH neonate—0.01] |
| Boric acid | Antimicrobial preservative Buffering agent | N.R. | 0.16 mg/kg bw/day for adults |
| Butylated hydroxyanisole | Antioxidant Antimicrobial preservative | N.R. | 1.25 mg/kg bw/day for children |
| Butylated hydroxytoluene | Antioxidant | N.R. | 0.25 mg/kg bw/day for adults |
| Butylparaben | Antimicrobial preservative | N.R. | Withdrawn 4 |
| Cresol | Antimicrobial preservative | N.R. | TDI of 50 µg/kg bw/day for adults |
| Azo dyes Quinoline dyes Triphenylmethane dyes Xanthene dyes | Coloring agents | Gastrointestinal intolerance Abdominal pain Vomiting Indigestion Hypersensitivity | To be avoided unless necessary |
| Edetic acid | Antimicrobial preservative Complexing agent | N.R. | 1.9 mg/kg of body weight for adults |
| Ethanol | Penetration enhancer Solvent Antimicrobial preservative | Acute intoxication in accidental overdose Chronic toxicity in routine use CNS depressant Respiratory/cardiovascular toxicities at high concentrations Long-term effects of low doses under discussion | 2.6 g/day for adults Blood ethanol levels should not exceed 1 mg/dL after a single dose containing ethanol (or a dose of 6 mg/kg/day) in children aged 2–6 years |
| Ethylparaben | Antimicrobial preservative | N.R. | 10 mg/kg bw for young children |
| Fructose | Sweetener | ↑ blood glucose concentration Laxative effects at high oral doses | Not in patients with diabetes, hypoglycaemia, hereditary fructose intolerance |
| γ-Cyclodextrin | Complexing agent Solubilizing agent | N.R. | Oral Tox [PDE 200 mg/kg/day; TH neonate—20] Parenteral [PDE—0.8 mg/kg/day; TH neonate—0.08] Dermal [safe amount ±0.1%; TH neonate—0.01] |
| Iron oxide | Colorant | N.R. | 0.5 mg/kg bw for adults |
| Lactose | Filler diluent in tablets and capsules | Severe prolonged diarrhoea Dehydration Metabolic acidosis in lactose intolerance | Intake of <3 g may provoke the described symptoms sensitivity to lactose varies in severity |
| Methacrylic acid/ethylacrylate copolymer | Coating material | Fibrosing colonopathy | N.R. |
| Methylparaben | Antimicrobial preservative | N.R. | 10 mg/kg bw for young children |
| Polyethylene glycol (PEG) | Solvent Diluent, lubricant (tablet, capsule) Ointment base Coating agent Suppository base Plasticizer | 10 mg/kg bw for adults | |
| Polysorbates | Solubilizing agent Wetting agent Dispersing agent Emulsifying agent Suspending agent Nonionic surfactant | Liver and kidney failure | 10 mg/kg bw for young children |
| Polyvynil pirrolidone (PVP) | Solubilizing agent | ADI 0–50 mg/kg/day | |
| Polypropylene glycol (PPG) | Plasticizer Stabilizing agent Antimicrobial preservative Disinfectant Solvent Humectant | Neurotoxic effects (Adolescents, schoolchildren) ↑ Death (Low-weight new-borns, preterm babies) Severe brain damage and life-long handicaps Metabolic acidosis Hyperosmolality and laxative effect (Limited metabolic pathway-alcohol dehydrogenase) | 1 mg/kg/day—neonates up to 28 days 50 mg/kg/day from 29 days up to 4 years 500 mg/kg—5 years up to 17 years and adults Not in paediatric dialysis patients |
| Propylparaben | Antimicrobial preservative | Agonistic activity at hormone receptors | 10 mg/kg bw for young children Up to 5 mg/kg/day for children >2 years with mature metabolic capacity Recently deleted from the list of permitted food additives in the EU |
| Saccharin | Sweetening agent | N.R. | 5 mg/kg bw for young children 5 |
| Sodium benzoate | Antimicrobial preservative Tablet and capsule lubricant | N.R. | 5 mg/kg bw |
| Sorbic acid | Antimicrobial preservative | N.R. | 25 mg/kg bw for young children |
| Sucralose | Sweetening agent | N.R. | 5 mg/kg bw/day |
| Sucrose | Sweetener | Decrease in dental plaque pH Dissolving tooth enamel Promoting dental caries | To be avoided for patients with hereditary fructose intolerance, diabetes For long-term therapy it should be replaced by sugar-free formulations |
| Rules and Thresholds in Flavors | Paediatric Age | |
|---|---|---|
| Conformity to the European laws | Conformity with Regulation EU 1334/2008 on flavorings and certain food ingredients with flavoring properties for use in and on foods and current implementation [32] | Required for all paediatric subpopulations |
| Use of flavoring substances permitted and present in the Union List (Annex I of Regulation 1334/2008) in 2012 with Regulation EU 872/2012 [32] | ||
| Conformity with Commission Implementing regulation EU 872/2012 adopting the list of flavoring substances provided for by Regulation EC 2232/96, introducing it in Annex I to Regulation EC 1334/2008 [33] | ||
| Contaminants | Conformity with Commission Regulation EU 1881/2006 setting maximum levels for certain contaminants in foodstuffs [34] | |
| Colorants in the flavor | Absent | |
| Pesticides | Conformity with Regulation EU 396/2005 on maximum residue levels of pesticides in or on food and feed of plant [35] | |
| Food allergens | Conformity with Annex II of Regulation EU 1169/2011 [36] | |
| Ethanol in liquid flavors | Absent | Infants/young children < 3 years |
| To be evaluated | Children/adolescents 4–18 years | |
| Carcinogenic mutagenic substances | Absent | Infants/young children < 3 years |
| To be evaluated | Children/adolescents 4–18 years | |
| Ethanol in liquid flavors | Absent | Infants/young children < 3 years |
| To be evaluated | Children/adolescents 4–18 years | |
| Diacetyl | Absent | Infants/young children < 3 years |
| To be evaluated | Children/adolescents 4–18 years | |
| Benzyl alcohol | Absent | Infants/young children < 3 years |
| <2.5 mg/kg/day | Children/adolescents 4–18 years | |
| Propylene glycol solvent in flavors | <5 mg/kg/day | <1 month |
| <25 mg/kg/day | 1–36 months | |
| <25 mg/kg/day | Children/adolescents 4–18 years |
| Mini-Tablets Features | |
|---|---|
| Pros | Cons |
| ↓ dose dumping | Price may be higher depending on production technology |
| ↓ inter-intra individual variability | Requirement of excellent powder flow due to the small dies |
| Good coating substrate | Coating may rupture by accidentally chewing |
| ↓ Local irritation | Limited drug loading capacity per tablet |
| ↓ Capping tendency | Multiple dosing might be necessary due to the limited drug load per single unit |
| Manufacturing w/o solvent or heating | Packaging or dosing technology platforms needs to be developed |
| Fine tuning of release rate | |
| Dose flexibility | |
| Allow coexistence of different/incompatible drugs | |
| Brand Name and Manufacturer | API | Dosage Form | Ingredients | Target Population | Therapeutic Indication |
|---|---|---|---|---|---|
| Creon® (Solvay Pharmaceuticals) | Pancreatic enzymes | MT (Delayed release) | Cetyl alcohol Dimethicone Hypromellose phthalate PEG Triethyl citrate | ≥6 ms | Chronic pancreatitis cystic fibrosis |
| Levetiracetam Desitin® (Desitin Arzneimittel GmbH) | Levetiracetam | 2 mm MTs (Stick pack) | Povidone K30 Microcrystalline cellulose (MCC) Silicon dioxide (SiO2) Magnesium (Mg) stearate Poly(vinyl alcohol) (PVA) Titanium dioxide (TiO2) Macrogol 3350 Talc | ≥6 years | Epilepsy |
| Kalideco® (Vertex) | Ivacaftor | 2 mm MTs (Stick pack) | SiO2 Croscarmellose sodium (Na) Hypromellose acetate succinate Lactose monohydrate Mg stearate Mannitol Sucralose Na lauryl sulfate | ≥6 years | Cystic fibrosis |
| Lamisil® (Novartis) | Terbinafine HCl | 2 mm MTs (Stick pack, capsule) | Basic butylated methacrylate copolymer SiO2 Dibutyl sebacate Hypromellose Mg stearate MCC PEG Na lauryl sulfate Na starch glycolate | ≥4 years | Antifungal treatment (tinea capitis) |
| Orfiril® Long (Desitin Arzneimittel GmbH) | Sodium Valproate | MTs (sachet/capsule for extended release) | Calcium (Ca) stearate Ethyl cellulose Colloidal SiO2 (methylated) Ammonium methacrylate copolymer (type B) Na dodecylsulphate Polysorbate 80 Oleic acid Dibutyldecandioate | ≥10 years | Epilepsy |
| Pancrease MT® (McNeil) | Pancreatic enzymes | 2 mm enteric-coated MTs (capsules for delayed release) | Methacrylic acid ethyl acrylate copolimer Cellulose Crospovidone Mg stearate SiO2 Triethyl citrate Talc Polydimethylsiloxane Wax Gelatin Iron oxide (Fe2O3) Polysorbate 80 Na lauryl sulfate TiO2 | From infancy | Chronic pancreatitis cystic fibrosis |
| Manufacturing Technique | Formulation (in % w/w) | Features | Ref. |
|---|---|---|---|
| Direct compression | Hydrochlorthiazide 15.5% Mannitol-Crospovidone-Polyvinylacetate dispersion (co-processed) 81%, Na stearyl fumarate 3.5% | Oro-dispersible Ø = 2 mm, Friability < 1%, WT = 3 s MU and DCU acceptable, AVs quite high | [52] |
| Hydrochlorthiazide 31%, Isomalt 62% Kollidon® CL 4%, SiO2 2% Mg stearate 1% | Oro-dispersible Ø = 2 mm, Angle of repose = 40° MU and DCU acceptable AVs quite high, DT < 30 s 60% of drug released in 5 min (pH 1.2) | [53] | |
| Enalapril maleate 1.6%, Isomalt 79%, Kollidon® CL-SF 4% Mg stearate 1% | Oro-dispersible Ø = 2 mm, Angle of repose = 34° MU and DCU acceptable, AVs quite high DT ≈ 30 s, 100% of drug released in 7 min at pH 6.8 | [54] | |
| Enalapril 1%, StarLac® 98%, Mg stearate 1% | Oro-dispersible Hardness 39 N, Friability < 1% Mean DC = 103.6%, WT = 23 s, DT = 28 s | [54] | |
| Risperidone 10%, Mannitol 46% MCC 34% Croscarmellose Na 10% Aerosil® 200/Aerosil® 300 (1/1, w/w) 1% Aspartame 0.5%, Peppermint oil 0.5% | Oro-dispersible Ø = 2 mm, Angle of repose = 29° Friability < 1%, Mean DC 98%, DT = 8 s | [55] | |
| Wet granulation (WG) + compression (rotary press) | Hydrocortisone 17%, MCC 22% Lactose monohydrate 52% HPMC 3% Croscarmellose Na 5%, SiO2 0.3%, Mg stearate 1% | Oro-dispersible Ø = 3 mm, Mean DC 102% Completed dissolution after 10 min | [56] |
| Direct compression | Loratadine 6.7%, MCC 80%, Corn starch 7.3% Croscarmellose Na 5%, Mg stearate 0.5%, SiO2 0.5% | Oro-dispersible Ø = 3 mm, DT 60 s, Hardness 32 N Friability = 0.4% Good MU, 80% of drug released in 3 min | [58] |
| Furosemide 10%, Ludipress® LCE 34% Skimmed milk powder 20%, Kollidon® CL-F 20% Mg stearate 1%, SiO2 1% Optarom® Cherry 14 Sucralose 0.5% | Uncoated/Oro-dispersible Ø = 4 mm, DT = 12 s, Hardness 31 N Friability < 1%, Mean DC 94.4% Completed dissolution in 30 min | [59] | |
| Furosemide 10%, Lactose monohydrate 45% Emdex® 22%, Kollidon® CL-F 20%, Mg stearate 1% SiO2 1%, Strawberry flavor 1% | Oro-dispersible Ø = 4 mm, DT 128 s, Hardness 19 N Friability < 1%, Mean DC 95.6% Completed dissolution within 30 min | [59] | |
| API cocrystallization + direct compression | Piroxicam 41%, Mannitol 19.5%, CMC 10% Croscarmellose Na 5%, Mg stearate 0.5% | Uncoated/Oro-dispersible Ø = 4 mm, Friability < 1% DT = 1 min, 80% drug released in 6 min | [60] |
| Direct compression | Lapatinib/HPMCP 1/3 spray dried 20% Croscarmellose Na 6%, CMC 71%, Mg stearate 1% SiO2 2% | Uncoated Ø = 2 mm, Friability < 1%, Hardness 25 N Mean DC 107% DT 15 min, 65% drug released Pharmacokinetic study in juvenile porcine model | [61] |
| Hot melt extrusion | Ketoprofen 40%, EPO 60% | Coated Ø = 5 mm, Friability < 1%, Mean DC 94–110% Completed dissolution in 20 min | [63] |
| Compressed electrospun nanofibers | Prednisolone 10%, PVP 90% | Uncoated Ø = 2 mm, Friability < 1%, Mean DC 96% Completed dissolution in 20 min | [64] |
| FDM printing + HME | Caffeine or Propranolol HCl 10% HPMC or HPC 79.5% PEG 6000 10%, Fumed silica 0.5% | Uncoated Ø = 1.5–2–3–4 mm, Mean DC not determined Passed MU, 80% drug released after 175 min | [69] |
| FDM printing + HME | Baclofen 10%, PVA 80%, Sorbitol 10% | Uncoated Mini-caplets L/W/H 7.5/4/2.5 mm, Hardness 450 N, Good MU DT > 20 min, 75% drug released in 60 min | [70] |
| Nifedipine 50%, HPC 34%, Kollidon® VA 64 10% PEG 4000 5%, Mg stearate 1% | Uncoated channeled Ø = 6 mm, Hardness 11 N, Friability < 1% Mean DC 50% 60% drug released after 4 h (burst release) | [71] | |
| FDM printing via passive diffusion drug loading | Nifedipine 4%, PVA-PEG 96% 1 | Uncoated Ø = 6 mm, Hardness 403 N, Friability < 1% Mean DC 4% Sustained drug release (6 h) | [71] |
| FDM printing + HME | Indomethacin 20%, HPMCAS 60%, PEG 6000 20% | Chewable soft dosage form Candy-like shapes, Negligible drug release in the mouth 80% drug released in 1 h (pH 7.4) | [73] |
| Embedded 3D printing | Ibuprofen/paracetamol 2, glycerol, gelatin Locust bean gum | Chewable soft dosage form Lego™-like bricks, 100% drug released in 2 h (pH 7.2) | [74] |
| Direct compression | Ibuprofen 14–25%, Spray-dried Mannitol/MCC ¼ Crospovidone 2%, SiO2 1%, Na stearyl fumarate 4% | Uncoated/Oro-dispersible Ø = 1.5–2.5 mm, 60–100 µm API particle size (D6.3) Friability < 1% MU and DCU passed, DT = 4–90 s | [76] |
| High shear wet granulation Dry API added to the powder blend | Intragranular: Ibuprofen 0.67%, MCC 10% Mannitol 81.3% Croscarmellose Na 2%, HPMC 2% Extra granular: Croscarmellose Na 2% Na stearyl fumarate 2% | Uncoated Ø = 1.2–1.5 mm, 0.67% w/w API loading 18 µm API particle size (D90), MU and DCU passed only for 30 units batch 70% API released in 20 min | [77] |
| High shear wet granulation API nanosuspension added in the granulation fluid | Intragranular: Ibersartam 0.01–0.5%, MCC 9.5%, Mannitol 81 Crospovidone 4%, HPMC 2% Extragranular: SiO2 0.5%, Na stearyl fumarate 3% | Uncoated Ø = 1.2 mm, 0.01–0.5%w/w API loading 380 nm API particle size Good MU and DCU per unit at every API loading 97% API released within 10 min | [78] |
| Direct compression of an interactive mixture | Na salicylate 1%, Mannitol SD 98%, Mg stearate 1% | Oro-dispersible Ø = 2 mm, 1% w/w API loading Good DC uniformity, DT 3 min | [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuccari, G.; Alfei, S.; Marimpietri, D.; Iurilli, V.; Barabino, P.; Marchitto, L. Mini-Tablets: A Valid Strategy to Combine Efficacy and Safety in Pediatrics. Pharmaceuticals 2022, 15, 108. https://doi.org/10.3390/ph15010108
Zuccari G, Alfei S, Marimpietri D, Iurilli V, Barabino P, Marchitto L. Mini-Tablets: A Valid Strategy to Combine Efficacy and Safety in Pediatrics. Pharmaceuticals. 2022; 15(1):108. https://doi.org/10.3390/ph15010108
Chicago/Turabian StyleZuccari, Guendalina, Silvana Alfei, Danilo Marimpietri, Valentina Iurilli, Paola Barabino, and Leonardo Marchitto. 2022. "Mini-Tablets: A Valid Strategy to Combine Efficacy and Safety in Pediatrics" Pharmaceuticals 15, no. 1: 108. https://doi.org/10.3390/ph15010108