

Cholesterol Is a Regulator of CAV1 Localization and Cell Migration in Oral Squamous Cell Carcinoma

 , , , , ,
, , , , ,  , , and
, , and 
Abstract
:1. Introduction
2. Results
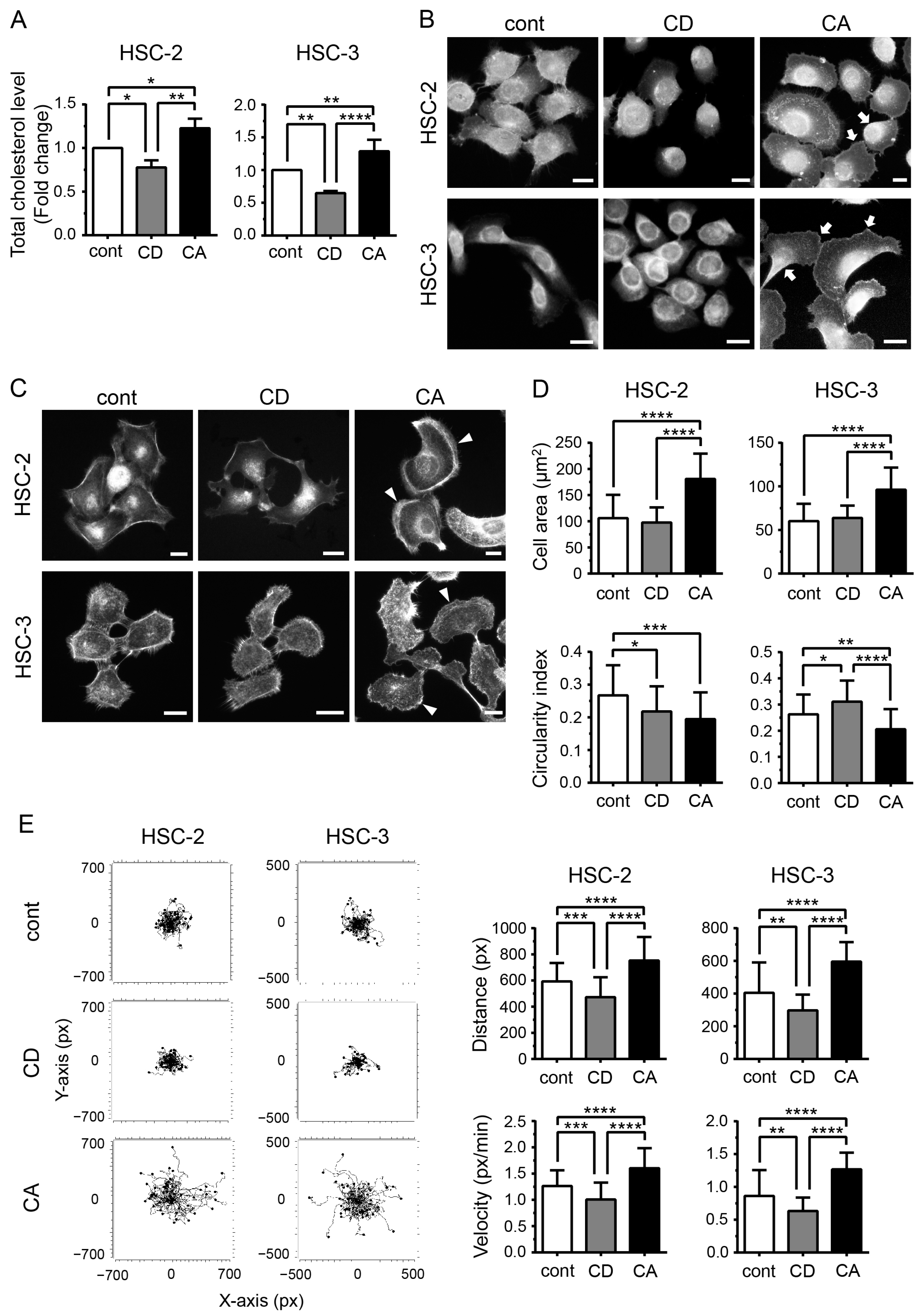
2.1. Cholesterol Influences the Morphology of OSCC Cells and Promotes Their Migration
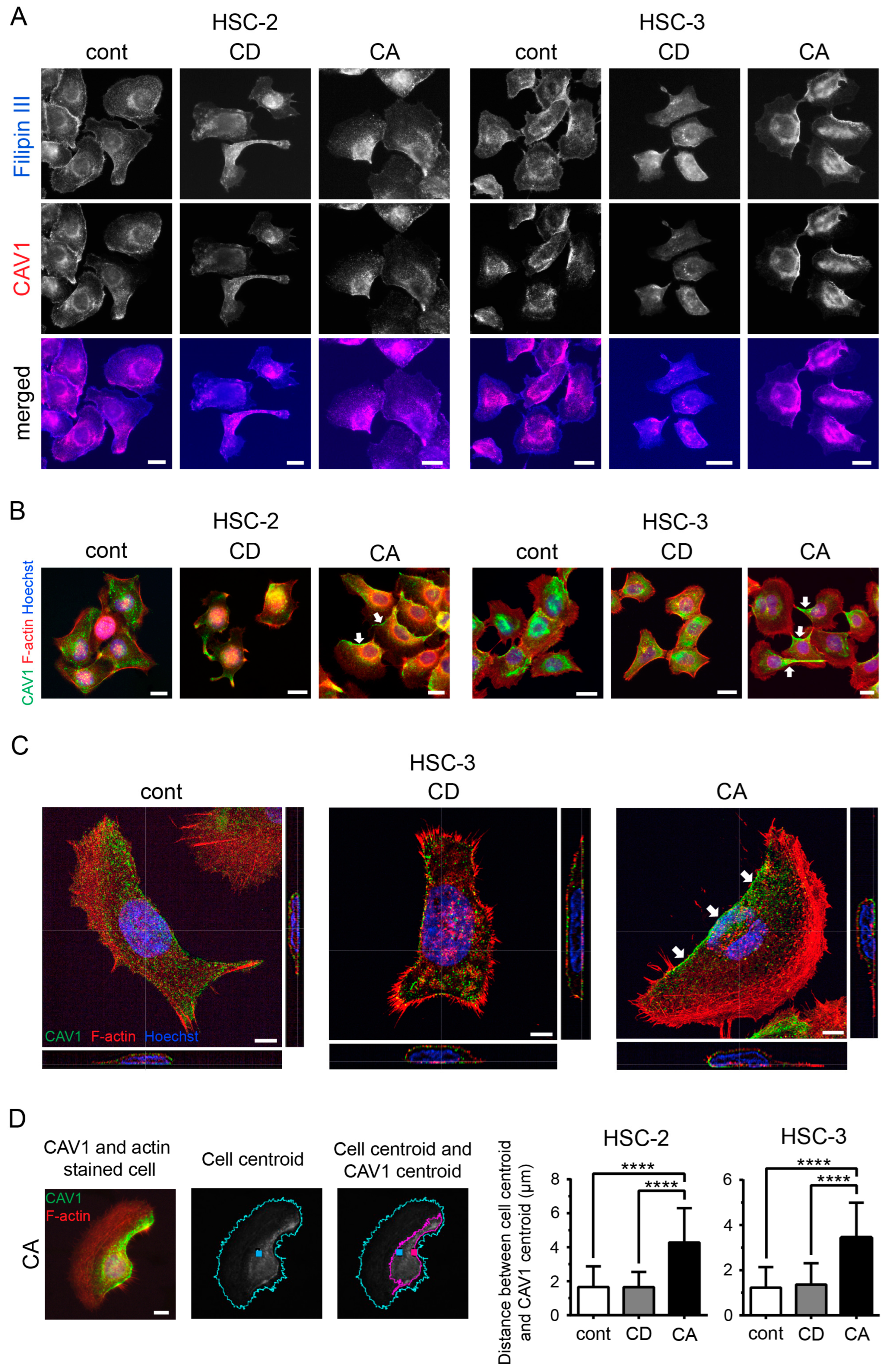
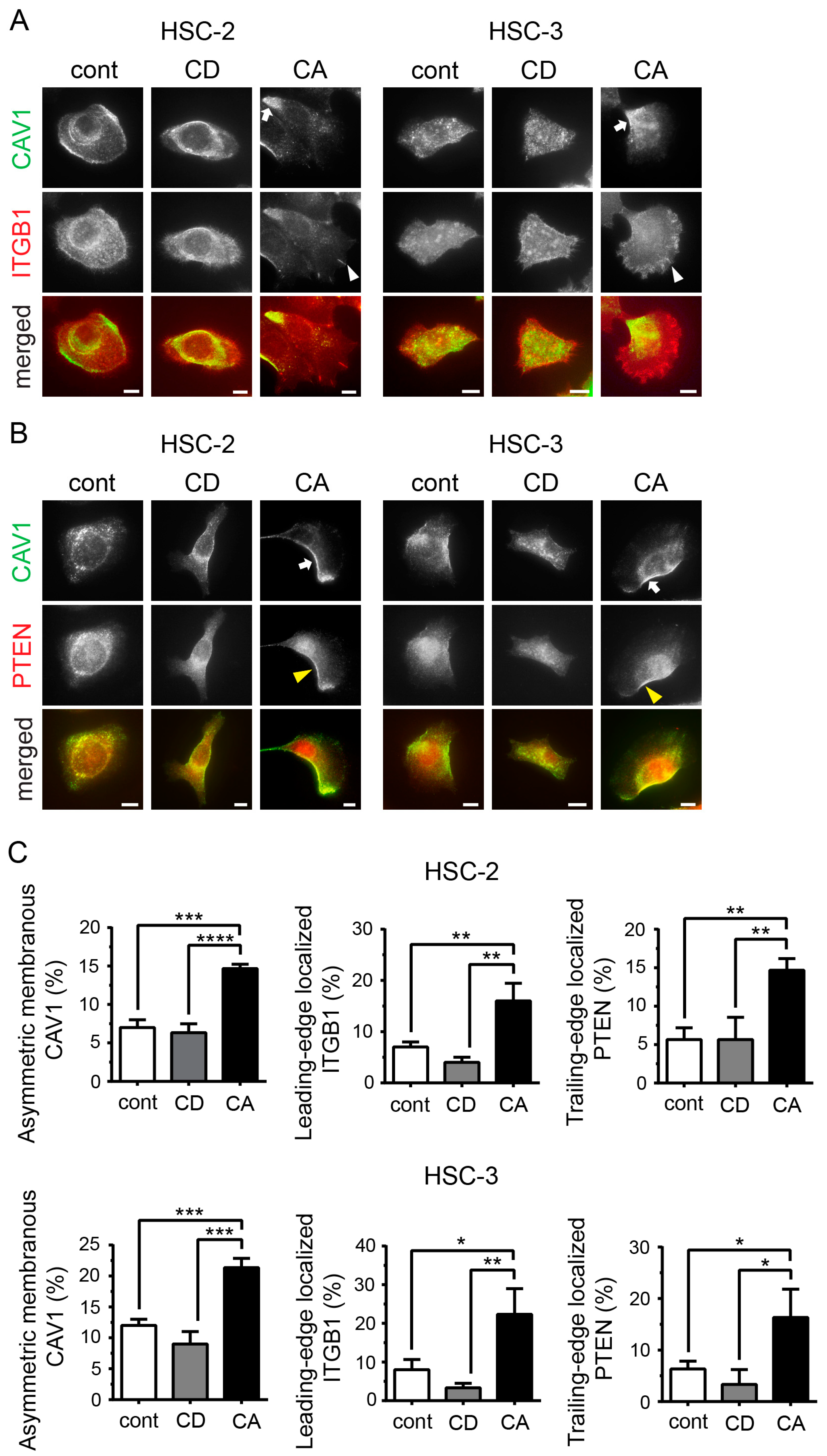
2.2. Cholesterol Leads to Cell Polarity by Promoting Asymmetric Membranous Localization of CAV1
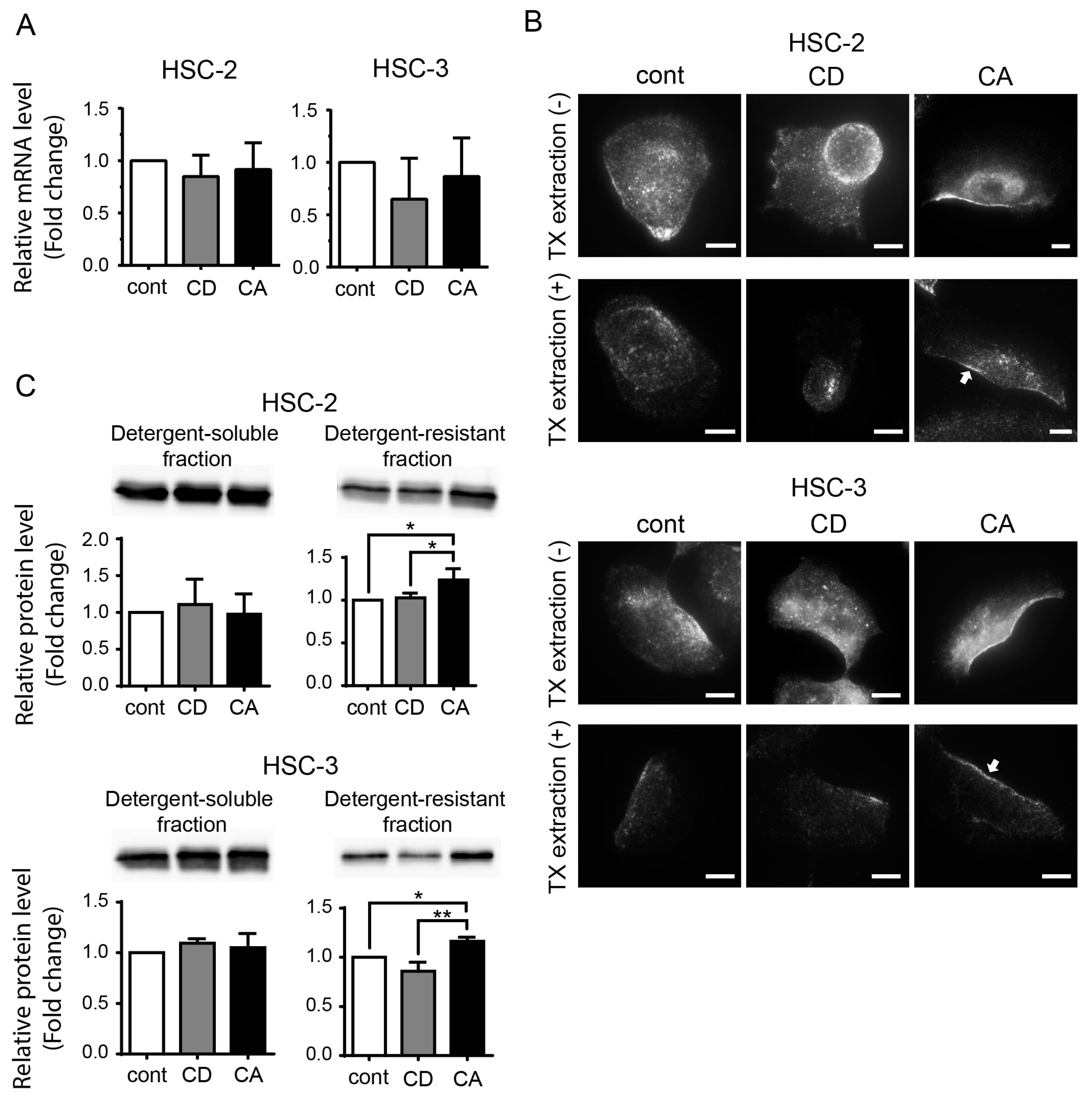
2.3. Cholesterol Increases the CAV1 Level in Cold Triton X-100 (TX)-Resistant Lipid Raft Region
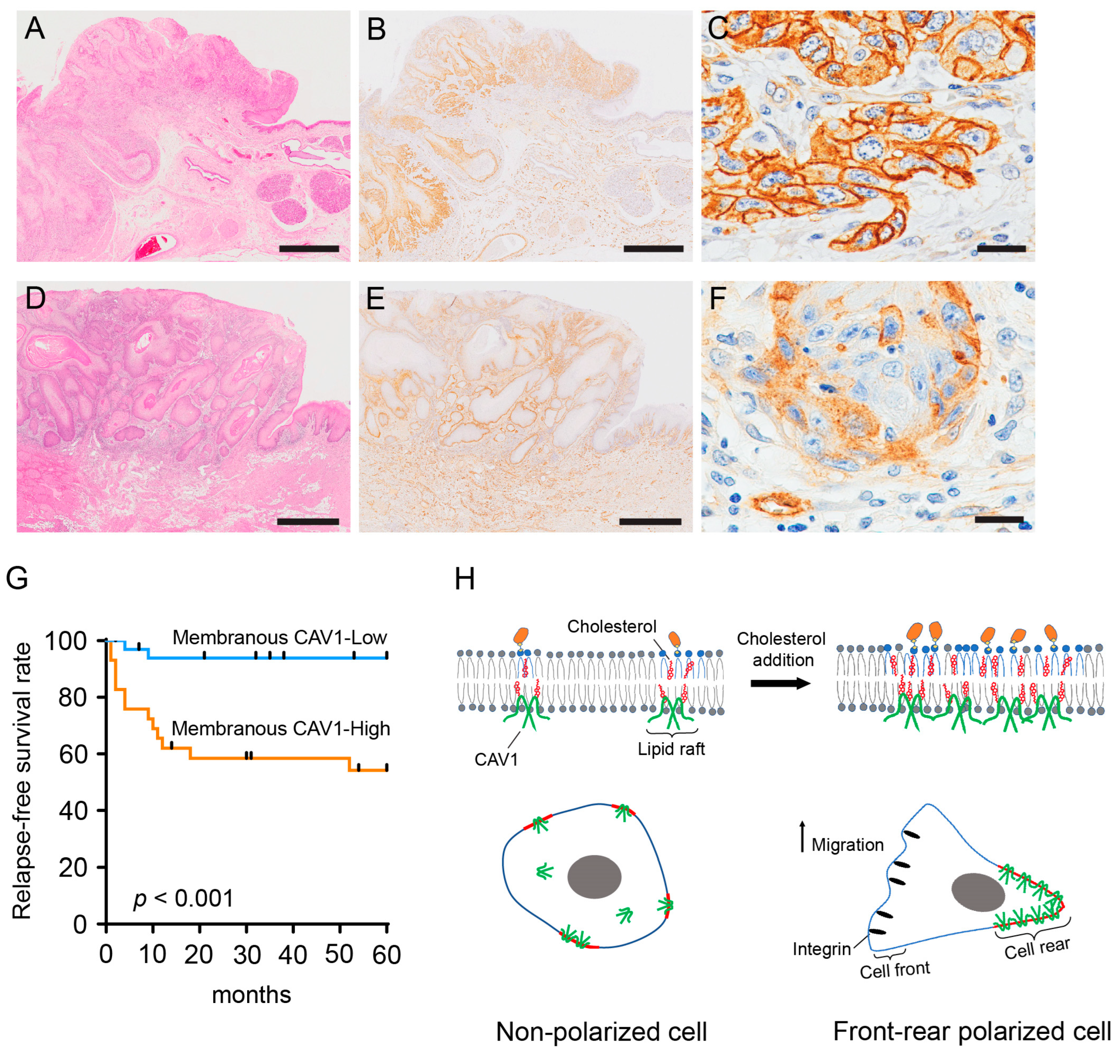
2.4. Membranous CAV1 Expression in OSCC Tissue Specimens Is Correlated with the Aggressiveness of the Tumor and Poor Clinical Outcomes
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Tissue Samples
4.3. Reagents and Antibodies
4.4. Cholesterol Manipulation
4.5. Cell Viability Assay
4.6. Quantification of Cellular Cholesterol
4.7. Fluorescence Staining
4.8. Image Analysis
4.9. Timelapse Imaging and Cell Tracking Analysis
4.10. Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.11. Western Blotting
4.12. Immunohistochemistry Staining for CAV1 in Tissue Sections
4.13. Immunohistochemistry Analysis
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Skotland, T.; Kavaliauskiene, S.; Sandvig, K. The Role of Lipid Species in Membranes and Cancer-Related Changes. Cancer Metastasis Rev. 2020, 39, 343–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Song, B.-L.; Xu, C. Cholesterol Metabolism in Cancer: Mechanisms and Therapeutic Opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.-C.; Huang, S.-F.; Lee, Y.-M.; Lai, H.-C.; Cheng, B.-H.; Cheng, W.-C.; Ho, J.Y.-P.; Jeng, L.-B.; Ma, W.-L. Cholesterol Import and Steroidogenesis Are Biosignatures for Gastric Cancer Patient Survival. Oncotarget 2017, 8, 692–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oni, T.E.; Biffi, G.; Baker, L.A.; Hao, Y.; Tonelli, C.; Somerville, T.D.D.; Deschênes, A.; Belleau, P.; Hwang, C.; Sánchez-Rivera, F.J.; et al. SOAT1 Promotes Mevalonate Pathway Dependency in Pancreatic Cancer. J. Exp. Med. 2020, 217, e20192389. [Google Scholar] [CrossRef] [PubMed]
- Hryniewicz-Jankowska, A.; Augoff, K.; Sikorski, A.F. Highlight Article: The Role of Cholesterol and Cholesterol-Driven Membrane Raft Domains in Prostate Cancer. Exp. Biol. Med. 2019, 244, 1053–1061. [Google Scholar] [CrossRef]
- Zhuang, L.; Kim, J.; Adam, R.M.; Solomon, K.R.; Freeman, M.R. Cholesterol Targeting Alters Lipid Raft Composition and Cell Survival in Prostate Cancer Cells and Xenografts. J. Clin. Investig. 2005, 115, 959–968. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Rong, X.; Palladino, E.N.D.; Wang, J.; Fogelman, A.M.; Martín, M.G.; Alrefai, W.A.; Ford, D.A.; Tontonoz, P. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 2018, 22, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, A.; Saraswat, M.; Joenväärä, S.; Agarwal, R.; Jyllikoski, D.; Wilkman, T.; Mäkitie, A.; Silén, S. Mass Spectrometry–Based Lipidomics of Oral Squamous Cell Carcinoma Tissue Reveals Aberrant Cholesterol and Glycerophospholipid Metabolism—A Pilot Study. Transl. Oncol. 2020, 13, 100807. [Google Scholar] [CrossRef]
- Sakurai, K.; Tomihara, K.; Yamazaki, M.; Heshiki, W.; Moniruzzaman, R.; Sekido, K.; Tachinami, H.; Ikeda, A.; Imaue, S.; Fujiwara, K.; et al. CD36 Expression on Oral Squamous Cell Carcinoma Cells Correlates with Enhanced Proliferation and Migratory Activity. Oral Dis. 2020, 26, 745–755. [Google Scholar] [CrossRef]
- Ganavi, B.S.; Patil, S.; Rao, R.S. Evaluation of Serum Lipids and Lipoproteins as Prognosticators in Leukoplakia. J. Contemp. Dent. Pract. 2015, 15, 294–299. [Google Scholar] [CrossRef]
- Acharya, S.; Rai, P.; Hallikeri, K.; Anehosur, V.; Kale, J. Serum Lipid Profile in Oral Squamous Cell Carcinoma: Alterations and Association with Some Clinicopathological Parameters and Tobacco Use. Int. J. Oral Maxillofac. Surg. 2016, 45, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Bailwad, S.; Singh, N.; Bailwad, S.A.; Jani, D.R.; Patil, P.; Singh, M.; Deep, G.; Singh, S. Alterations in serum lipid profile patterns in oral cancer: Correlation with histological grading and tobacco abuse. Oral Health Dent. Manag. 2014, 13, 573–579. [Google Scholar]
- Kumar, P.; Augustine, J.; Urs, A.B.; Arora, S.; Gupta, S.; Mohanty, V.R. Serum Lipid Profile in Oral Cancer and Leukoplakia: Correlation with Tobacco Abuse and Histological Grading. J. Cancer Res. Ther. 2012, 8, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Codini, M.; Garcia-Gil, M.; Albi, E. Cholesterol and Sphingolipid Enriched Lipid Rafts as Therapeutic Targets in Cancer. Int. J. Mol. Sci. 2021, 22, 726. [Google Scholar] [CrossRef]
- Dongoran, R.A.; Wang, K.H.; Lin, T.J.; Yuan, T.C.; Liu, C.H. Anti-Proliferative Effect of Statins Is Mediated by DNMT1 Inhibition and P21 Expression in OSCC Cells. Cancers 2020, 12, 2084. [Google Scholar] [CrossRef]
- Xu, B.; Muramatsu, T.; Inazawa, J. Suppression of Met Signaling Mediated by Pitavastatin and Capmatinib Inhibits Oral and Esophageal Cancer Cell Growth. Mol. Cancer Res. 2021, 19, 585–597. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, S.; Tang, Q.; Xia, H.; Bi, F. Cholesterol Metabolism: New Functions and Therapeutic Approaches in Cancer. Biochim. Et Biophys. Acta—Rev. Cancer 2020, 1874, 188394. [Google Scholar] [CrossRef]
- Ketteler, J.; Klein, D. Caveolin-1, Cancer and Therapy Resistance. Int. J. Cancer 2018, 14, 2092–2104. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Rudick, M.; Anderson, R.G.W. Multiple Functions of Caveolin-1. J. Biol. Chem. 2002, 277, 41295–41298. [Google Scholar] [CrossRef] [Green Version]
- Ikonen, E.; Parton, R.G. Caveolins and Cellular Cholesterol Balance. Traffic 2000, 1, 212–217. [Google Scholar] [CrossRef]
- Simón, L.; Campos, A.; Leyton, L.; Quest, A.F.G. Caveolin-1 Function at the Plasma Membrane and in Intracellular Compartments in Cancer. Cancer Metastasis Rev. 2020, 39, 435–453. [Google Scholar] [CrossRef]
- Surguchov, A. Caveolin: A New Link between Diabetes and AD. Cell Mol. Neurobiol. 2020, 40, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.F.; Lin, S.C.; Liu, C.J.; Chang, C.S.; Chang, K.W.; Kao, S.Y. The Biphasic Differential Expression of the Cellular Membrane Protein, Caveolin-1, in Oral Carcinogenesis. J. Oral Pathol. Med. 2003, 32, 461–467. [Google Scholar] [CrossRef]
- Xue, J.; Chen, H.; Diao, L.; Chen, X.; Xia, D. Expression of Caveolin-1 in Tongue Squamous Cell Carcinoma by Quantum Dots. Eur. J. Histochem. 2010, 54, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Miyazawa, H.; Kobayashi, H.; Noguchi, N.; Lambert, D.; Kawashiri, S. Caveolin-1 Expression at Metastatic Lymph Nodes Predicts Unfavorable Outcome in Patients with Oral Squamous Cell Carcinoma. Pathol. Oncol. Res. 2020, 26, 2105–2113. [Google Scholar] [CrossRef]
- Auzair, L.B.M.; Vincent-Chong, V.K.; Ghani, W.M.N.; Kallarakkal, T.G.; Ramanathan, A.; Lee, C.E.; Rahman, Z.A.A.; Ismail, S.M.; Abraham, M.T.; Zain, R.B. Caveolin 1 (Cav-1) and Actin-Related Protein 2/3 Complex, Subunit 1B (ARPC1B) Expressions as Prognostic Indicators for Oral Squamous Cell Carcinoma (OSCC). Eur. Arch. Otorhinolaryngol. 2016, 273, 1885–1893. [Google Scholar] [CrossRef]
- Huang, C.-F.; Yu, G.-T.; Wang, W.-M.; Liu, B.; Sun, Z.-J. Original Article Prognostic and Predictive Values of SPP1, PAI and Caveolin-1 in Patients with Oral Squamous Cell Carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 6032–6039. [Google Scholar] [PubMed]
- Nakatani, K.; Wada, T.; Nakamura, M.; Uzawa, K.; Tanzawa, H.; Fujita, S. Expression of Caveolin-1 and Its Correlation with Cisplatin Sensitivity in Oral Squamous Cell Carcinoma. J. Cancer Res. Clin. Oncol. 2005, 131, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Mahammad, S.; Parmryd, I. Cholesterol Depletion Using Methyl-β-Cyclodextrin. Methods Mol. Biol. 2015, 1232, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Zidovetzki, R.; Levitan, I. Use of Cyclodextrins to Manipulate Plasma Membrane Cholesterol Content: Evidence, Misconceptions and Control Strategies. Biochim. Biophys. Acta 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [Green Version]
- Orlando, K.; Guo, W. Membrane Organization and Dynamics in Cell Polarity. Cold Spring Harb. Perspect. Biol. 2009, 1, a001321. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell Migration: Integrating Signals from Front to Back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Seveau, S.; Eddy, R.J.; Maxfield, F.R.; Pierini, L.M. Cytoskeleton-Dependent Membrane Domain Segregation during Neutrophil Polarization. Mol. Biol. Cell. 2001, 12, 3550–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, F.S.; da Silva Sampaio, L.; Konig, S.; Bonamino, M.; Rossi, M.I.D.; Costa, M.L.; Fernandes, P.; Mermelstein, C. Membrane Cholesterol Depletion Reduces Breast Tumor Cell Migration by a Mechanism That Involves Non-Canonical Wnt Signaling and IL-10 Secretion. Transl. Med. Commun. 2016, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Irungbam, K.; Kataria, M. Depletion of Membrane Cholesterol Compromised Caspase-8 Imparts in Autophagy Induction and Inhibition of Cell Migration in Cancer Cells. Cancer Cell Int. 2018, 18, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, C.; Nagao, T.; Grosheva, I.; Maxfield, F.R.; Pierini, L.M. Elevated Plasma Membrane Cholesterol Content Alters Macrophage Signaling and Function. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 372–378. [Google Scholar] [CrossRef] [Green Version]
- Nagao, T.; Qin, C.; Grosheva, I.; Maxfield, F.R.; Pierini, L.M. Elevated Cholesterol Levels in the Plasma Membranes of Macrophages Inhibit Migration by Disrupting Rhoa Regulation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1596–1602. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Deng, Y.; Deng, G.; Chen, P.; Wang, Y.; Wu, H.; Ji, Z.; Yao, Z.; Zhang, X.; Yu, B.; et al. High Cholesterol Induces Apoptosis and Autophagy through the ROS-Activated AKT/FOXO1 Pathway in Tendon-Derived Stem Cells. Stem. Cell Res. Ther. 2020, 11, 131. [Google Scholar] [CrossRef] [Green Version]
- Lomakin, A.J.; Lee, K.C.; Han, S.J.; Bui, D.A.; Davidson, M.; Mogilner, A.; Danuser, G. Competition for Actin between Two Distinct F-Actin Networks Defines a Bistable Switch for Cell Polarization. Nat. Cell Biol. 2015, 17, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Isshiki, M.; Ando, J.; Yamamoto, K.; Fujita, T.; Ying, Y.; Anderson, R.G.W. Sites of Ca2+ wave initiation move with caveolae to the trailing edge of migrating cells. J. Cell. Sci. 2001, 115, 475–484. [Google Scholar] [CrossRef]
- Parat, M.O.; Anand-Apte, B.; Fox, P.L. Differential Caveolin-1 Polarization in Endothelial Cells during Migration in Two and Three Dimensions. Mol. Biol. Cell 2003, 14, 3156–3168. [Google Scholar] [CrossRef] [PubMed]
- Beardsley, A.; Fang, K.; Mertz, H.; Castranova, V.; Friend, S.; Liu, J. Loss of Caveolin-1 Polarity Impedes Endothelial Cell Polarization and Directional Movement. J. Biol. Chem. 2005, 280, 3541–3547. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.H.; Flynn, D.C.; Castranova, V.; Millecchia, L.L.; Beardsley, A.R.; Liu, J. Identification of a Novel Domain at the N Terminus of Caveolin-1 That Controls Rear Polarization of the Protein and Caveolae Formation. J. Biol. Chem. 2007, 282, 7232–7241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentini, D.; Guzzi, F.; Pimpinelli, F.; Zaninetti, R.; Cassetti, A.; Coco, S.; Maggi, R.; Parenti, M. Polarization of Caveolins and Caveolae during Migration of Immortalized Neurons. J. Neurochem. 2008, 104, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.M.; Daud, N.H.; Aung, C.S.; Loo, D.; Martin, S.; Murphy, S.; Black, D.M.; Barry, R.; Simpson, F.; Liu, L.; et al. Co-Regulation of Cell Polarization and Migration by Caveolar Proteins PTRF/Cavin-1 and Caveolin-1. PLoS ONE 2012, 7, e43041. [Google Scholar] [CrossRef]
- Grande-García, A.; Echarri, A.; de Rooij, J.; Alderson, N.B.; Waterman-Storer, C.M.; Valdivielso, J.M.; del Pozo, M.A. Caveolin-1 Regulates Cell Polarization and Directional Migration through Src Kinase and Rho GTPases. J. Cell Biol. 2007, 177, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Gao, J.; Jordan, R.; Naumann, C.A. Changes in Cholesterol Level Alter Integrin Sequestration in Raft-Mimicking Lipid Mixtures. Biophys. J. 2018, 114, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Ehehalt, R. Cholesterol, Lipid Rafts, and Disease. J. Clin. Investig. 2002, 110, 597–603. [Google Scholar] [CrossRef]
- Schmidt, M.L.; Davis, J.H. Liquid Disordered-Liquid Ordered Phase Coexistence in Lipid/Cholesterol Mixtures: A Deuterium 2D NMR Exchange Study. Langmuir 2017, 33, 1881–1890. [Google Scholar] [CrossRef]
- London, E.; Brown, D.A. Insolubility of Lipids in Triton X-100: Physical Origin and Relationship to Sphingolipid/Cholesterol Membrane Domains (Rafts). Biochim. Biophys. Acta 2000, 1508, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Fra, A.M.; Williamson, E.; Simons, K.; Parton, R.G. De Novo Formation of Caveolae in Lymphocytes by Expression of VIP21-Caveolin. Proc. Natl. Acad. Sci. USA 1995, 92, 8655–8659. [Google Scholar] [CrossRef] [Green Version]
- Frank, P.G.; W-C Cheung, M.; Pavlides, S.; Llaverias, G.; Park, D.S.; Lisanti, M.P.; Pav-lides, S. Caveolin-1 and Regulation of Cellular Cholesterol Homeostasis. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, 677–686. [Google Scholar] [CrossRef] [Green Version]
- Smart, E.J.; Ying, Y.S.; Donzell, W.C.; Anderson, R.G.W. A Role for Caveolin in Transport of Cholesterol from Endoplasmic Reticulum to Plasma Membrane. J. Biol. Chem. 1996, 271, 29427–29435. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Liao, H.L.; Niu, X.L.; Yuan, Y.; Lin, T.; Verna, L.; Stemerman, M.B. Low Density Lipoprotein Induces eNOS Translocation to Membrane Caveolae: The Role of RhoA Activation and Stress Fiber Formation. Biochim. Biophys. Acta 2003, 1635, 117–126. [Google Scholar] [CrossRef]
- Sanyour, H.J.; Li, N.; Rickel, A.P.; Torres, H.M.; Anderson, R.H.; Miles, M.R.; Childs, J.D.; Francis, K.R.; Tao, J.; Hong, Z. Statin-Mediated Cholesterol Depletion Exerts Coordinated Effects on the Alterations in Rat Vas-cular Smooth Muscle Cell Biomechanics and Migration. J. Physiol. 2020, 598, 1505–1522. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, A.P.; Jozic, I.; Stone, R.C.; Pastar, I.; Egger, A.N.; Stojadinovic, O.; Glinos, G.D.; Kirsner, R.S.; Tom-ic-Canic, M. Mevastatin Promotes Healing by Targeting Caveolin-1 to Restore EGFR Signaling. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaafari-Ashkavandi, Z.; Aslani, E. Caveolin-1 Expression in Oral Lichen Planus, Dysplastic Lesions and Squamous Cell Carcinoma. Pathol. Res. Pract. 2017, 213, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Cassoni, P.; Senetta, R.; Castellano, I.; Ortolan, E.; Bosco, M.; Magnani, I.; Ducati, A. Caveolin-1 Expression Is Variably Displayed in Astroglial-Derived Tumors and Absent in Oligodendrogliomas: Concrete Premises for a New Reliable Diagnostic Marker in Gliomas. Am. J. Surg. Pathol. 2007, 31, 760–769. [Google Scholar] [CrossRef]
- Hoque Apu, E.; Akram, S.U.; Rissanen, J.; Wan, H.; Salo, T. Desmoglein 3—Influence on Oral Carcinoma Cell Migration and Invasion. Exp. Cell Res. 2018, 370, 353–364. [Google Scholar] [CrossRef]
- Haga, K.; Yamazaki, M.; Maruyama, S.; Kawaharada, M.; Suzuki, A.; Hoshikawa, E.; Chan, N.N.; Funayama, A.; Mi-kami, T.; Kobayashi, T.; et al. Crosstalk between Oral Squamous Cell Carcinoma Cells and Can-cer-Associated Fibroblasts via the TGF-β/SOX9 Axis in Cancer Progression. Transl. Oncol. 2021, 14. [Google Scholar] [CrossRef]
- Salo, T.; Sutinen, M.; Hoque Apu, E.; Sundquist, E.; Cervigne, N.K.; de Oliveira, C.E.; Akram, S.U.; Ohlmeier, S.; Suomi, F.; Eklund, L.; et al. A Novel Human Leiomyoma Tissue Derived Matrix for Cell Culture Studies. BMC Cancer 2015, 15, 981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brierley, J.D.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumors, 8th ed.; John Wiley & Sons: Oxford, UK, 2017; pp. 17–21. [Google Scholar]
- Conrad, P.A.; Nederlof, M.A.; Herman, I.M.; Lansing Taylor, D. Correlated Distribution of Actin, Myosin, and Microtubules at the Leading Edge of Migrating Swiss 3T3 Fibroblasts. Cell Motil. Cytoskeleton. 1989, 14, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, Z.; Guo, Y.; Shan, X.; Li, X.; Lin, J. High-Efficiency Procedure to Characterize, Segment, and Quantify Complex Multicellularity in Raw Micrographs in Plants. Plant Methods 2020, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | N | Membranous CAV1 Expression | p-Value | |
|---|---|---|---|---|
| High (≥15%) | Low (<15%) | |||
| Age | ||||
| ≤Median (72) | 32 | 9 | 23 | |
| >Median (72) | 31 | 20 | 11 | <0.01 |
| Gender | ||||
| Male | 35 | 14 | 21 | |
| Female | 28 | 15 | 13 | 0.31 |
| T-factor | ||||
| T1/T2 | 54 | 22 | 32 | |
| T3/T4 | 9 | 7 | 2 | 0.07 |
| Primary site | ||||
| Tongue | 58 | 28 | 30 | |
| Buccal mucosa | 4 | 1 | 3 | 0.43 |
| Gingiva | 1 | 0 | 1 | |
| Lymph node metastasis | ||||
| Negative | 57 | 23 | 34 | |
| Positive | 6 | 6 | 0 | <0.01 |
| Clinical stage | ||||
| I/II | 51 | 19 | 32 | |
| III/IV | 12 | 10 | 2 | <0.01 |
| Mode of invasion | ||||
| 1/2/3 | 41 | 15 | 26 | |
| 4C/4D | 22 | 14 | 8 | 0.06 |
| Local recurrence | ||||
| Negative | 61 | 27 | 34 | 0.21 |
| Positive | 2 | 2 | 0 | |
| Regional recurrence | ||||
| Negative | 50 | 18 | 32 | |
| Positive | 13 | 11 | 2 | <0.01 |
| Variable | Category | N | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|---|---|
| RR | p-Value | RR | p-Value | |||
| Age | ≤72 >72 | 33 30 | 0.59 1.68 | 0.32 | ||
| Gender | Male Female | 36 27 | 0.65 1.53 | 0.41 | ||
| T-factor | T1/T2 T3/T4 | 54 9 | 0.38 2.64 | 0.13 | ||
| Lymph node metastasis | Negative Positive | 57 6 | 0.35 2.86 | 0.15 | ||
| Stage | I/II III/IV | 51 12 | 0.4 2.53 | 0.11 | ||
| Mode of invasion | 1/2/3 4C/4D | 41 22 | 0.15 6.58 | <0.001 | 0.22 4.51 | <0.01 |
| Membranous CAV1 | High Low | 29 34 | 9.16 0.1 | <0.001 | 6.47 0.15 | <0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, N.N.; Yamazaki, M.; Maruyama, S.; Abé, T.; Haga, K.; Kawaharada, M.; Izumi, K.; Kobayashi, T.; Tanuma, J.-i. Cholesterol Is a Regulator of CAV1 Localization and Cell Migration in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 6035. https://doi.org/10.3390/ijms24076035
Chan NN, Yamazaki M, Maruyama S, Abé T, Haga K, Kawaharada M, Izumi K, Kobayashi T, Tanuma J-i. Cholesterol Is a Regulator of CAV1 Localization and Cell Migration in Oral Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2023; 24(7):6035. https://doi.org/10.3390/ijms24076035
Chicago/Turabian StyleChan, Nyein Nyein, Manabu Yamazaki, Satoshi Maruyama, Tatsuya Abé, Kenta Haga, Masami Kawaharada, Kenji Izumi, Tadaharu Kobayashi, and Jun-ichi Tanuma. 2023. "Cholesterol Is a Regulator of CAV1 Localization and Cell Migration in Oral Squamous Cell Carcinoma" International Journal of Molecular Sciences 24, no. 7: 6035. https://doi.org/10.3390/ijms24076035