
Design, Molecular Docking, Synthesis, Anticancer and Anti-Hyperglycemic Assessments of Thiazolidine-2,4-diones Bearing Sulfonylthiourea Moieties as Potent VEGFR-2 Inhibitors and PPARγ Agonists

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
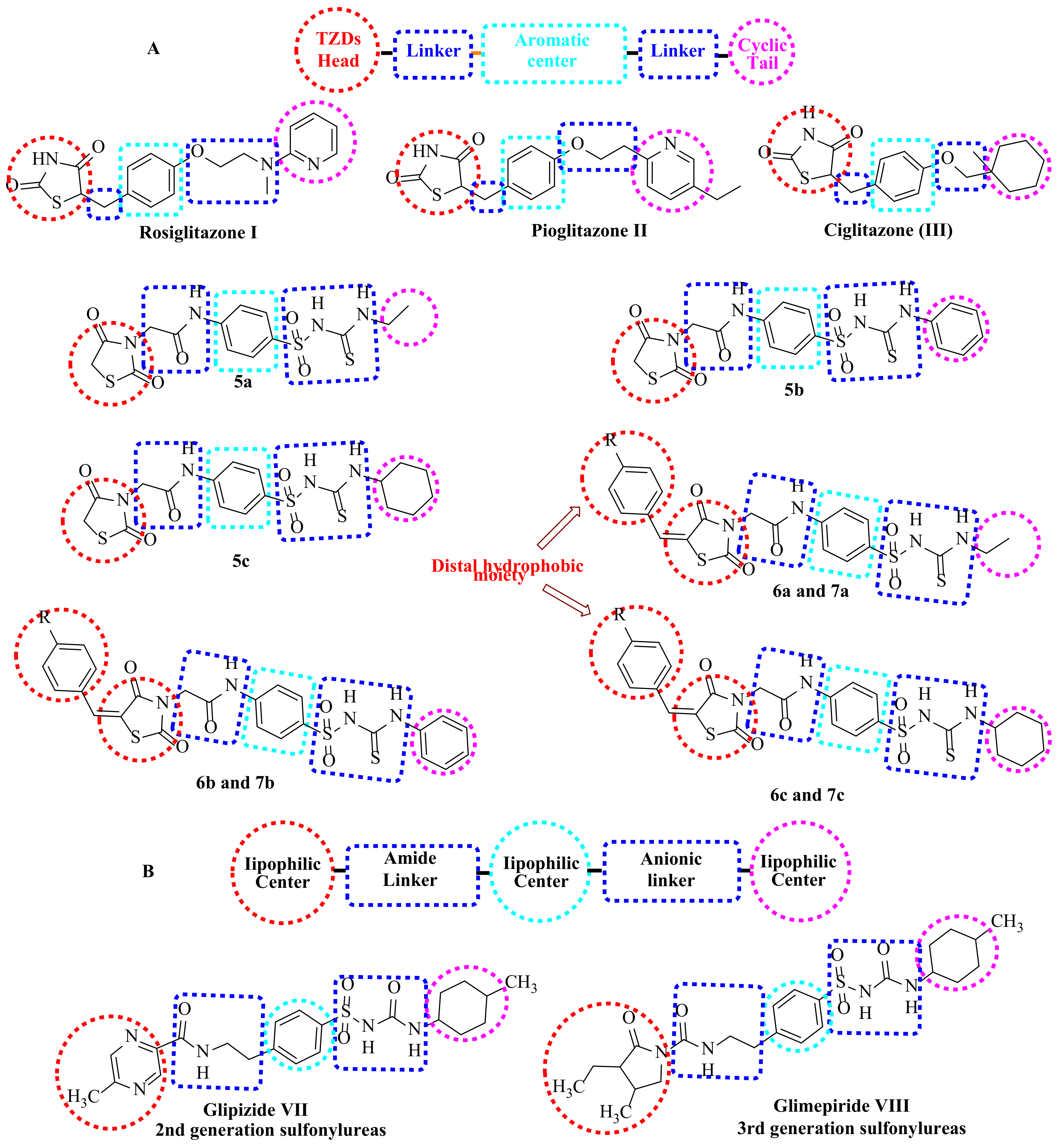
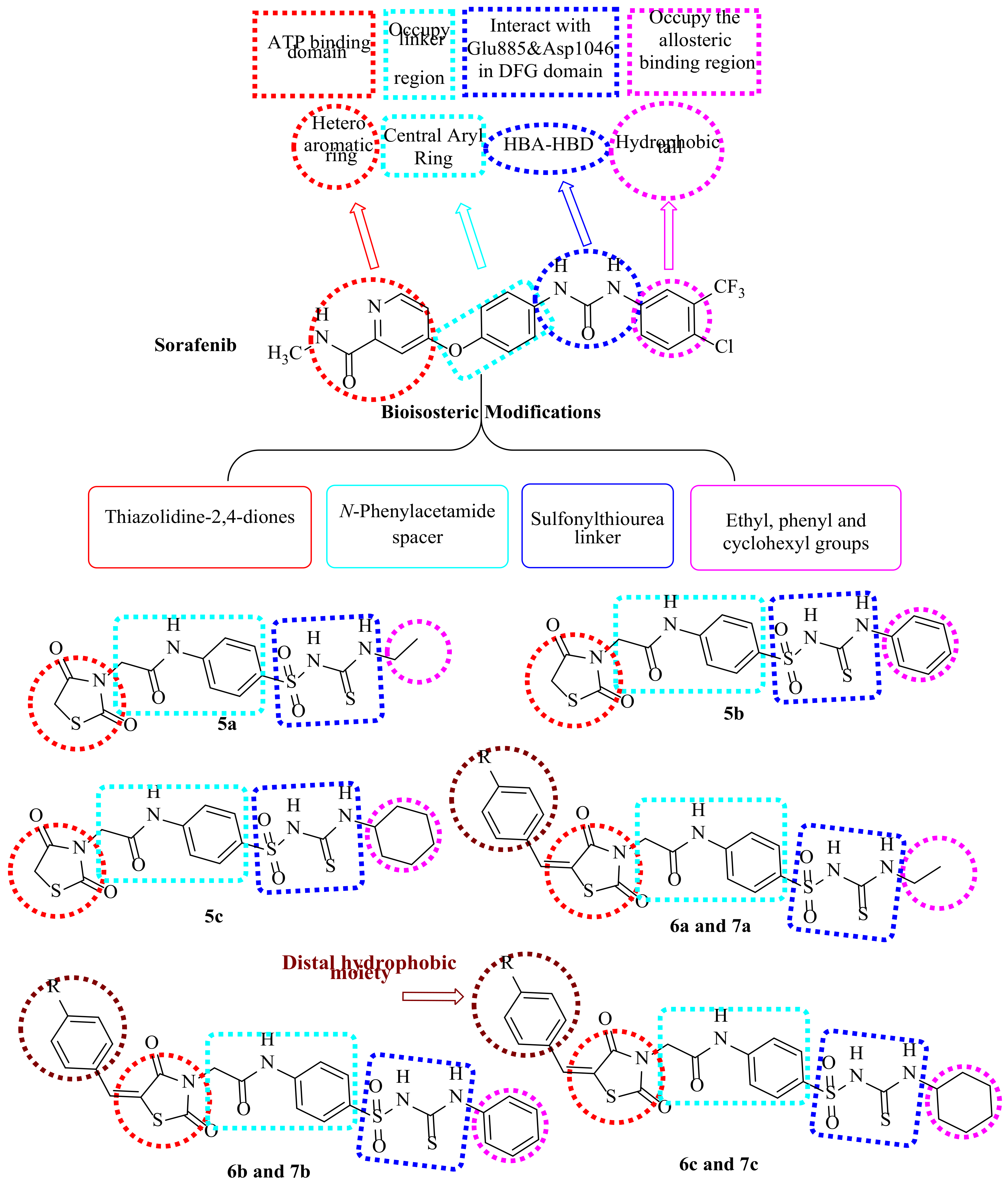
1.1. Structure-Based Design Rationale
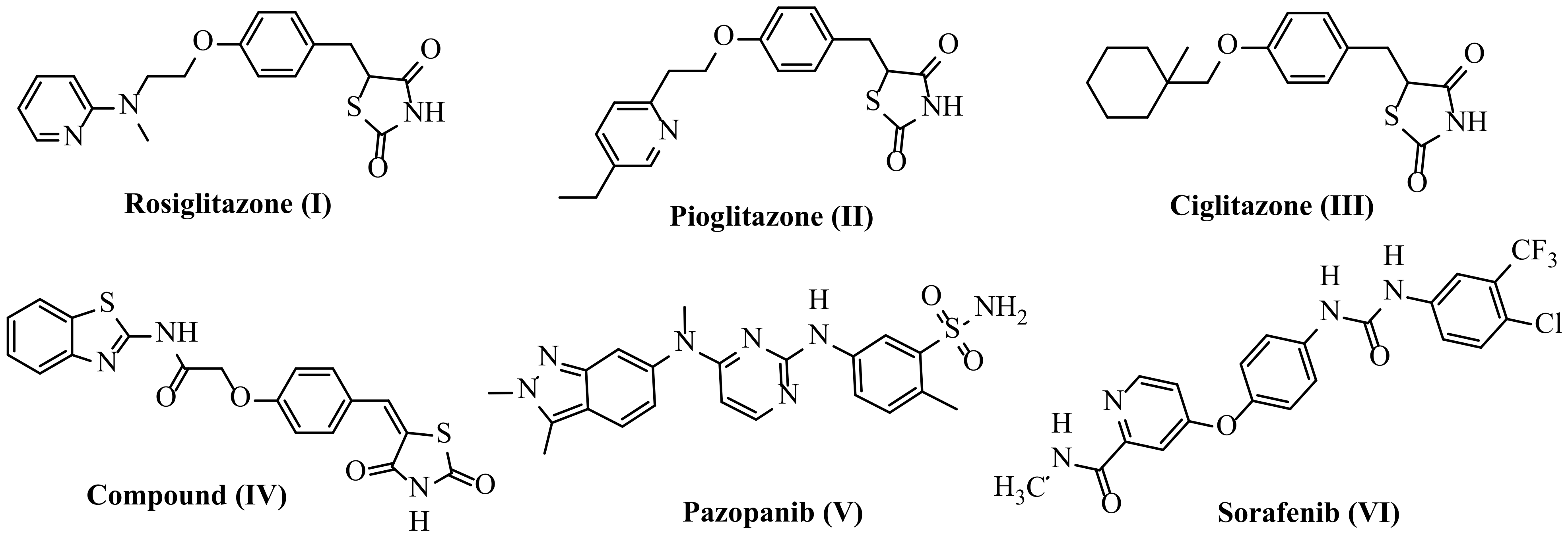
1.1.1. As Anticancer Agents
1.1.2. As Antidiabetic Agents
2. Results and Discussion
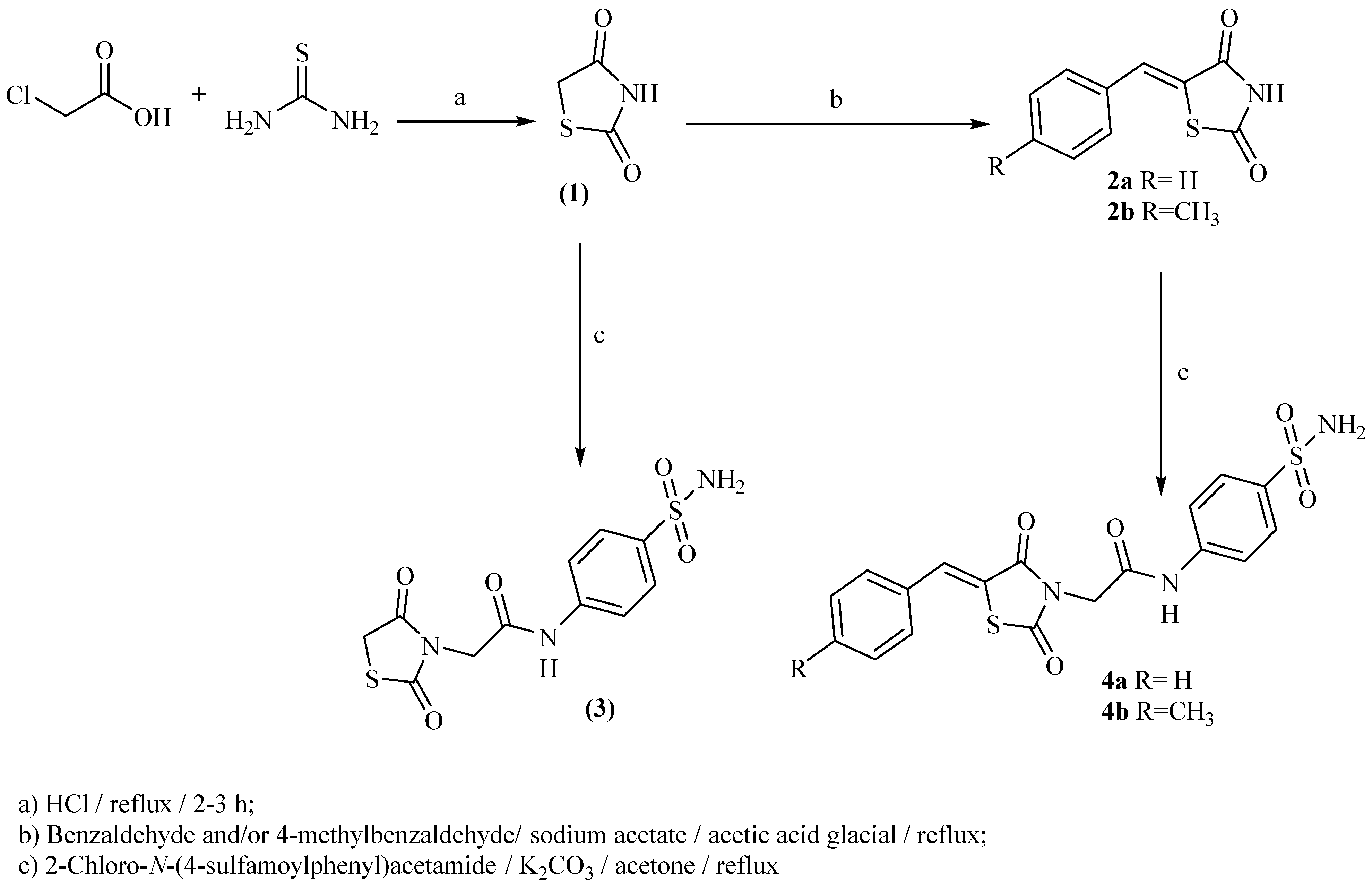
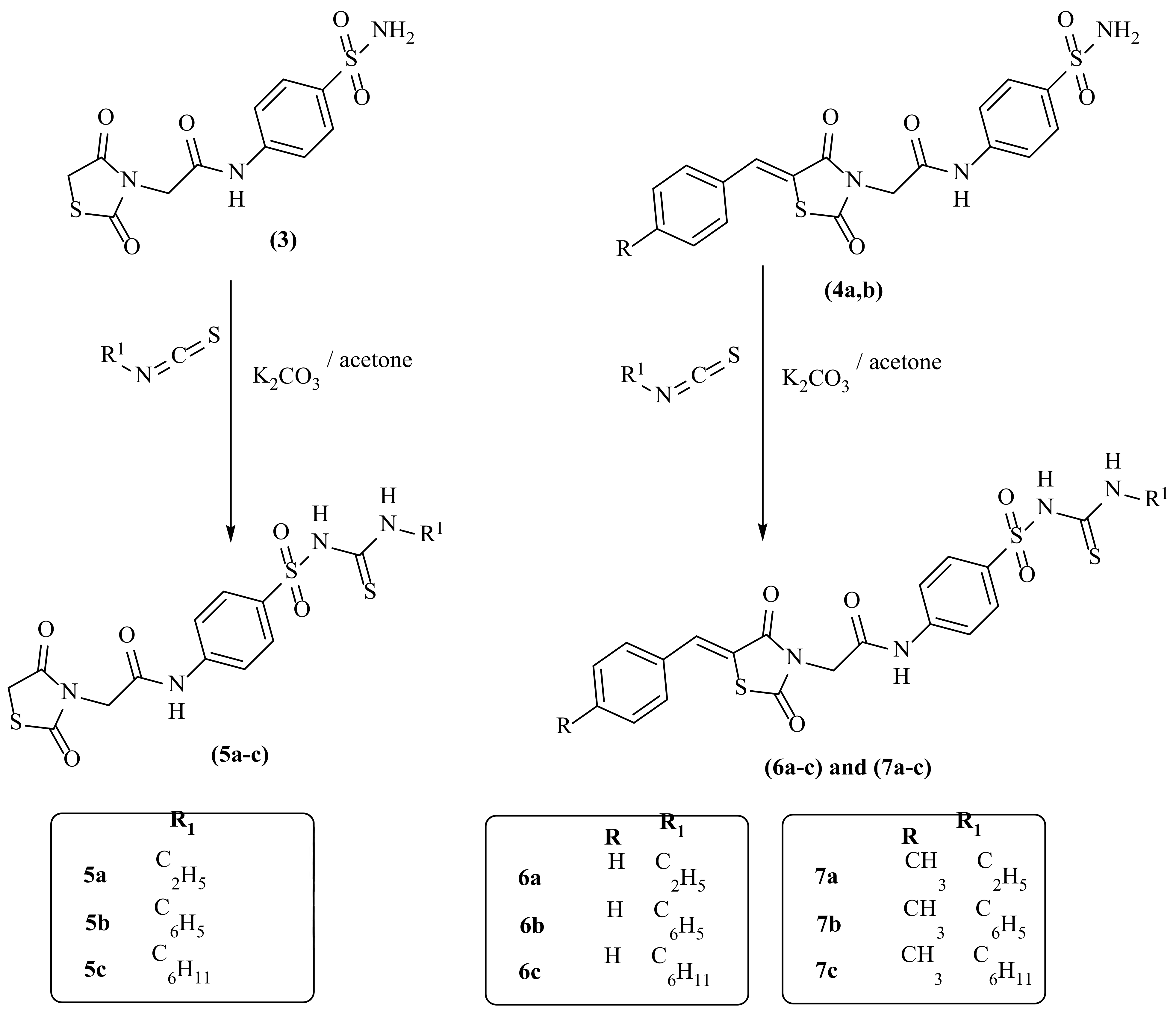
2.1. Chemistry
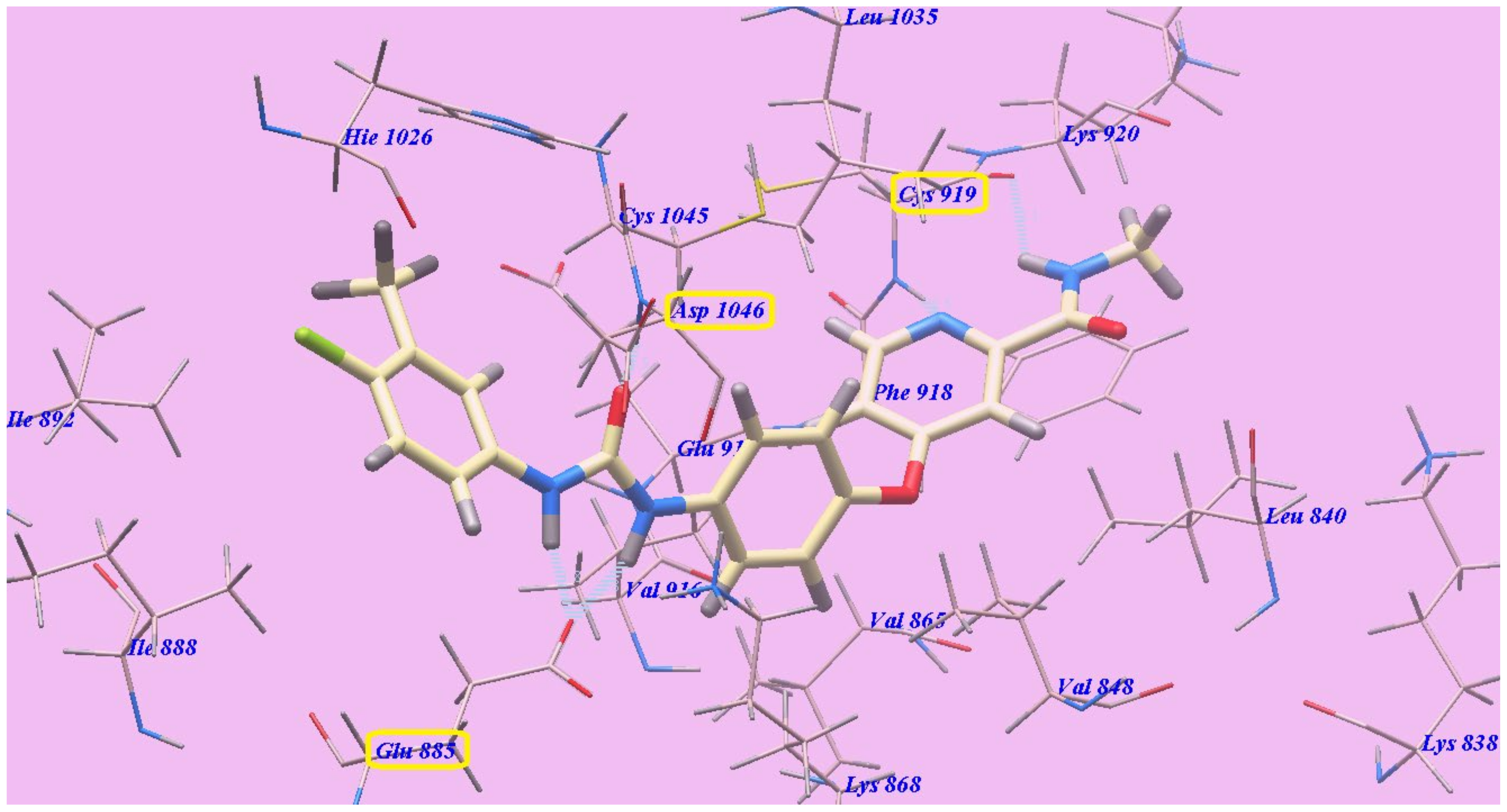
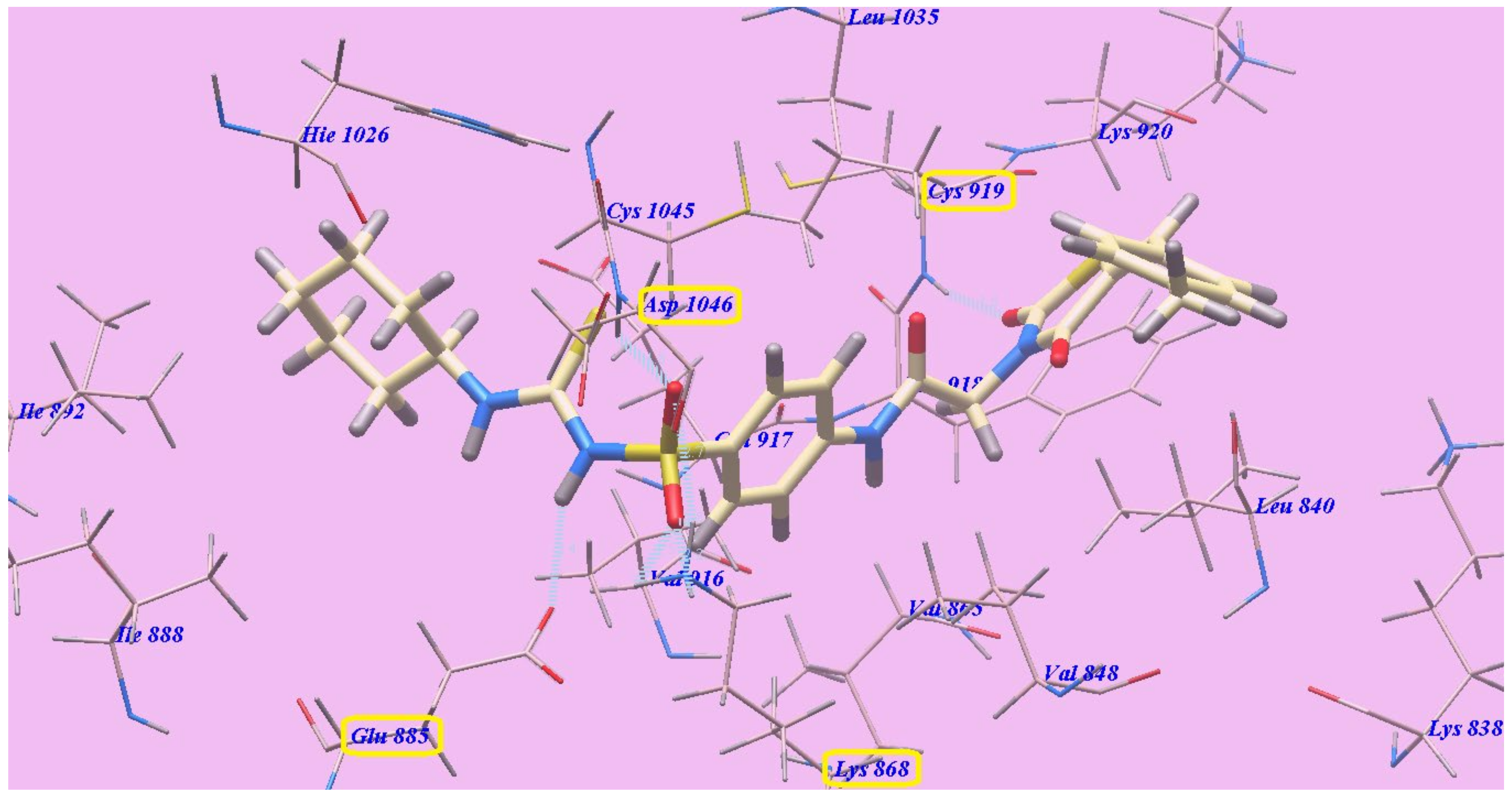
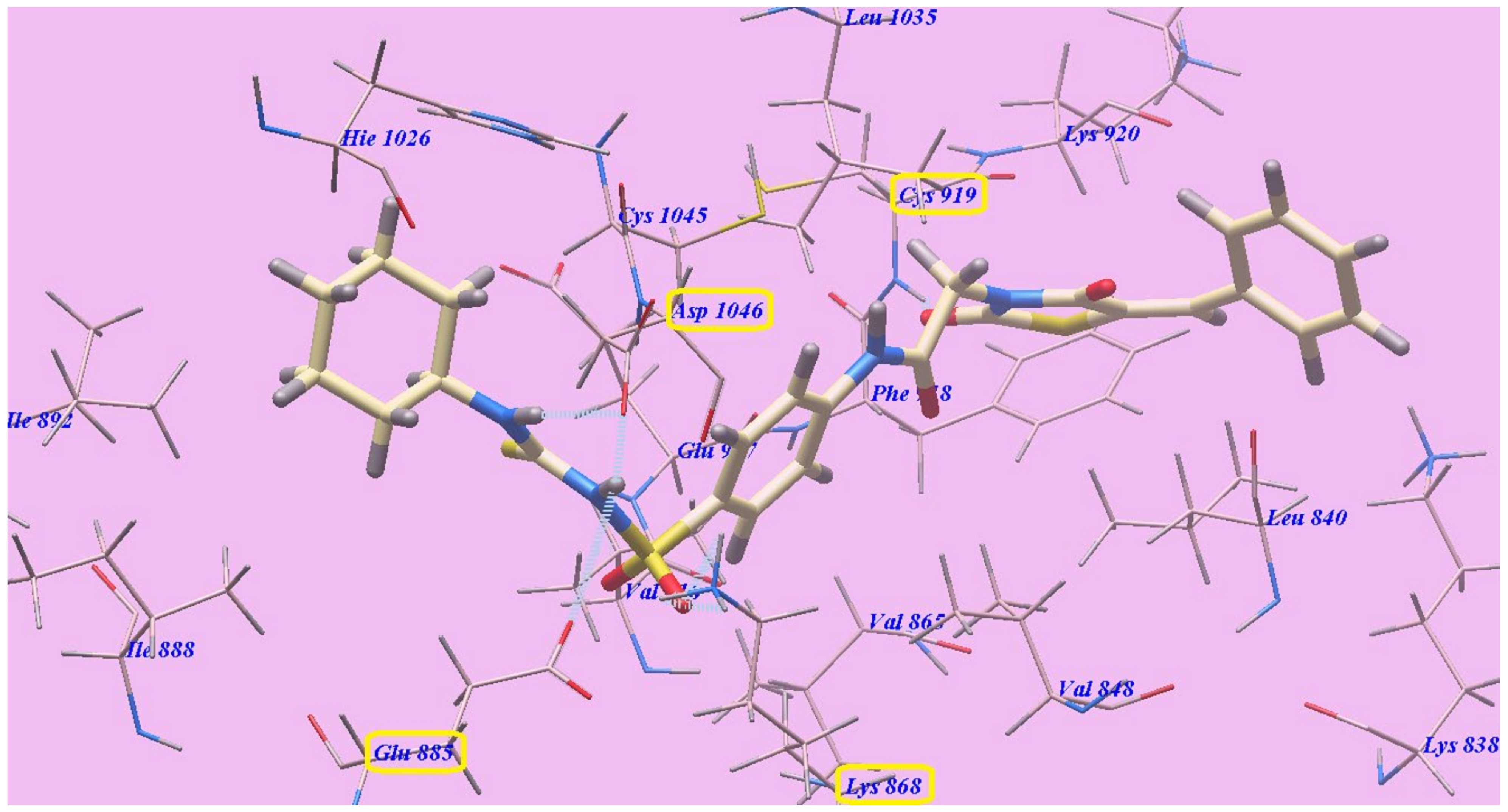
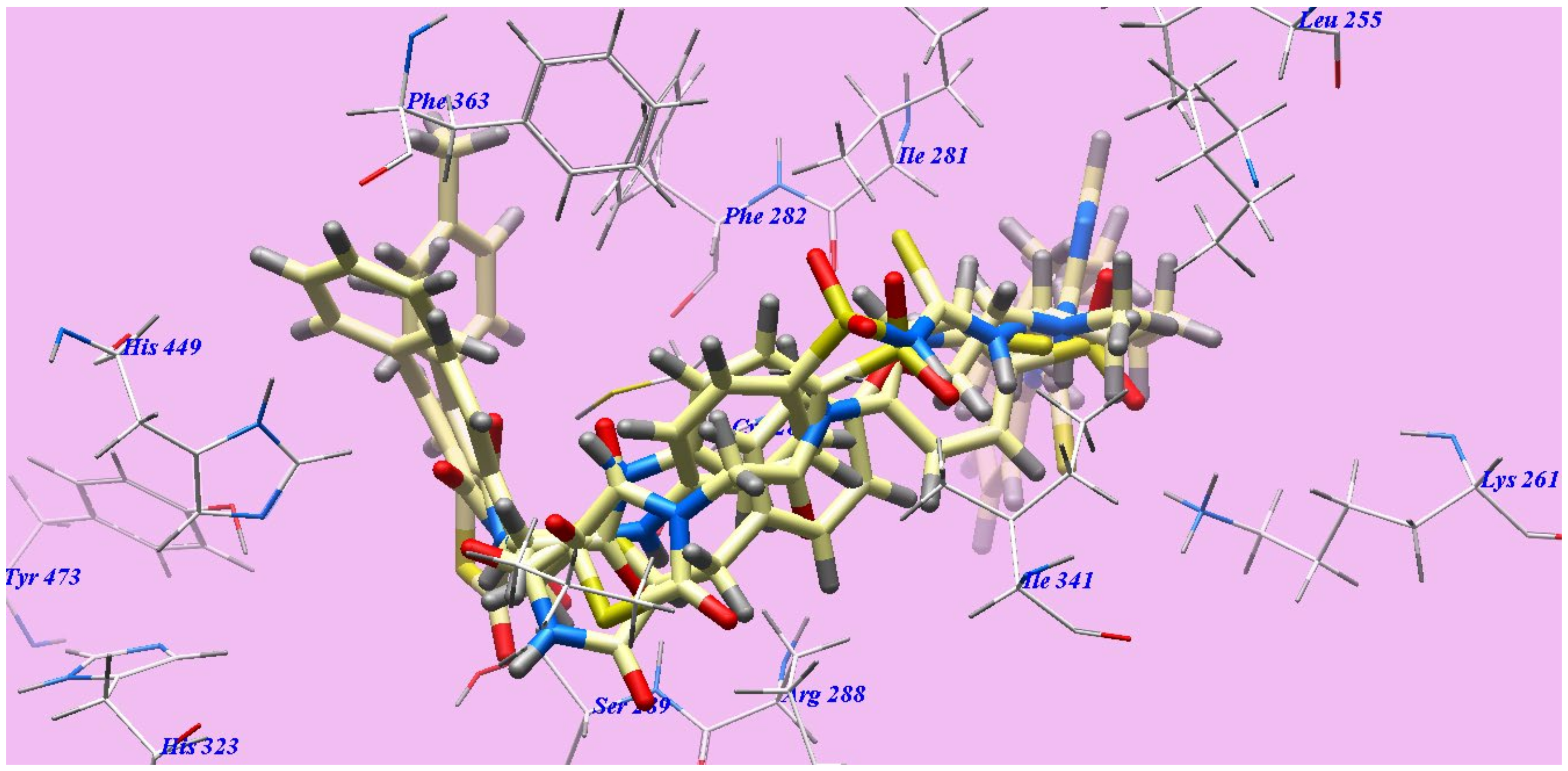
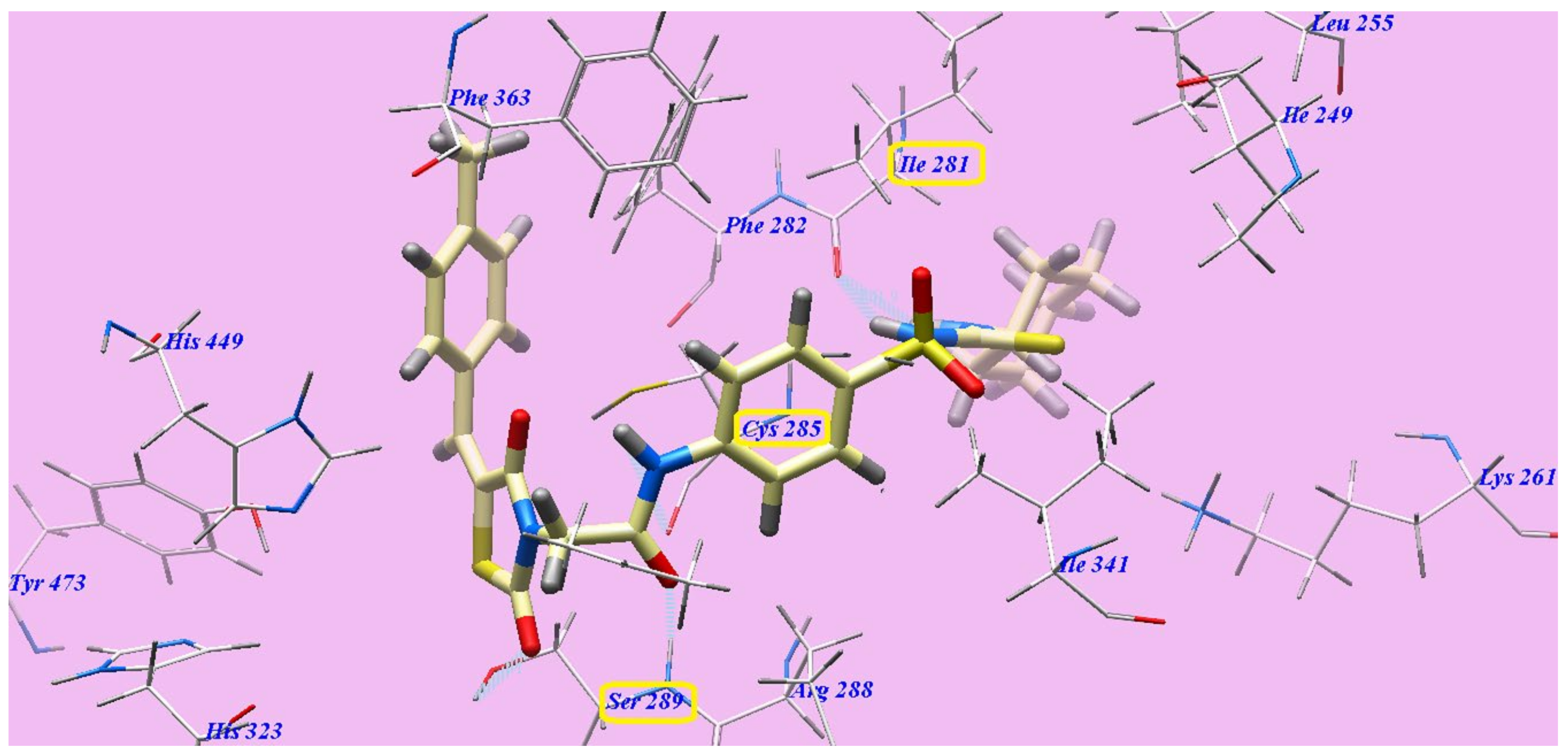
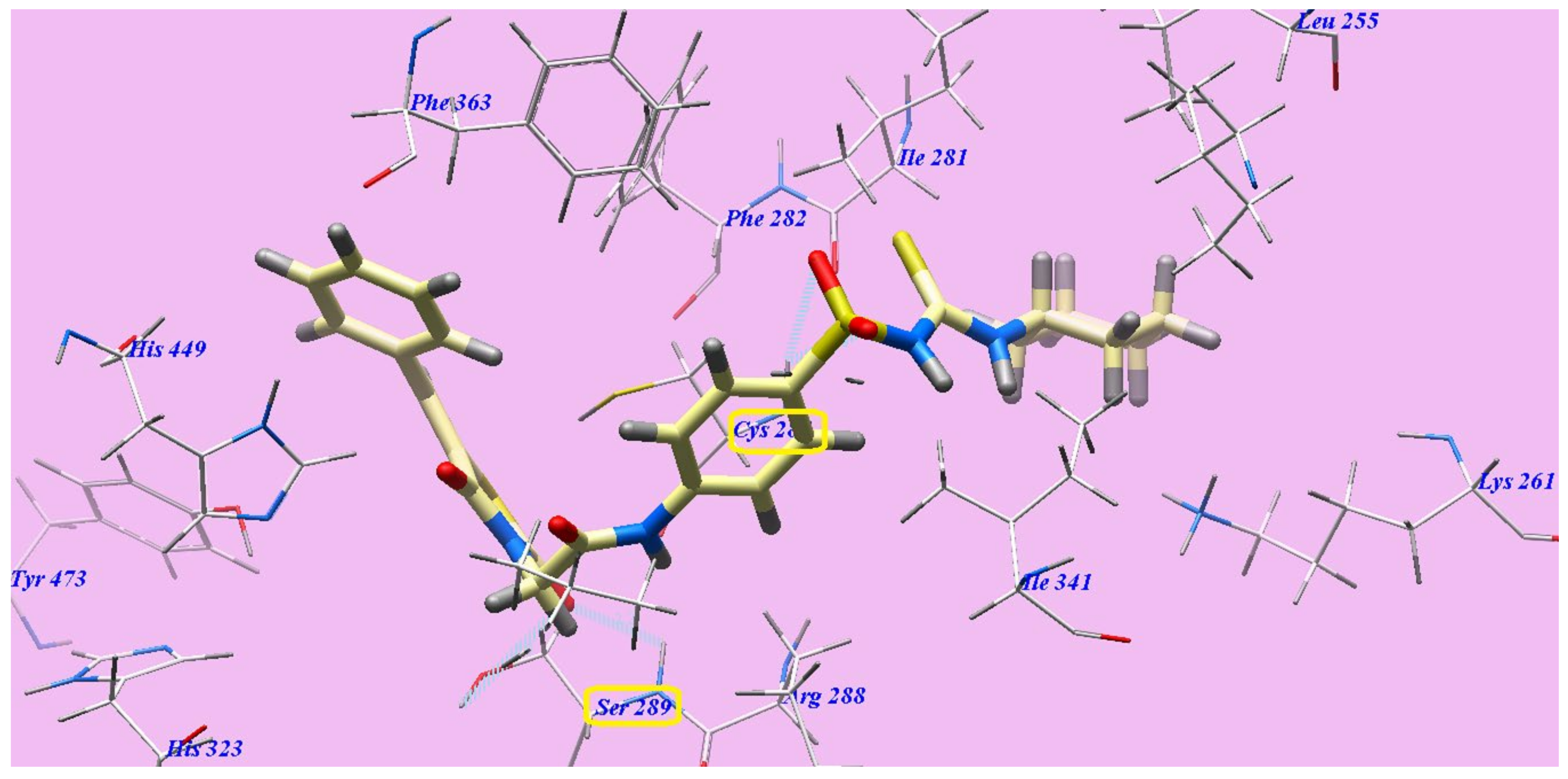
2.2. Docking Studies
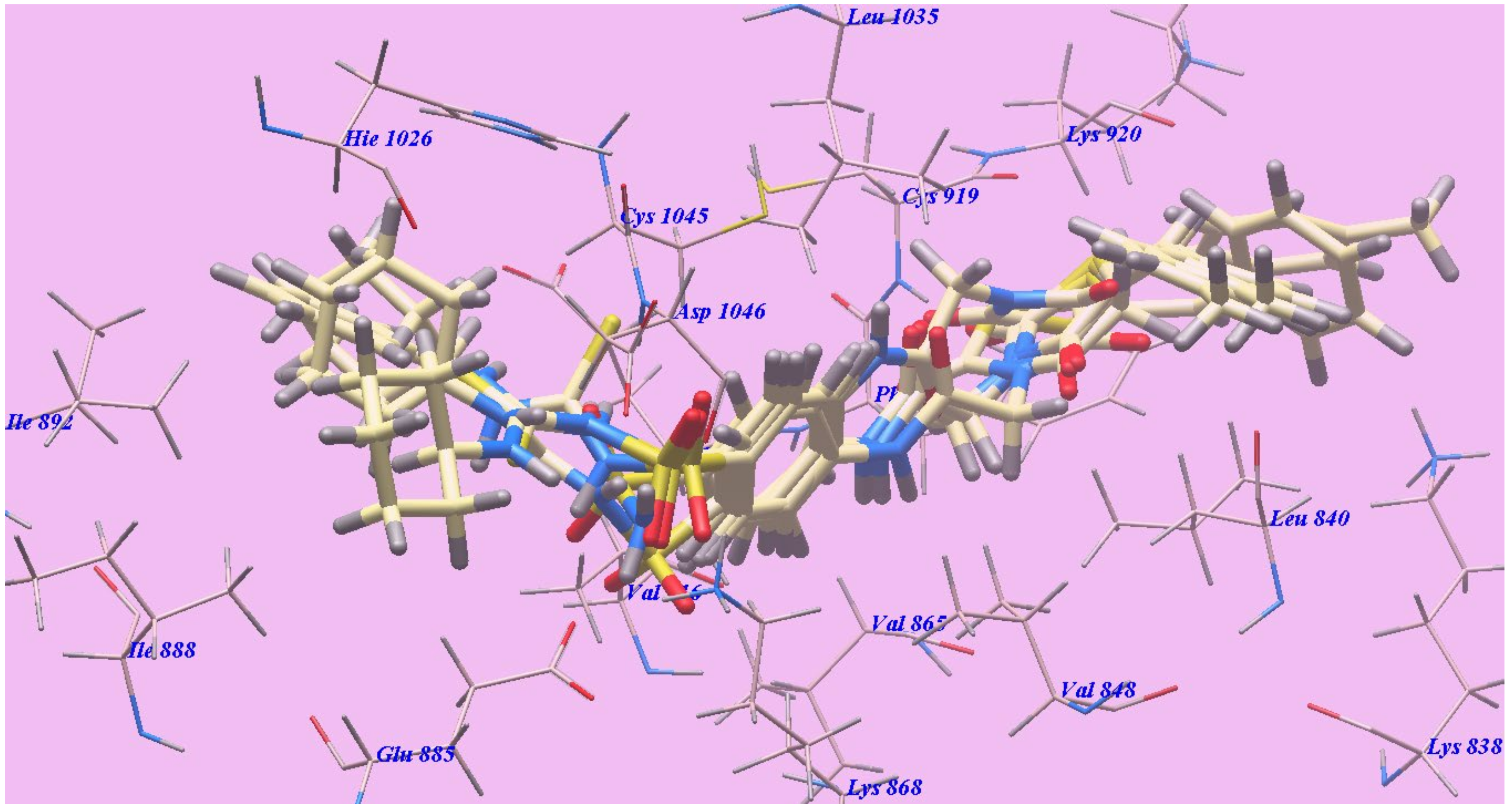
2.2.1. Docking Studies as VEGFR-2 Inhibitors
2.2.2. Docking Studies as PPARγ Agonists
2.3. In Vitro Cytotoxic Activity
2.4. In Vitro Assay of VEGFR-2 Kinase
2.5. In Vitro Binding Assay of PPARγ Ligand
2.6. In Vitro Insulin Assay
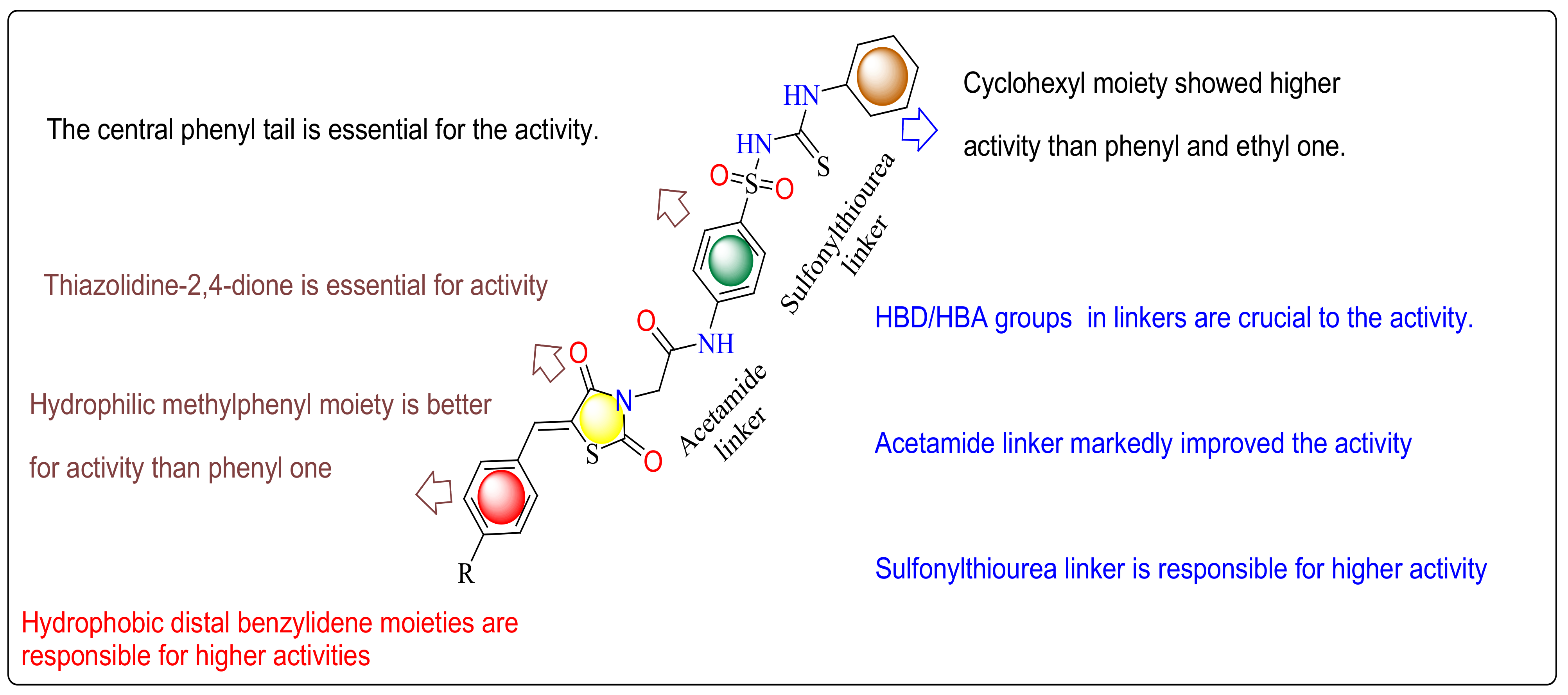
2.7. SAR (Structure Activity Relationship)
2.8. ADMET, in Silico Studies Profile
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. General Procedure for Synthesis of 2-(2,4-Dioxothiazolidin-3-yl)-N-(4-Sulfamoylphenyl)-Acetamide (3)
3.1.3. General Procedure for Synthesis of Compounds (4a,b)
2-(5-Benzylidene-2,4-dioxothiazolidin-3-yl)-N-(4-sulfamoylphenyl)acetamide (4a)
2-(5-(4-Methylbenzylidene)-2,4-dioxothiazolidin-3-yl)-N-(4-sulfamoylphenyl)acetamide (4b)
3.1.4. General Procedure for Syntheses of Compounds (5a–c)
2-(2,4-Dioxothiazolidin-3-yl)-N-(4-(N-(ethylcarbamothioyl)sulfamoyl)phenyl)acetamide (5a)
2-(2,4-Dioxothiazolidin-3-yl)-N-(4-(N-(phenylcarbamothioyl)sulfamoyl)phenyl)acetamide (5b)
N-(4-(N-(Cyclohexylcarbamothioyl)sulfamoyl)phenyl)-2-(2,4-dioxothiazolidin-3-yl)acetamide (5c)
3.1.5. General Procedure for Syntheses of Compounds (6a–c)
2-(5-Benzylidene-2,4-dioxothiazolidin-3-yl)-N-(4-(N-(ethylcarbamothioyl)sulfamoyl)phenyl)acetamide (6a)
2-(5-Benzylidene-2,4-dioxothiazolidin-3-yl)-N-(4-(N-(phenylcarbamothioyl)sulfamoyl)phenyl)-acetamide (6b)
2-(5-Benzylidene-2,4-dioxothiazolidin-3-yl)-N-(4-(N-(cyclohexylcarbamothioyl)sulfamoyl)phenyl)-acetamide (6c)
3.1.6. General Procedure for Synthesis of Compounds (7a–c)
N-(4-(N-(Ethylcarbamothioyl)sulfamoyl)phenyl)-2-(5-(4-methylbenzylidene)-2,4-dioxothiazolidin-3-yl)acetamide (7a)
2-(5-(4-Methylbenzylidene)-2,4-dioxothiazolidin-3-yl)-N-(4-(N-(phenylcarbamothioyl)sulfamoyl)-phenyl)acetamide (7b)
N-(4-(N-(Cyclohexylcarbamothioyl)sulfamoyl)phenyl)-2-(5-(4-methylbenzylidene)-2,4-dioxothiazolidin-3-yl)acetamide (7c)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; Hu, X.; Zhang, Q.; Zou, R. Diabetes mellitus and risk of fall in older adult: A systematic review and meta-analysis. Age Ageing 2016, 45, 761–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, U.; Vanjari, Y.; Laxmikeshav, K.; Tokala, R.; Niggula, P.K.; Kumar, M.; Talla, V.; Kamal, A.; Shankaraiah, N. Synthesis and in Vitro Cytotoxicity Evaluation of Phenanthrene Linked 2,4-Thiazolidinediones as Potential Anticancer Agents. Anti-Cancer Agents Med. Chem. 2021, 21, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Tahlan, S.S.; Verma, P.K. Biological potential of thiazolidinedione derivatives of synthetic origin. Chem. Cent. J. 2017, 11, 130. [Google Scholar] [CrossRef]
- Hussein, A.H.; Moghimi, A.; Roohbakhsh, A. Anticonvulsant and ameliorative effects of pioglitazone on cognitive deficits, inflammation and apoptosis in the hippocampus of rat pups exposed to febrile seizure. Iran. J. Basic Med. Sci. 2019, 22, 267–276. [Google Scholar] [CrossRef]
- Sutinen, J. The Effects of Thiazolidinediones on Metabolic Complications and Lipodystrophy in HIV-Infected Patients. PPAR Res. 2009, 2009, 373524. [Google Scholar] [CrossRef]
- Rekha, S.; Shantharam, U.; Vineet, C. Synthesis and evaluation of novel thiazolidinedione for anti-inflammatory activity. Int. Res. J. Pharm. 2011, 2, 81–84. [Google Scholar]
- Sameeh, M.Y.; Khowdiary, M.M.; Nassar, H.S.; Abdelall, M.M.; Amer, H.H.; Hamed, A.; Elhenawy, A.A. Thiazolidinedione Derivatives: In Silico, In Vitro, In Vivo, Antioxidant and Anti-Diabetic Evaluation. Molecules 2022, 27, 830. [Google Scholar] [CrossRef]
- Panigrahy, D.; Singer, S.; Shen, L.Q.; Butterfield, C.E.; Freedman, D.A.; Chen, E.J.; Moses, M.A.; Kilroy, S.; Duensing, S.; Fletcher, C.; et al. PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J. Clin. Investig. 2002, 110, 923–932. [Google Scholar] [CrossRef]
- Joshi, H.; Pal, T.; Ramaa, C. A new dawn for the use of thiazolidinediones in cancer therapy. Expert Opin. Investig. Drugs 2014, 23, 501–510. [Google Scholar] [CrossRef]
- Bhanushali, U.; Rajendran, S.; Sarma, K.; Kulkarni, P.; Chatti, K.; Chatterjee, S.; Ramaa, C. 5-Benzylidene-2, 4-thiazolidenedione derivatives: Design, synthesis and evaluation as inhibitors of angiogenesis targeting VEGR-2. Bioorg. Chem. 2016, 67, 139–147. [Google Scholar] [CrossRef]
- Rashid, M.; Shrivastava, N.; Husain, A. Synthesis and sar strategy of thiazolidinedione: A novel approach for cancer treatment. J. Chil. Chem. Soc. 2020, 65, 4817–4832. [Google Scholar] [CrossRef]
- Han, S.W.; Roman, J. Anticancer actions of PPARγ ligands: Current state and future perspectives in human lung cancer. World J. Biol. Chem. 2010, 1, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Bo, Q.F.; Sun, X.M.; Liu, J.; Sui, X.M.; Li, G.X. Antitumor action of the peroxisome proliferator-activated receptor-γ agonist rosiglitazone in hepatocellular carcinoma. Oncol. Lett. 2015, 10, 1979–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanquicett, C.; Roman, J.; Hart, C.M. Thiazolidinediones as anti-cancer agents. Cancer Ther. 2008, 6, 25–34. [Google Scholar] [PubMed]
- Yoshizaki, T.; Motomura, W.; Tanno, S.; Kumei, S.; Yoshizaki, Y.; Tanno, S.; Okumura, T. Thiazolidinediones enhance vascular endothelial growth factor expression and induce cell growth inhibition in non-small-cell lung cancer cells. J. Exp. Clin. Cancer Res. 2010, 29, 22. [Google Scholar] [CrossRef] [Green Version]
- Corigliano, D.M.; Syed, R.; Messineo, S.; Lupia, A.; Patel, R.; Reddy, C.V.R.; Dubey, P.K.; Colica, C.; Amato, R.; De Sarro, G.; et al. Indole and 2,4-Thiazolidinedione conjugates as potential anticancer modulators. PeerJ 2018, 6, e5386. [Google Scholar] [CrossRef]
- Ha, Y.M.; Park, Y.J.; Kim, J.A.; Park, D.; Park, J.Y.; Lee, H.J.; Lee, J.Y.; Moon, H.R.; Chung, H.Y. Design and synthesis of 5-(substituted benzylidene) thiazolidine-2, 4-dione derivatives as novel tyrosinase inhibitors. Eur. J. Med. Chem. 2012, 49, 245–252. [Google Scholar] [CrossRef]
- Li, Q.; Al-Ayoubi, A.; Guo, T.; Zheng, H.; Sarkar, A.; Nguyen, T.; Eblen, S.T.; Grant, S.; Kellogg, G.E.; Zhang, S. Structure–activity relationship (SAR) studies of 3-(2-amino-ethyl)-5-(4-ethoxy-benzylidene)-thiazolidine-2, 4-dione: Development of potential substrate-specific ERK1/2 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6042–6046. [Google Scholar] [CrossRef]
- Nastasă, C.; Tamaian, R.; Oniga, O.; Tiperciuc, B. 5-Arylidene (chromenyl-methylene)-thiazolidinediones: Potential New Agents against Mutant Oncoproteins K-Ras, N-Ras and B-Raf in Colorectal Cancer and Melanoma. Medicina 2019, 55, 85. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.K.; Menon, K.M.J.; Cabrera, L.M.; Vahratian, A.; Kavoussi, S.K.; Lebovic, D.I. Thiazolidinediones decrease vascular endothelial growth factor (VEGF) production by human luteinized granulosa cells in vitro. Fertil. Steril. 2010, 93, 2042–2047. [Google Scholar] [CrossRef]
- El-Adl, K.; El-Helby, A.-G.A.; Sakr, H.; Eissa, I.H.; El-Hddad, S.S.; Shoman, F.M. Design, synthesis, molecular docking and anticancer evaluations of 5-benzylidenethiazolidine-2,4-dione derivatives targeting VEGFR-2 enzyme. Bioorg. Chem. 2020, 102, 104059. [Google Scholar] [CrossRef] [PubMed]
- El-Adl, K.; Sakr, H.; El-Hddad, S.S.A.; El-Helby, A.A.; Nasser, M.; Abulkhair, H.S. Design, synthesis, docking, ADMET profile, and anticancer evaluations of novel thiazolidine-2,4-dione derivatives as VEGFR-2 inhibitors. Arch. Pharm. 2021, 354, e2000491. [Google Scholar] [CrossRef] [PubMed]
- El-Adl, K.; Sakr, H.; Nasser, M.; Alswah, M.; Shoman, F.M.A. 5-(4-Methoxybenzylidene)thiazolidine-2, 4-dione-derived VEGFR-2 inhibitors: Design, synthesis, molecular docking, and anticancer evaluations. Arch. Pharm. 2020, 353, e2000079. [Google Scholar] [CrossRef] [PubMed]
- El-Adl, K.; El-Helby, A.A.; Sakr, H.; Ayyad, R.R.; Mahdy, H.A.; Nasser, M.; Abulkhair, H.S.; El-Hddad, S.S.A. Design, synthesis, molecular docking, anticancer evaluations, and in silico pharmacokinetic studies of novel 5-[(4-chloro/2,4-dichloro)benzylidene]thiazolidine-2,4-dione derivatives as VEGFR-2 inhibitors. Arch. Pharm. 2020, 354, e2000279. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biol. Pharm. Bull. 2011, 34, 17851788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kankanala, J.; Latham, A.; Johnson, A.; Homer-Vanniasinkam, S.; Fishwick, C.; Ponnambalam, S. A combinatorial in silico and cellular approach to identify a new class of compounds that target VEGFR2 receptor tyrosine kinase activity and angiogenesis. Br. J. Pharmacol. 2012, 166, 737–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Pircher, A.; Hilbe, W.; Heidegger, I.; Drevs, J.; Tichelli, A.; Medinger, M. Biomarkers in tumor angiogenesis and anti-angiogenic therapy. Int. J. Mol. Sci. 2011, 12, 7077–7099. [Google Scholar] [CrossRef] [Green Version]
- Herbst, K.J.; Coltharp, C.; Amzel, L.M.; Zhang, J. Direct activation of Epac by sulfonylurea is isoform selective. Chem. Biol. 2011, 18, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Faidallah, H.M.; Al-Mohammadi, M.M.; Alamry, K.A.; Khan, K.A. Synthesis and biological evaluation of fluoropyrazolesulfonylurea and thiourea derivatives as possible antidiabetic agents. J. Enzym. Inhib. Med. Chem. 2016, 31, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila-Carriles, W.H.; Zhao, G.; Bryan, J. Defining a binding pocket for sulfonylureas in ATP-sensitive potassium channels. FASEB J. 2007, 21, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Fukuen, S.; Iwaki, M.; Yasui, A.; Makishima, M.; Matsuda, M.; Shimomura, I. Sulfonylurea agents exhibit peroxisome proliferator-activated receptor γ agonistic activity. J. Biol. Chem. 2015, 280, 23653–23659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inukai, K.; Watanabe, M.; Nakashima, Y.; Takata, N.; Isoyama, A.; Sawa, T.; Kurihara, S.; Awata, T.; Katayama, S. Glimepiride enhances intrinsic peroxisome proliferator-activated receptor-γ activity in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2005, 328, 484–490. [Google Scholar] [CrossRef]
- Arrault, A.; Rocchi, S.; Picard, F.; Maurois, P.; Pirotte, B.; Vamecq, J. A short series of antidiabetic sulfonylureas exhibit multiple ligand PPARγ-binding patterns. Biomed. Pharmacother. 2009, 63, 56–62. [Google Scholar] [CrossRef]
- El-Adl, K.; El-Helby, A.-G.A.; Sakr, H.; Elwan, A. Design, synthesis, molecular docking and anti-proliferative evaluations of [1,2,4]Triazolo[4,3-a]quinoxaline and [1,2,4]triazolo[4,3-a]quinoxaline-1-thiol derived DNA intercalators: Design, synthesis, molecular docking, in silico ADMET profile and antiproliferative evaluations. New J. Chem. 2021, 45, 881–897. [Google Scholar] [CrossRef]
- Eissa, I.H.; Metwaly, A.M.; Belal, A.; Mehany, A.B.; Ayyad, R.R.; El-Adl, K.; Mahdy, H.A.; Taghour, M.S.; El-Gamal, K.M.; El-Sawah, M.E.; et al. Discovery and anti-proliferative evaluation of new quinoxalines as potential DNA intercalators and topoisomerase II inhibitors. Arch. Pharm. 2019, 352, e1900123. [Google Scholar] [CrossRef]
- El-Adl, K.; Ibrahim, M.K.; Alesawy, M.S.I.; Eissa, I.H. [1,2,4]Triazolo[4,3-c]quinazoline and bis([1,2,4]triazolo)[4,3-a:4’,3’-c]quinazoline derived DNA intercalators: Design, synthesis, in silico ADMET profile, molecular docking and anti-proliferative evaluation studies. Bioorg. Med. Chem. 2021, 30, 115958. [Google Scholar] [CrossRef]
- El-Adl, K.; El-Helby, A.-G.A.; Sakr, H.; Elwan, A. Design, synthesis, molecular docking and anti-proliferative evaluations of [1,2,4]triazolo[4,3-a]quinoxaline derivatives as DNA intercalators and Topoisomerase II inhibitors. Bioorg. Chem. 2020, 105, 104399. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Ibrahim, M.-K.; Eissa, I.H.; El-Adl, K. Design, synthesis, in silico ADMET, docking, and antiproliferative evaluations of [1,2,4]triazolo[4,3-c]quinazolines as classical DNA intercalators. Arch. Pharm. 2022, e2100412. [Google Scholar] [CrossRef]
- El-Zahabi, M.A.; Sakr, H.; El-Adl, K.; Zayed, M.; Abdelraheem, A.S.; Eissa, S.I.; Elkady, H.; Eissa, I.H. Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents. Bioorg. Chem. 2020, 104, 104218. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.; Li, W.; Qin, Y.; Yu, T.; Liu, Z.; Zhou, M.; Liu, C.; Qiao, T.; Li, X.; Yousef, R.G.; et al. Inhibition of Vascular Smooth Muscle and Cancer Cell Proliferation by New VEGFR Inhibitors and Their Immunomodulator Effect: Design, Synthesis, and Biological Evaluation. Oxidative Med. Cell. Longev. 2021, 2021, 8321400. [Google Scholar] [CrossRef] [PubMed]
- El-Helby, A.A.; Sakr, H.; Ayyad, R.R.; Mahdy, H.A.; Khalifa, M.M.; Belal, A.; Rashed, M.; El-Sharkawy, A.; Metwaly, A.M.; Elhendawy, M.A.; et al. Design, synthesis, molecular modeling, in vivo studies and anticancer activity evaluation of new phthalazine derivatives as potential DNA intercalators and topoisomerase II inhibitors. Bioorg. Chem. 2020, 103, 104233. [Google Scholar] [CrossRef] [PubMed]
- Sakr, H.; Ayyad, R.R.; El-Helby, A.A.; Khalifa, M.M.; Mahdy, H.A. Discovery of novel triazolophthalazine derivatives as DNA intercalators and topoisomerase II inhibitors. Arch. Pharm. 2021, 354, e2000456. [Google Scholar] [CrossRef]
- Eissa, I.H.; El-Helby, A.A.; Mahdy, H.A.; Khalifaa, M.M.; Elnagar, H.A.; Mehany, A.B.M.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; et al. Discovery of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorg. Chem. 2020, 105, 104380. [Google Scholar] [CrossRef]
- Saleh, N.M.; El-Gaby, M.S.A.; El-Adl, K.; El-Sattar, N.E.A.A. Design, green synthesis, molecular docking and anticancer evaluations of diazepam bearing sulfonamide moieties as VEGFR-2 inhibitors. Bioorg. Chem. 2020, 104, 104350. [Google Scholar] [CrossRef]
- El-Adl, K.; El-Helby, A.A.; Ayyad, R.R.; Mahdy, H.A.; Khalifa, M.M.; Elnagar, H.A.; Mehany, A.B.M.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; et al. Design, synthesis, and anti-proliferative evaluation of new quinazolin-4(3H)-ones as potential VEGFR-2 inhibitors. Bioorg. Med. Chem. 2020, 29, 115872. [Google Scholar] [CrossRef]
- El-Helby, A.A.; Sakr, H.; Eissa, I.H.; Al-Karmalawy, A.A.; El-Adl, K. Benzoxazole/benzothiazole-derived VEGFR-2 inhibitors: Design, synthesis, molecular docking, and anticancer evaluations. Arch. Pharm. 2019, 352, 1900178. [Google Scholar] [CrossRef]
- El-Helby, A.-G.A.; Ayyad, R.R.A.; Sakr, H.; El-Adl, K.; Ali, M.M.; Khedr, F. Design, Synthesis, Molecular Docking, and Anticancer Activity of Phthalazine Derivatives as VEGFR-2 Inhibitors. Arch. Pharm. 2017, 350, 1700240. [Google Scholar] [CrossRef]
- El-Helby, A.-G.A.; Ayyad, R.R.A.; Sakr, H.; El-Adl, K.; Ali, M.M.; Khedr, F. Design, Synthesis, In Vitro Anti-cancer Activity, ADMET Profile and Molecular Docking of Novel Triazolo[3,4-a]phthalazine Derivatives Targeting VEGFR-2 Enzyme. Anti-cancer Agents Med. Chem. 2018, 18, 1184. [Google Scholar] [CrossRef]
- El-Helby, A.A.; Sakr, H.; Eissa, I.H.; Abulkhair, H.; Al-Karmalawy, A.A.; El-Adl, K. Design, synthesis, molecular docking, and anticancer activity of benzoxazole derivatives as VEGFR-2 inhibitors. Arch. Pharm. 2019, 352, 1900113. [Google Scholar] [CrossRef] [PubMed]
- Turky, A.; Bayoumi, A.H.; Sherbiny, F.F.; El-Adl, K.; Abulkhair, H.S. Unravelling the anticancer potency of 1,2,4-triazole-N-arylamide hybrids through inhibition of STAT3: Synthesis and in silico mechanistic studies. Mol. Divers. 2021, 25, 403. [Google Scholar] [CrossRef] [PubMed]
- El-Helby, A.G.A.; Ayyad, R.R.; Sakr, H.M.; Abdelrahim, A.S.; El-Adl, K.; Sherbiny, F.S.; Eissa, I.H.; Khalifa, M.M. Design, synthesis, molecular modeling and biological evaluation of novel 2,3-dihydrophthalazine-1,4-dione derivatives as potential anticonvulsant agents. J. Mol. Struct. 2017, 1130, 333. [Google Scholar] [CrossRef]
- El-Shershaby, M.H.; El-Gamal, K.M.; Bayoumi, A.H.; El-Adl, K.; Ahmed, H.E.A.; Abulkhair, H.S. Synthesis, antimicrobial evaluation, DNA gyrase inhibition, and in silico pharmacokinetic studies of novel quinoline derivatives. Arch. Pharm. 2021, 354, e2000277. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Jeong, K.-W.; Lee, Y.; Song, J.Y.; Kim, M.S.; Lee, G.S.; Kim, Y. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur. J. Med. Chem. 2010, 45, 542–547. [Google Scholar] [CrossRef]
- Machado, V.A.; Peixoto, D.; Costa, R.; Froufe, H.J.; Calhelha, R.C.; Abreu, R.M.; Ferreira, I.C.; Soares, R.; Queiroz, M.-J.R. Synthesis, antiangiogenesis evaluation and molecular docking studies of 1-aryl-3-[(thieno[3,2-b] pyridin-7-yl- thio)phenyl]ureas: Discovery of a new substitution pattern for type II VEGFR-2 Tyr kinase inhibitors. Bioorg. Med. Chem. 2015, 23, 6497–6509. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, J.; Hulme, C.; Hurley, L.H. The design, synthesis, and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: A structural analysis of the binding interactions of Gleevec®, Nexavar®, and BIRB-796. Bioorg. Med. Chem. 2010, 18, 5738–5748. [Google Scholar] [CrossRef]
- Garofalo, A.; Goossens, L.; Six, P.; Lemoine, A.; Ravez, S.; Farce, A.; Depreux, P. Impact of aryloxy-linked quinazolines: A novel series of selective VEGFR-2 receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2106–2112. [Google Scholar] [CrossRef]
- Aziz, M.A.; Serya, R.A.; Lasheen, D.S.; Abdel-Aziz, A.K.; Esmat, A.; Mansour, A.M.; Singab, A.N.B.; Abouzid, K.A. Discovery of Potent VEGFR-2 Inhibitors based on Furopyrimidine and Thienopyrimidne Scaffolds as Cancer Targeting Agents. Sci. Rep. 2016, 6, 24460. [Google Scholar] [CrossRef] [Green Version]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2007, 1771, 915–925. [Google Scholar] [CrossRef]
- Groop, L.; Neugebauer, G. Clinical Pharmacology of Sulfonylureas. In Oral Antidiabetics; Kuhlmann, J., Puls, W., Eds.; Springer: Berlin/Heidelberg, Germany, 1996; 199p. [Google Scholar]
- Ibrahim, M.-K.; Eissa, I.H.; Alesawy, M.S.; Metwaly, A.M.; Radwan, M.M.; ElSohly, M.A. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of quinazolin-4(3H)-one derivatives as potential PPARc and SUR agonists. Bioorg. Med. Chem. 2017, 25, 4723–4744. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, W.A.; Heidelberger, M. Methods for the acylation of aromatic amino compounds and ureas, with especial reference to chloroacetylation. J. Am. Chem. Soc. 1917, 39, 1439–1447. [Google Scholar] [CrossRef] [Green Version]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazreen, S. Design, synthesis, and molecular docking studies of thiazolidinediones as PPAR-γ agonists and thymidylate synthase inhibitors. Arch. Pharm. 2021, 354, e2100021. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Abou-Seri, S.M.; Eldehna, W.M.; Ali, M.M.; Abou El Ella, D.A. 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: Synthesis and in vitro biological evaluation. Eur. J. Med. Chem. 2016, 107, 165–179. [Google Scholar] [CrossRef]
- Saleh, N.M.; Abdel-Rahman, A.A.-H.; Omar, A.M.; Khalifa, M.M.; El-Adl, K. Pyridine-derived VEGFR-2 inhibitors: Rational design, synthesis, anticancer evaluations, in silico ADMET profile, and molecular docking. Arch. Pharm. 2021, 354, e2100085. [Google Scholar] [CrossRef]
- Jameson, D.M.; Mocz, G. Fluorescence polarization/anisotropy approaches to study protein-ligand interactions. In Protein-Ligand Interactions: Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2005; pp. 301–322. [Google Scholar]
- Schmidt, S.D.; Mazzella, M.J.; Nixon, R.A.; Mathews, P.M. Aβ measurement by enzyme-linked immunosorbent assay. In Amyloid Proteins: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2012; pp. 507–527. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Beig, A.; Agbaria, R.; Dahan, A. Oral Delivery of Lipophilic Drugs: The Tradeoff between Solubility Increase and Permeability Decrease When Using Cyclodextrin-Based Formulations. PLoS ONE 2013, 8, e68237. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ∆G [kcal mol−1] | Compound | ∆G [kcal mol−1] |
|---|---|---|---|
| 3 | −90.00 | 6b | −126.72 |
| 4a | −92.62 | 6c | −130.36 |
| 4b | −98.37 | 7a | −125.48 |
| 5a | −115.75 | 7b | −129.68 |
| 5b | −121.80 | 7c | −138.79 |
| 5c | −125.96 | Sorafenib | −110.12 |
| 6a | −118.64 |
| Compound | ∆G [kcal mol−1] | Compound | ∆G [kcal mol−1] |
|---|---|---|---|
| 3 | −84.12 | 6b | −118.03 |
| 4a | −92.97 | 6c | −128.20 |
| 4b | −91.92 | 7a | −125.77 |
| 5a | −108.58 | 7b | −125.42 |
| 5b | −111.97 | 7c | −132.78 |
| 5c | −119.97 | Rosiglitazone | −94.38 |
| 6a | −115.47 |
| Compound | IC50 (µM) a | ||||
|---|---|---|---|---|---|
| HepG2 | HCT116 | MCF-7 | VERO | VEGFR-2 | |
| 3 | 58.55 ± 5.1 | 61.48 ± 5.1 | 60.18 ± 5.1 | b NT | 0.93± 0.06 |
| 4a | 52.87 ± 5.1 | 55.12 ± 5.1 | 54.99 ± 5.1 | b NT | 0.92 ± 0.06 |
| 4b | 48.56 ± 5.1 | 57.87 ± 5.1 | 62.43 ± 5.1 | b NT | 0.89 ± 0.06 |
| 5a | 24.49 ± 2.2 | 40.11 ± 2.2 | 28.79 ± 2.2 | b NT | 0.46 ± 0.05 |
| 5b | 21.99 ± 2.0 | 25.68 ± 2.0 | 23.24 ± 2.0 | b NT | 0.44 ± 0.05 |
| 5c | 20.75 ± 2.6 | 23.56 ± 2.6 | 24.59 ± 2.6 | b NT | 0.44 ± 0.05 |
| 6a | 14.16 ± 2.3 | 17.65 ± 2.3 | 16.47 ± 2.3 | 48.31 ± 0.22 | 0.17 ± 0.02 |
| 6b | 10.67 ± 1.6 | 13.78 ± 1.2 | 12.95 ± 1.2 | 40.88 ± 0.22 | 0.15 ± 0.02 |
| 6c | 8.99 ± 1.2 | 7.11 ± 1.7 | 8.15 ± 1.6 | 49.26 ± 0.22 | 0.08 ± 0.02 |
| 7a | 12.05 ± 1.5 | 16.79 ± 1.5 | 16.66 ± 1.5 | 60.12 ± 0.18 | 0.14 ± 0.02 |
| 7b | 9.65 ± 1.7 | 13.48 ± 1.6 | 12.89 ± 1.7 | 52.61 ± 0.22 | 0.11 ± 0.02 |
| 7c | 8.82 ± 1.9 | 5.77 ± 1.9 | 7.78 ± 1.9 | 68.25 ± 0.21 | 0.08 ± 0.02 |
| Sorafenib | 9.18 ± 0.6 | 5.47 ± 0.3 | 7.26 ± 0.3 | b NT | 0.10 ± 0.02 |
| Doxorubicin | 7.94 ± 0.6 | 8.07 ± 0.8 | 6.75 ± 0.4 | b NT | b NT |
| Comp. | In Vitro a | |
|---|---|---|
| IC50 (µM) b PPARγ Binding Affinity | EC50 (µM) c Insulin Secreting Activity | |
| 5a | 0.393 | 1.20 |
| 5b | 0.377 | 1.13 |
| 5c | 0.360 | 1.00 |
| 6a | 0.323 | 0.87 |
| 6b | 0.308 | 0.78 |
| 6c | 0.300 | 0.70 |
| 7a | 0.320 | 0.81 |
| 7b | 0.305 | 0.75 |
| 7c | 0.296 | 0.70 |
| Rosiglitazone | 0.292 | NT d |
| Glimiperide | NT d | 0.73 |
| Parameter | 6c | 7b | 7c | Rosiglit. | Sorafenib |
|---|---|---|---|---|---|
| Physicochemical properties | |||||
| Molecular Weight | 558.69 | 566.67 | 572.72 | 357.43 | 464.82 |
| LogP | 3.8471 | 4.34532 | 4.15552 | 2.4909 | 5.5497 |
| Rotatable Bonds | 10 | 10 | 10 | 7 | 9 |
| Acceptors | 5 | 5 | 5 | 4 | 7 |
| Donors | 3 | 3 | 3 | 1 | 3 |
| TPSA | 190.45 Å2 | 190.45 Å2 | 190.45 Å2 | 96.83 Å2 | 92.35 Å2 |
| Absorption | |||||
| Water solubility | −4.149 | −3.637 | −4.204 | −3.841 −5.284 | −4.822 |
| Caco2 permeability | 0.714 | 0.699 | 0.623 | 0.934 | 0.689 |
| Human Intest. absorption | 74.229 | 76.168 | 74.687 | 95.437 | 89.043 |
| Skin Permeability | −2.738 | −2.735 | −2.738 | 95.437 | −2.767 |
| Substrate for P-glycoprotein | + | + | + | - | + |
| Inhibitor of P-glycoprotein I | + | + | + | - | + |
| Inhibitor of P-glycoprotein II | + | + | + | - | + |
| Distribution | |||||
| VDss (human) | −0.192 | −0.008 | −0.168 | −0.264 | −0.29 |
| Human unbound fraction | 0.07 | 0.064 | 0.06 | 0.188 | 0.065 |
| Permeability throughout BBB | −1.618 | −1.594 | −1.648 | −0.618 | −1.684 |
| Permeability to CNS | −2.529 | −2.254 | −2.454 | −2.72 | −2.007 |
| Metabolism | |||||
| CYP2D6 substrate | - | - | - | - | - |
| CYP3A4 substrate | + | + | + | + | + |
| Inhibition of CYP3A4 | - | + | - | - | + |
| Inhibition of CYP2D6 | - | - | - | - | - |
| Inhibition of CYP2C9 | - | + | - | - | + |
| Inhibition of CYP2C19 | - | + | - | - | + |
| Inhibition of CYP1A2 | - | + | - | - | + |
| Excretion | |||||
| Clearance | −0.538 | −0.670 | −0.594 | 0.104 | −0.219 |
| Renal OCT2 substrate | - | - | - | - | - |
| Toxicity | |||||
| AMES toxicity | - | - | - | - | - |
| Human Max. tolerated dose | −0.512 | 0.558 | −0.546 | −0.460 | 0.549 |
| Inhibitor of hERG I | - | - | - | - | - |
| hERG II inhibitor | + | + | + | - | + |
| Acute Toxic activity (LD50) | 2.708 | 2.306 | 2.773 | 2.877 | 2.538 |
| Chronic Toxic activity (LOAEL) | 0.826 | 1.465 | 0.739 | 1.313 | 1.198 |
| Hepatotoxic effect | + | + | + | + | + |
| Skin Sensitization | - | - | - | - | - |
| T. Pyriformis toxicity | 0.337 | 0.29 | 0.337 | 1.194 | 0.383 |
| Minnow toxic activity | 0.214 | 1.337 | 0.543 | 0.609 | 0.189 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelgawad, M.A.; El-Adl, K.; El-Hddad, S.S.A.; Elhady, M.M.; Saleh, N.M.; Khalifa, M.M.; Khedr, F.; Alswah, M.; Nayl, A.A.; Ghoneim, M.M.; et al. Design, Molecular Docking, Synthesis, Anticancer and Anti-Hyperglycemic Assessments of Thiazolidine-2,4-diones Bearing Sulfonylthiourea Moieties as Potent VEGFR-2 Inhibitors and PPARγ Agonists. Pharmaceuticals 2022, 15, 226. https://doi.org/10.3390/ph15020226
Abdelgawad MA, El-Adl K, El-Hddad SSA, Elhady MM, Saleh NM, Khalifa MM, Khedr F, Alswah M, Nayl AA, Ghoneim MM, et al. Design, Molecular Docking, Synthesis, Anticancer and Anti-Hyperglycemic Assessments of Thiazolidine-2,4-diones Bearing Sulfonylthiourea Moieties as Potent VEGFR-2 Inhibitors and PPARγ Agonists. Pharmaceuticals. 2022; 15(2):226. https://doi.org/10.3390/ph15020226
Chicago/Turabian StyleAbdelgawad, Mohamed A., Khaled El-Adl, Sanadelaslam S. A. El-Hddad, Mostafa M. Elhady, Nashwa M. Saleh, Mohamed M. Khalifa, Fathalla Khedr, Mohamed Alswah, AbdElAziz A. Nayl, Mohammed M. Ghoneim, and et al. 2022. "Design, Molecular Docking, Synthesis, Anticancer and Anti-Hyperglycemic Assessments of Thiazolidine-2,4-diones Bearing Sulfonylthiourea Moieties as Potent VEGFR-2 Inhibitors and PPARγ Agonists" Pharmaceuticals 15, no. 2: 226. https://doi.org/10.3390/ph15020226