
Investigating the Diversity of Wolbachia across the Spiny Ants (Polyrhachis)

Abstract
:1. Introduction
2. Materials and Methods
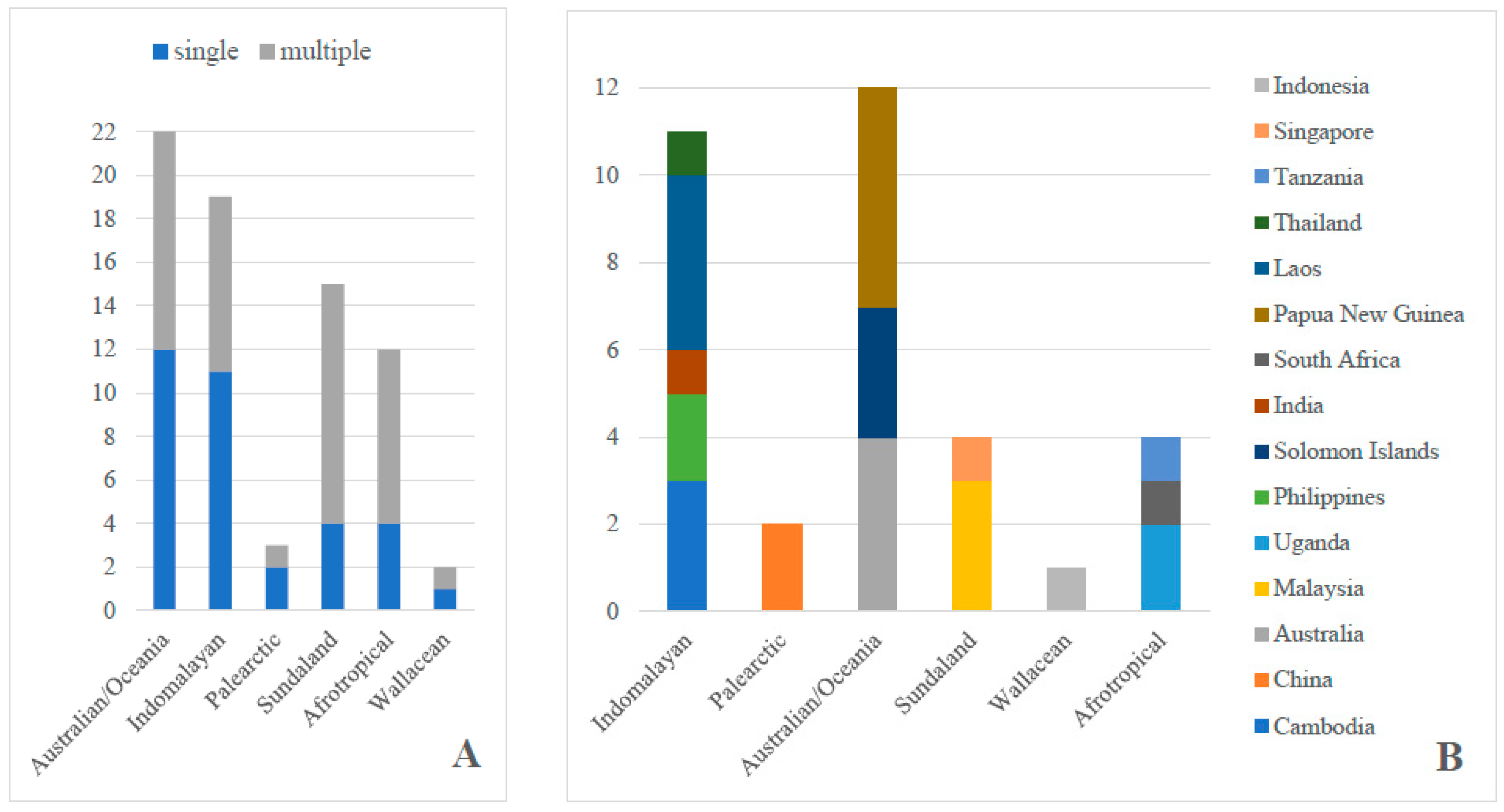
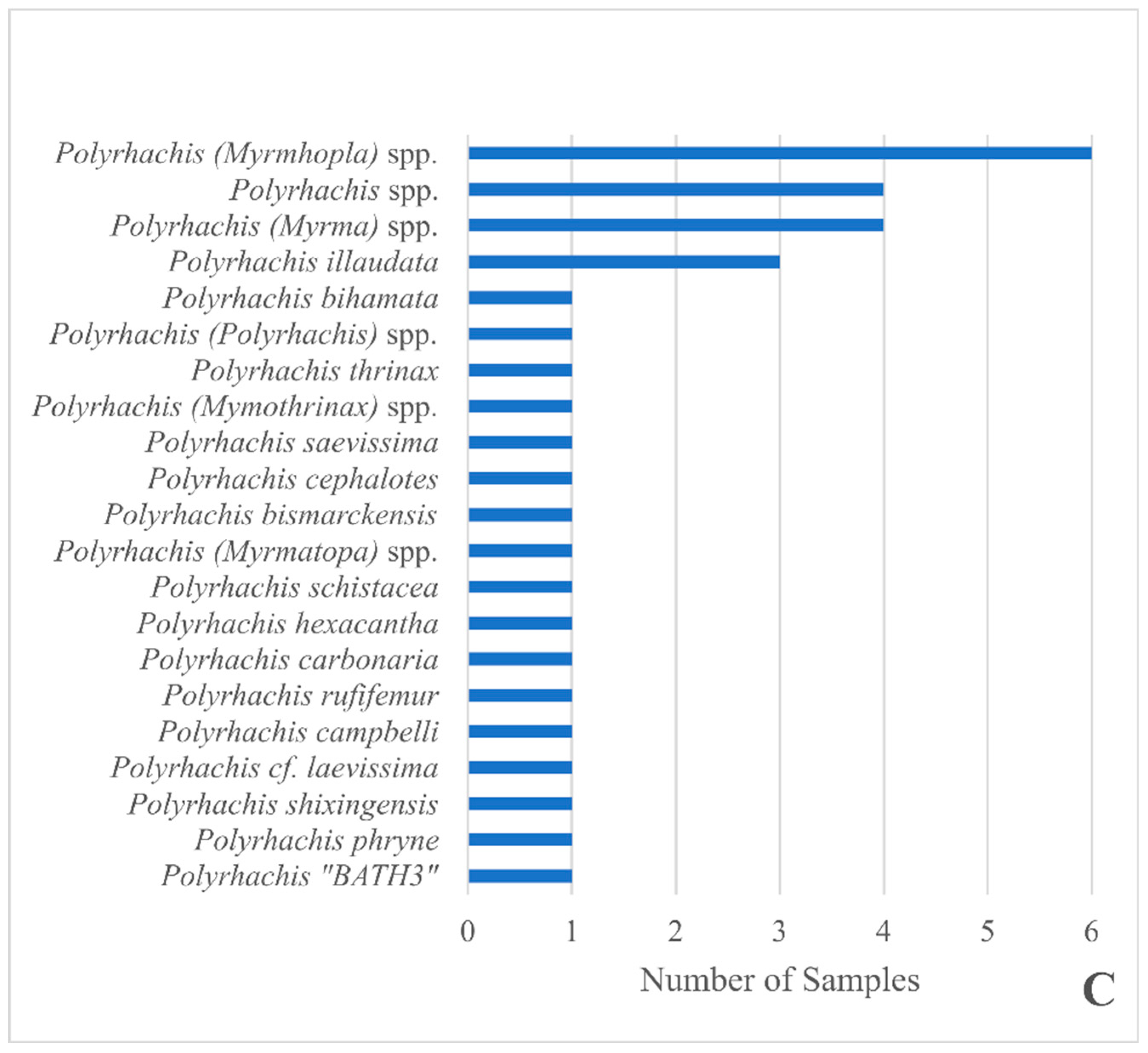
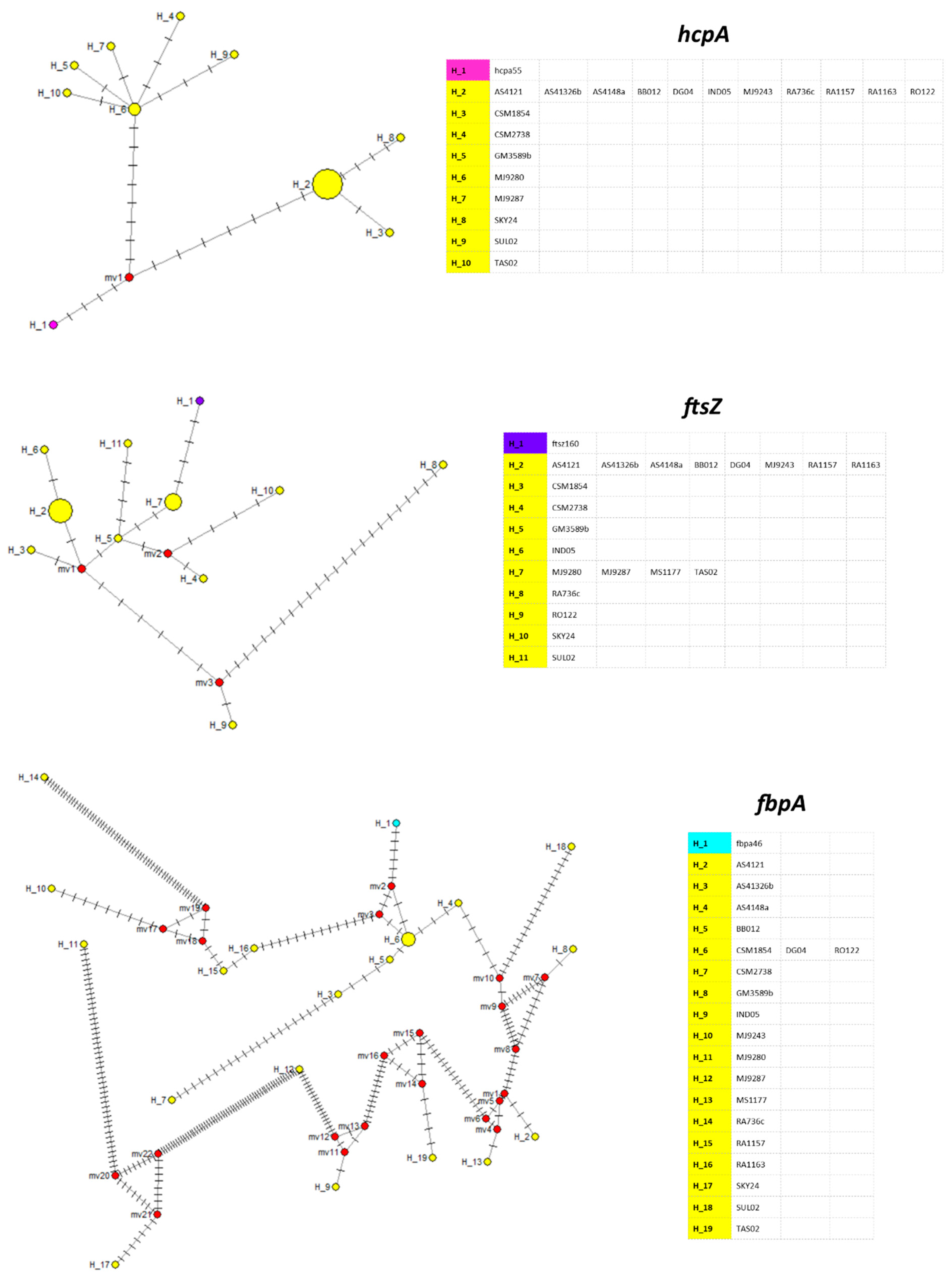
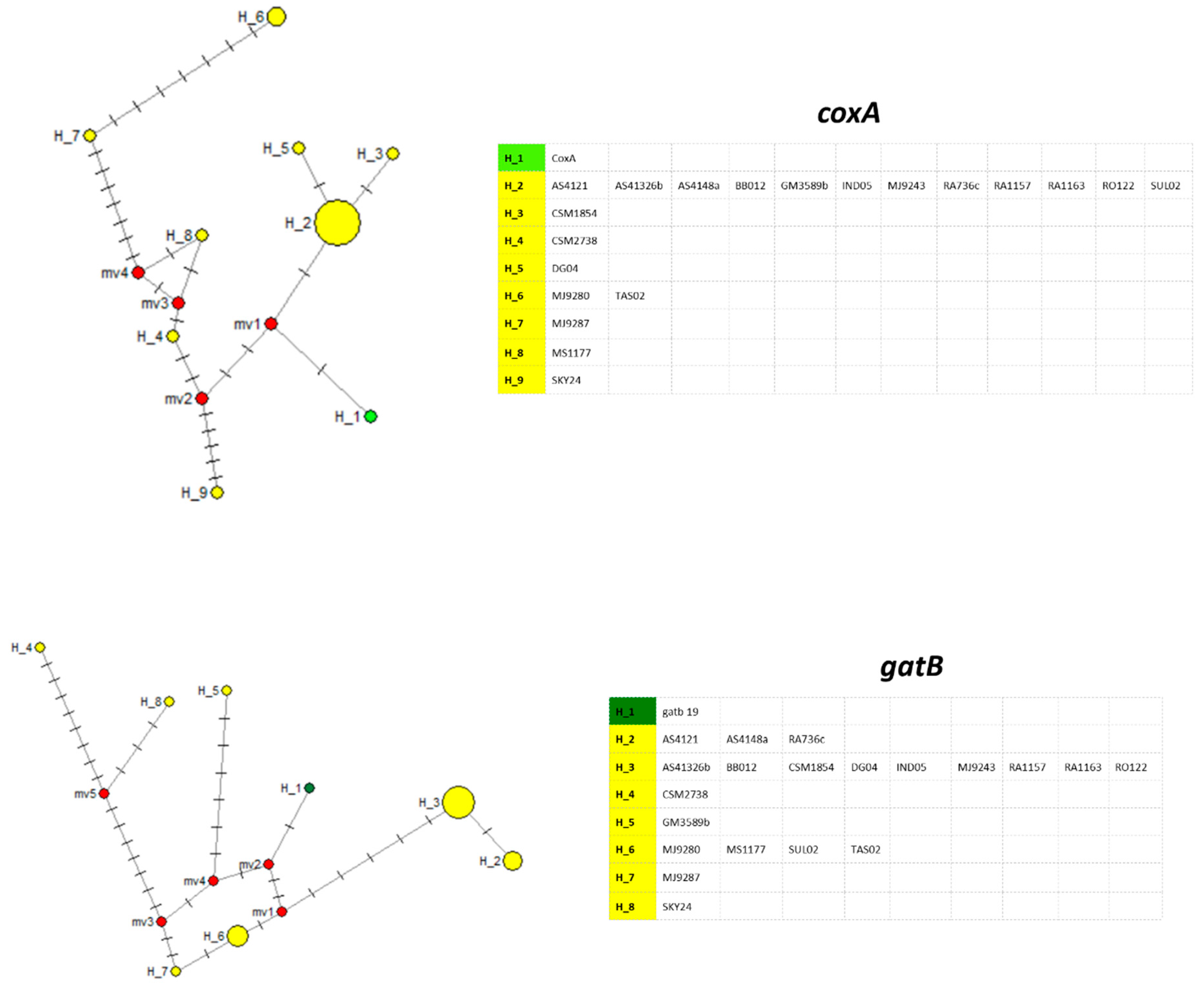
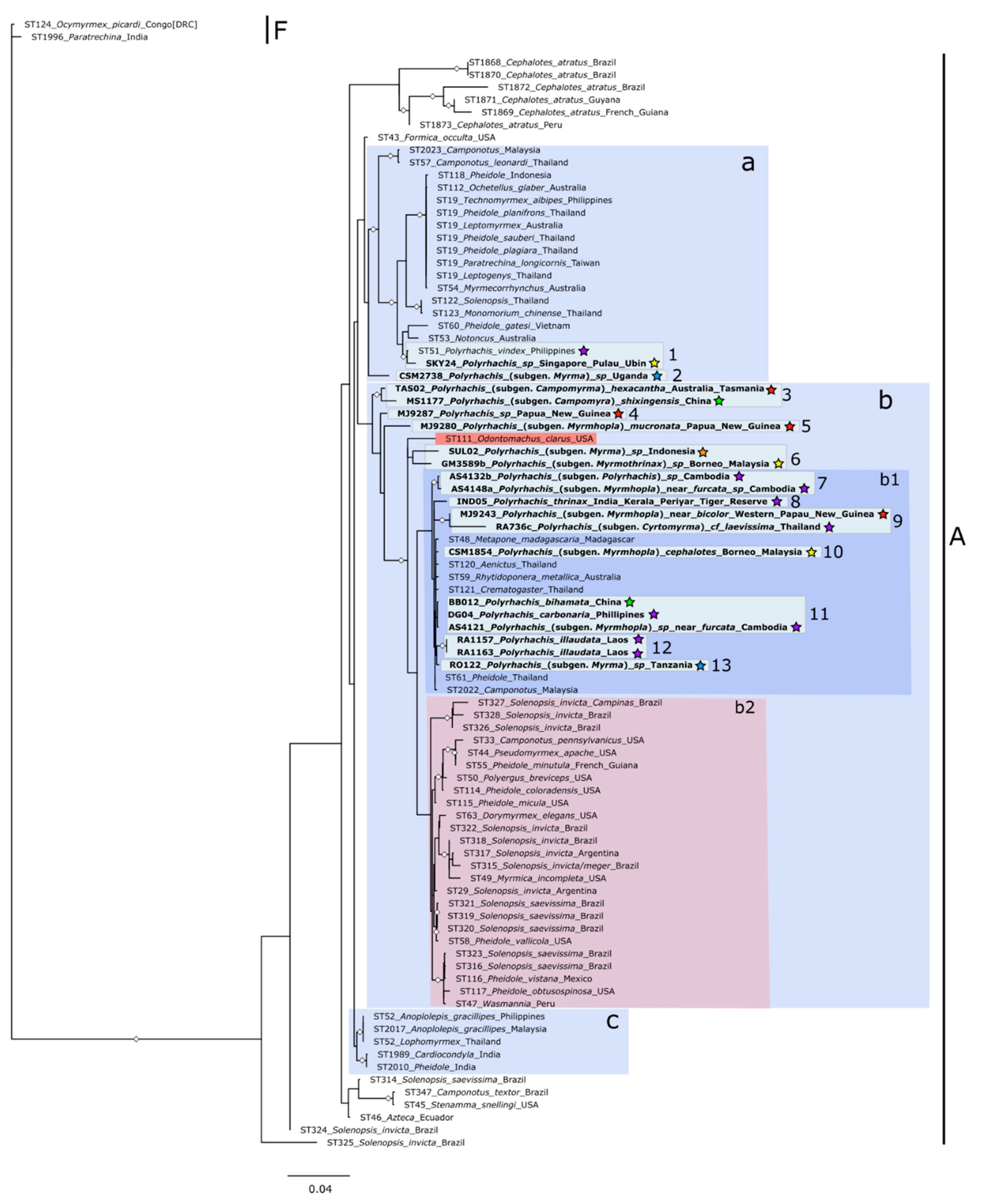
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moreau, C.S. Symbioses among ants and microbes. Curr. Opin. Insect Sci. 2020, 39, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sanders, J.G.; Łukasik, P.; D’Amelio, C.L.; Millar, J.S.; Vann, D.R.; Lan, Y.; Newton, J.A.; Schotanus, M.; Kronauer, D.J.; et al. Herbivorous turtle ants obtain essential nutrients from a conserved nitrogen-recycling gut microbiome. Nat. Commun. 2018, 9, 964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, J.A. The ants (Hymenoptera: Formicidae) are unique and enigmatic hosts of prevalent Wolbachia (Alphaproteobacteria) symbionts. Myrmecol. News Myrmecol. News 2012, 16, 7–23. [Google Scholar]
- Werren, J.H.; Windsor, D.M. Wolbachia infection frequencies in insects: Evidence of a global equilibrium? Proc. R. Soc. B Biol. Sci. 2000, 267, 1277–1285. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, M.O.; Vieira, A.S.; Pereira, M.C.; Moreau, C.S.; Bueno, O.C. Transovarian Transmission of Blochmannia and Wolbachia Endosymbionts in the Neotropical Weaver Ant Camponotus textor (Hymenoptera, Formicidae). Curr. Microbiol. 2018, 75, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Baldo, L.; Dunning Hotopp, J.C.; Jolley, K.A.; Bordenstein, S.R.; Biber, S.A.; Choudhury, R.R.; Hayashi, C.; Maiden, M.C.J.; Tettelin, H.; Werren, J.H. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl. Environ. Microbiol. 2006, 72, 7098–7110. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Linksvayer, T.A. Wolbachia-infected ant colonies have increased reproductive investment and an accelerated life cycle. J. Exp. Biol. 2020, 223, jeb220079. [Google Scholar] [CrossRef]
- Tseng, S.P.; Wetterer, J.K.; Suarez, A.V.; Lee, C.Y.; Yoshimura, T.; Shoemaker, D.W.; Yang, C.C.S. Genetic Diversity and Wolbachia Infection Patterns in a Globally Distributed Invasive Ant. Front. Genet. 2019, 10, 838. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, M.O.; Moreau, C.S. The evolution and biogeography of wolbachia in ants (Hymenoptera: Formicidae). Diversity 2020, 12, 426. [Google Scholar] [CrossRef]
- Ramalho, M.D.O.; Kim, Z.; Wang, S.; Moreau, C.S. Wolbachia across Social Insects: Patterns and Implications. Ann. Entomol. Soc. Am. 2021, 114, 206–218. [Google Scholar] [CrossRef]
- Baldo, L.; Werren, J.H. Revisiting Wolbachia supergroup typing based on WSP: Spurious lineages and discordance with MLST. Curr. Microbiol. 2007, 55, 81–87. [Google Scholar] [CrossRef]
- Gerth, M. Classification of Wolbachia (Alphaproteobacteria, Rickettsiales): No evidence for a distinct supergroup in cave spiders. Infect. Genet. Evol. 2016, 43, 378–380. [Google Scholar] [CrossRef] [PubMed]
- Glowska, E.; Dragun-Damian, A.; Dabert, M.; Gerth, M. New Wolbachia supergroups detected in quill mites (Acari: Syringophilidae). Infect. Genet. Evol. 2015, 30, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.A.; Goldman-Huertas, B.; Moreau, C.S.; Baldo, L.; Stahlhut, J.K.; Werren, J.H.; Pierce, N.E. Specialization and geographic isolation among Wolbachia symbionts from ants and lycaenid butterflies. Evolution 2009, 63, 624–640. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Rousset, F.; O’Neill, S. Phylogeny and PCR–based classification of Wolbachia strains using wsp gene sequences. Proc. R. Soc. London B Biol. Sci. 1998, 265, 509–515. [Google Scholar] [CrossRef] [Green Version]
- Baldo, L.; Lo, N.; Werren, J.H. Mosaic nature of the wolbachia surface protein. J. Bacteriol. 2005, 187, 5406–5418. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Norris, D.E.; Rasgon, J.L. Distribution and molecular characterization ofWolbachia endosymbionts and larial nematodes inMaryland populations of the lone star tick (Amblyomma americanum). FEMS Microbiol. Ecol. 2011, 77, 50–56. [Google Scholar] [CrossRef]
- Salunke, B.K.; Salunkhe, R.C.; Dhotre, D.P.; Walujkar, S.A.; Khandagale, A.B.; Chaudhari, R.; Chandode, R.K.; Ghate, H.V.; Patole, M.S.; Werren, J.H.; et al. Determination of Wolbachia diversity in butterflies from Western Ghats, India, by a multigene approach. Appl. Environ. Microbiol. 2012, 78, 4458–4467. [Google Scholar] [CrossRef] [Green Version]
- Kelly, M.; Price, S.L.; de Oliveira Ramalho, M.; Moreau, C.S. Diversity of Wolbachia Associated with the Giant Turtle Ant, Cephalotes atratus. Curr. Microbiol. 2019, 76, 1330–1337. [Google Scholar] [CrossRef]
- Ramalho, M.O.; Martins, C.; Silva, L.M.R.; Martins, V.G.; Bueno, O.C. Intracellular symbiotic bacteria of Camponotus textor, Forel (Hymenoptera, Formicidae). Curr. Microbiol. 2017, 74, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Martins, C.; de Oliveira Ramalho, M.; Silva, L.M.R.; de Souza, R.F.; Bueno, O.C. New Strains of Wolbachia Unveiling the Complexity of This Symbiotic Interaction in Solenopsis (Hymenoptera: Formicidae). Microbiol. Res. 2021, 12, 40. [Google Scholar] [CrossRef]
- Guénard, B.; Weiser, M.D.; Gómez, K.; Narula, N.; Economo, E.P. The Global Ant Biodiversity Informatics (GABI) database: Synthesizing data on the geographic distribution of ant species (Hymenoptera: Formicidae). Myrmecol. News 2017, 24, 83–89. [Google Scholar]
- Blanchard, B.D.; Moreau, C.S. Defensive spines are associated with large geographic range but not diversification in spiny ants (Hymenoptera: Formicidae: Polyrhachis). Syst. Entomol. 2022, 1–13. [Google Scholar] [CrossRef]
- Mezger, D.; Moreau, C.S. Out of South-East Asia: Phylogeny and biogeography of the spiny ant genus Polyrhachis Smith (Hymenoptera: Formicidae). Syst. Entomol. 2016, 41, 369–378. [Google Scholar] [CrossRef]
- Blanchard, B.D.; Nakamura, A.; Cao, M.; Chen, S.T.; Moreau, C.S. Spine and dine: A key defensive trait promotes ecological success in spiny ants. Ecol. Evol. 2020, 10, 5852–5863. [Google Scholar] [CrossRef]
- Robson, S.K.A.; Kohout, R.J.; Beckenbach, A.T.; Moreau, C.S. Evolutionary transitions of complex labile traits: Silk weaving and arboreal nesting in Polyrhachis ants. Behav. Ecol. Sociobiol. 2015, 69, 449–458. [Google Scholar] [CrossRef]
- van Zweden, J.S.; Carew, M.E.; Henshaw, M.T.; Robson, S.K.A.; Crozier, R.H. Social and genetic structure of a supercolonial weaver ant, Polyrhachis robsoni, with dimorphic queens. Insectes Soc. 2007, 54, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Wernegreen, J.J.; Kauppinen, S.N.; Brady, S.G.; Ward, P.S. One nutritional symbiosis begat another: Phylogenetic evidence that the ant tribe Camponotini acquired Blochmannia by tending sap-feeding insects. BMC Evol. Biol. 2009, 9, 292. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, M.O.; Bueno, O.C.; Moreau, C.S. Microbial composition of spiny ants (Hymenoptera: Formicidae: Polyrhachis) across their geographic range. BMC Evol. Biol. 2017, 17, 96. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, M.O.; Bueno, O.C.; Moreau, C.S. Species-specific signatures of the microbiome from Camponotus and Colobopsis ants across developmental stages. PLoS ONE 2017, 12, e0187461. [Google Scholar] [CrossRef] [Green Version]
- Braig, H.R.; Zhou, W.; Dobson, S.L.; O’Neill, S.L. Cloning and characterization of a gene encoding the major surface protein of the bacterial endosymbiont Wolbachia pipientis. J. Bacteriol. 1998, 180, 2373–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, C.S. A practical guide to DNA extraction, PCR, and gene-based DNA sequencing in insects. Halteres 2014, 5, 32–42. [Google Scholar]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications [version 1; referees: 2 approved]. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Chernomor, O.; Von Haeseler, A.; Minh, B.Q. Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B. The vegan package. Community Ecol. 2007, 10, 631–637. [Google Scholar]
- Salunke, B.K.; Salunkhe, R.C.; Dhotre, D.P.; Khandagale, A.B.; Walujkar, S.A.; Kirwale, G.S.; Ghate, H.V.; Patole, M.S.; Shouche, Y.S. Diversity of Wolbachia in Odontotermes spp. (Termitidae) and Coptotermes heimi (Rhinotermitidae) using the multigene approach. FEMS Microbiol. Lett. 2010, 307, 55–64. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | MLST Allele Number | ST | Country | Polyrhachis | ||||
|---|---|---|---|---|---|---|---|---|
| coxA | fbpA | ftsZ | gatB | hcpA | Host Species | |||
| AS4121 | 2 | 51 | 45 | 20 | 47 | 61 * | Cambodia | P. (Myrmhopla) sp. |
| AS4132b | 2 | 51 | 45 | 20 | 47 | 61 * | Cambodia | P. (Polyrhachis) sp. |
| AS4148a | 2 | 51 | 45 | 20 | 47 | 61 * | Cambodia | P. (Myrmhopla) sp. |
| BB012 | 2 | 51 | 45 | 20 | 47 | 61 * | China | P. bihamata |
| CSM1854 | 2 | 51 | 45 | 20 | 47 | 61 * | Malaysia | P. cephalotes |
| CSM2738 | 33 | 61 | 47 | 34 | 195 | ** | Uganda | P. (Myrma) sp. |
| DG04 | 2 | 51 | 45 | 20 | 47 | 61 * | Philippines | P. carbonaria |
| GM3589b | 2 | 356 | 258 | 22 | 343 | 52 * | Malaysia | P. (Mymothrinax) sp. |
| IND05 | 2 | 52 | 45 | 20 | 47 | 61 | India | P. thrinax |
| MJ9243 | 2 | 51 | 45 | 20 | 47 | 61 * | Papua New Guinea | P. (Myrmhopla) sp. |
| MJ9280 | 33 | 465 | 17 | 3 | 343 | ** | Papua New Guinea | P. (Myrmhopla) sp. |
| MJ9287 | 33 | 463 | 17 | 130 | 343 | ** | Papua New Guinea | Polyrhachis sp. |
| MS1177 | 33 | 463 | 17 | 3 | 343 | ** | China | P. shixingensis |
| RA1157 | 2 | 51 | 45 | 20 | 47 | 61 * | Laos | P. illaudata |
| RA1163 | 2 | 51 | 45 | 20 | 47 | 61 * | Laos | P. illaudata |
| RA736c | 2 | 51 | 45 | 20 | 47 | 61 * | Thailand | P. cf. laevissima |
| RO122 | 2 | 51 | 45 | 20 | 47 | 61 * | Tanzania | P. (Myrma) sp. |
| SKY24 | 32 | 48 | 6 | 57 | 50 | 51 * | Singapore | Polyrhachis sp. |
| SUL02 | 296 | 97 | 258 | 3 | 343 | ** | Indonesia | P. (Myrma) sp. |
| TAS02 | 33 | 277 | 17 | 3 | 343 | 481 * | Australia | P. hexacantha |
| CSM0655 | 32 | - | 6 | 57 | 50 | - | Australia | P. rufifemur |
| DG11 | 2 | - | 258 | 22 | 343 | - | Philippines | P. saevissima |
| EMS2584 | 218 | 6 | - | 158 | 141 | - | Solomon Islands | P. campbelli |
| EMS2617 | 33 | - | 17 | 3 | 343 | - | Solomon Islands | P. bismarckensis |
| FH1101 | 2 | - | 261 | 20 | 47 | - | Uganda | P. (Myrma) sp. |
| GM894 | 32 | - | 6 | 57 | 50 | - | Malaysia | P. (Myrmhopla) sp. |
| KATE02 | - | - | - | - | - | - | South Africa | P. schistacea |
| MJ8291 | - | - | - | - | - | - | Papua New Guinea | Polyrhachis sp. |
| MJ9286 | 109 | - | 261 | 191 | 83 | - | Papua New Guinea | Polyrhachis sp. |
| RA0755 | 32 | - | - | 57 | 50 | - | Australia | Polyrhachis “BATH3” |
| RA0784 | 33 | - | 17 | 3 | 343 | - | Solomon Islands | P. (Myrmatopa) sp. |
| RA1158 | 2 | - | 258 | 182 | 343 | - | Laos | P. (Myrmhopla) sp. |
| RA1160 | 2 | - | 45 | 20 | 47 | - | Laos | P. illaudata |
| TAS03 | 33 | - | 17 | 3 | 343 | - | Australia | P. phryne |
| Sample ID | Species | Country | Sample ID | Species | Country |
|---|---|---|---|---|---|
| DG06 | (Polyrhachis (Myrmatopa) sp. | Phillipines | RA0766 | Polyrhachis flavibasis | Australia |
| ISR_06 | Polyrhachis (Myrma) sp. | Thailand | SUL02 | Polyrhachis (Myrma) sp. 1 | Indonesia |
| GM 894 | Polyrhachis (Myrmhopla) sp. 2 | Malaysia | SKY20 | Polyrhachis sp. | Singapore |
| GM3990 | Polyrhachis (Myrmhopla) sp. 4 | Malaysia | SL_28_2 | Polyrhachis illaudata | Malaysia |
| GM3589b | Polyrhachis (Myrmothrinax) sp. | Malaysia | SKY24 | Polyrhachis sp. | Singapore |
| AS4132a | Polyrhachis (Polyrhachis) sp. | Cambodia | LEA04 | Polyrachis schistaceae | Mozambique |
| CSM0776 | Polyrhachis abbreviata | Australia | MS1177 | Polyrhachis shixingensis | China |
| DG10 | Polyrhachis armata | Philippines | RA0784 | Polyrhachis sp. | Solomon Islands |
| DG14 | Polyrhachis armata | Phillipines | RA1157 | Polyrhachis illaudata | Laos |
| CSM0761 | Polyrhachis australis | Australia | MJ9286 | Polyrhachis sp. | Papua New Guinea |
| DG26 | Polyrhachis bicolor | Philippines | RA1163 | Polyrhachis illaudata | Laos |
| BB012 | Polyrhachis bihamata | China | MJ 8277 | Polyrhachis sp. | Papua New Guinea |
| CSM1806a | Polyrhachis bihamata | Malaysia | PH09 | Polyrhachis afrc_cd03 | Democratic Republic of the Congo |
| CSM1806b | Polyrhachis bihamata | Malaysia | PH11 | Polyrhachis laboriosa | Democratic Republic of the Congo |
| DG08 | Polyrhachis bihamata | Phillipines | RA0769 | Polyrhachis “chario5” | Australia |
| CSM1846 | Polyrhachis boltoni | Malaysia | PH14 | Polyrhachis gagates | South Africa |
| EMS2584 | Polyrhachis campbelli | Solomon Islands | RA736a | Polyrhachis dives-group sp. | Thailand |
| DG04 | Polyrhachis carbonaria | Phillipines | RA0765 | Polyrhachis ammon | Australia |
| CSM1854 | Polyrhachis cephalotes | Malaysia | PH15 | Polyrhachis afr_cd01 | Democratic Republic of the Congo |
| EMS2617 | Polyrhachis cf. bismarckensis | Solomon Islands | RA736c | Polyrhachis cf. laevissima | Thailand |
| CSM1841 | Polyrhachis danum | Malaysia | PH12 | Polyrhachis revoili | Democratic Republic of the Congo |
| BB28 | Polyrhachis hippomanes | China | MJ 9243 | Polyrhachis sp. near bicolor | Papua New Guinea |
| JRNG01 | Polyrhachis hookeri | Australia | TAS 02 | Polyrhachis hexacantha | Australia |
| DG03 | Polyrhachis illaudata | Phillipines | RA0755 | Polyrhachis “BATH3” | Australia |
| GM3551 | Polyrhachis illaudata | Malaysia | SKY21 | Polyrhachis nigropilosa | Singapore |
| DG25 | Polyrhachis inermis | Philippines | RA1162 | Polyrhachis illaudata | Laos |
| EMS2637 | Polyrhachis kaipi | Solomon Islands | RO 122 | Polyrhachis sp. | Tanzania |
| ISR_03 | Polyrhachis lacteipennis | Israel | SOH 02 | Polyrhachis beccari | Singapore |
| CSM1868 | Polyrhachis lepida | Malaysia | PH21 | Polyrhachis schistacea | Mozambique |
| DG16 | Polyrhachis near lilianae | Philippines | RA1158 | Polyrhachis mucronata-group sp. | Laos |
| BB48 | Polyrhachis proxima | China | MJ 9280 | Polyrhachis mucronata-group sp. | |
| CSM0655 | Polyrhachis rufifemur | Australia | MJ8280 | Polyrhachis sp. | Papua New Guinea |
| CSM0740 | Polyrhachis rufifemur | Australia | MJ9242 | Polyrhachis sexspi-sa group | Papua New Guinea |
| DG11 | Polyrhachis saevissima | Phillipines | RA1154 | Polyrhachis mucronata-group sp. | Laos |
| DG17 | Polyrhachis saevissima | Phillipines | RA1160 | Polyrhachis illaudata? | Laos |
| KATE02 | Polyrhachis schistacea | South Africa | TAS04 | Polyrhachis semipolita | Australia |
| AS4132b | Polyrhachis sp. | Cambodia | LEA05 | Polyrachis schistaceae | Mozambique |
| BB026 | Polyrhachis sp. | China | MJ 8282 | Polyrhachis sexspi-sa group | Papua New Guinea |
| CSM1860 | Polyrhachis sp. | Malaysia | PH16 | Polyrhachis latharis | Democratic Republic of the Congo |
| CSM2632 | Polyrhachis sp. | Uganda | PH22 | Polyrhachis schistacea | Tanzania |
| CSM2738 | Polyrhachis sp. | Uganda | PSW5403 | Polyrhachis andromache | Australia |
| CSM2745 | Polyrhachis sp. | Uganda | PSW6454 | Polyrhachis obesior | Malaysia |
| CSM2831 | Polyrhachis sp. | Australia | RA0735 | Polyrhachis abdominalis | Singapore |
| FH1085 | Polyrhachis sp. | Uganda | RO538 | Polyrhachis sp. | Tanzania |
| FH1101 | Polyrhachis sp. | Uganda | SKY05 | Polyrhachis frustorferi | Indonesia |
| FH205 | Polyrhachis sp. | Kenya | SKY11 | Polyrhachis lamellidens | Japan |
| FH987 | Polyrhachis sp. | Uganda | SKY17 | Polyrhachis hector | Indonesia |
| JCM120P | Polyrhachis sp. | Palau | TAS 01 | Polyrhachis hexacantha | Australia |
| JRNG02 | Polyrhachis sp. | Australia | TAS03 | Polyrhachis phryne | Australia |
| LD01 | Polyrhachis sp. | Ghana | LEA03 | Polyrachis schistaceae | Mozambique |
| AS4121 | Polyrhachis sp. near furcata | Cambodia | MJ 8263 | Polyrhachis sp. | Papua New Guinea |
| AS4148a | Polyrhachis sp. near furcata | Cambodia | MJ 8291 | Polyrhachis sp. | Papua New Guinea |
| BB_075 | Polyrhachis sp. near sixspi-sa | China | MJ9275 | Polyrhachis sp. | Papua New Guinea |
| CSM0746 | Polyrhachis thais | Australia | SL32 | Polyrhachis furcata | Malaysia |
| IND05 | Polyrhachis thrinax | India | MJ 9287 | Polyrhachis sp. | Papua New Guinea |
| CAB01 | Polyrhachis ypsilon | Malaysia |
| Partition | Gene(s) | Position in Concatenation (bp) | Length of Gene(s) in Partition (bp) | Model |
|---|---|---|---|---|
| 1 | coxA | 1–403 | 403 | HKY + F+G4 |
| 2 | fbpA | 404–840 | 437 | HKY + F+G4 |
| 3 | hcpA, ftsZ | 1651–2098, 841–1277 | 448, 437 | TIM3 + F+I+G4 |
| 4 | gatB | 1278–1650 | 373 | TIM + F+I+G4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webb, J.L.; Graber, L.C.; Ramalho, M.O.; Moreau, C.S. Investigating the Diversity of Wolbachia across the Spiny Ants (Polyrhachis). Diversity 2023, 15, 348. https://doi.org/10.3390/d15030348
Webb JL, Graber LC, Ramalho MO, Moreau CS. Investigating the Diversity of Wolbachia across the Spiny Ants (Polyrhachis). Diversity. 2023; 15(3):348. https://doi.org/10.3390/d15030348
Chicago/Turabian StyleWebb, Jenna L., Leland C. Graber, Manuela O. Ramalho, and Corrie S. Moreau. 2023. "Investigating the Diversity of Wolbachia across the Spiny Ants (Polyrhachis)" Diversity 15, no. 3: 348. https://doi.org/10.3390/d15030348