
Identification of Mir-182-3p/FLI-1 Axis as a Key Signaling in Immune Response in Cervical Cancer: A Comprehensive Bioinformatic Analysis

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Mir-182-3p Expression Increases in Cervical Cancer and Could Regulate the FLI-1 Expression
2.2. FLI-1 Expression Decreases in Cervical Cancer
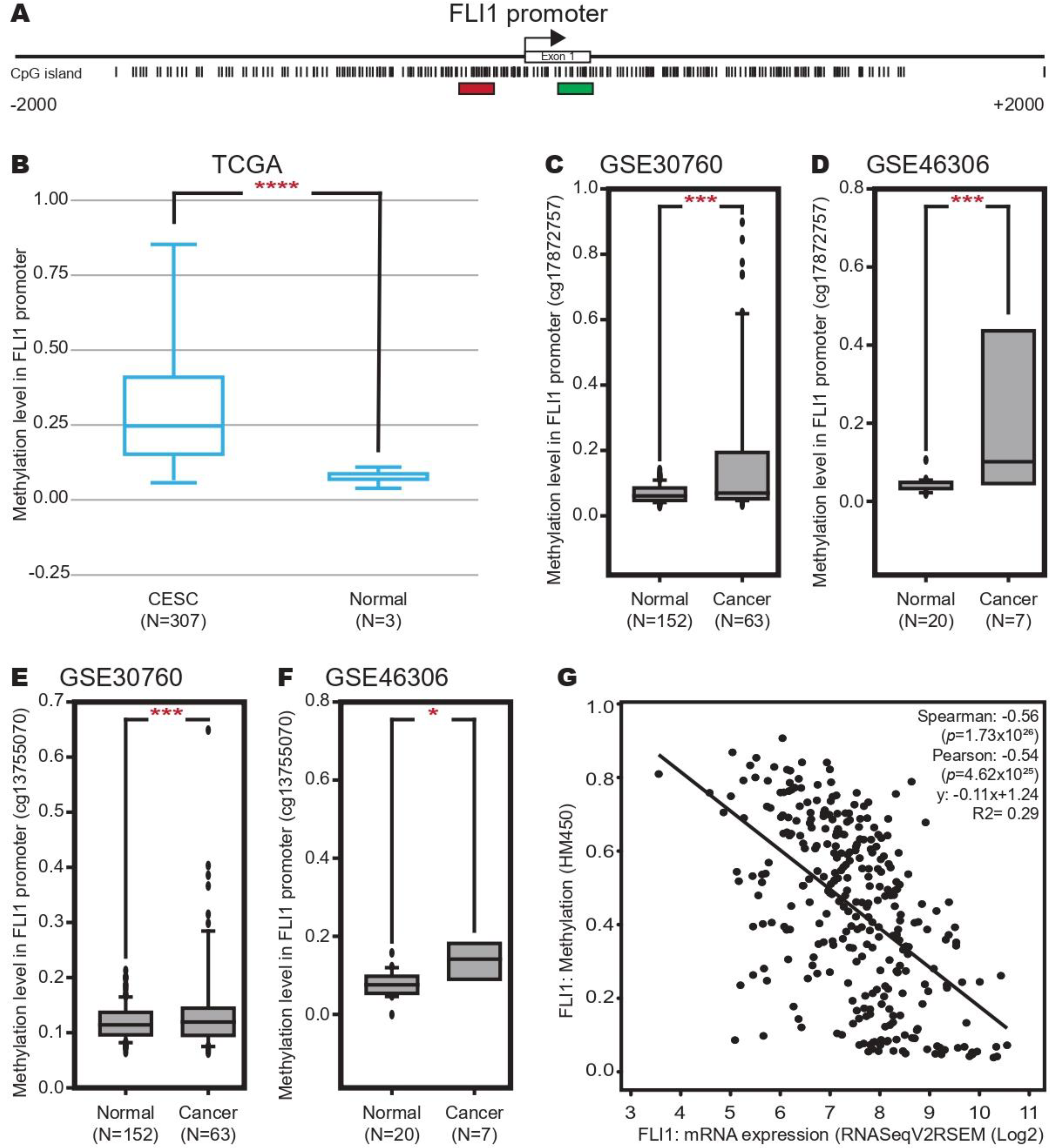
2.3. Methylation in FLI-1 Promoter Increases in Cervical Cancer
2.4. AP2α Expression Increases in Cervical Cancer and Could Downregulate FLI-1 Expression
2.5. FLI-1 as a Possible Key Gene in Immune Response in Cervical Cancer
3. Discussion
4. Materials and Methods
4.1. Expression Analysis
4.2. Receiver Operating Characteristic (ROC) Curve Analysis
4.3. Survival Analysis
4.4. Identification Analysis of Target mRNAs
4.5. Correlation Analysis
4.6. Methylation Analysis
4.7. Identification Analysis of Transcription Factors Binding Sites
4.8. Estimation Analysis of Immune Cell Recruitment
4.9. PPI, Pathway Enrichment, and GSEA Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Xu, W.; Xu, M.; Wang, L.; Zhou, W.; Xiang, R.; Shi, Y.; Zhang, Y.; Piao, Y. Integrative analysis of DNA methylation and gene expression identified cervical cancer-specific diagnostic biomarkers. Signal Transduct. Target. Ther. 2019, 4, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, L.M.; Li, W.T.; Shende, N.; Tsai, J.C.; Ma, J.; Chakladar, J.; Gnanasekar, A.; Qu, Y.; Dereschuk, K.; Wang-Rodriguez, J.; et al. Analysis of the immune landscape in virus-induced cancers using a novel integrative mechanism discovery approach. Comput. Struct. Biotechnol. J. 2021, 19, 6240–6254. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Menon, A.; Abd-Aziz, N.; Khalid, K.; Poh, C.L.; Naidu, R. miRNA: A Promising Therapeutic Target in Cancer. Int. J. Mol. Sci. 2022, 23, 11502. [Google Scholar] [CrossRef]
- Chen, G.; Yu, W.; Li, Z.; Wang, Q.; Yang, Q.; Du, Z.; Zhang, G.; Song, Y. Potential Regulatory Effects of miR-182-3p in Osteosarcoma via Targeting EBF2. Biomed. Res. Int. 2019, 2019, 4897905. [Google Scholar] [CrossRef]
- Zhu, Z.; Shi, Y.; Gong, X.; Li, J.; Zhang, M. LINC00511 Knockdown Suppresses Resistance to Cisplatin in Lung Adenocarcinoma by Interacting with miR-182-3p and BIRC5. Mol. Biotechnol. 2022, 64, 252–262. [Google Scholar] [CrossRef]
- Guo, J.; Su, Y.; Zhang, M. Circ_0000140 restrains the proliferation, metastasis and glycolysis metabolism of oral squamous cell carcinoma through upregulating CDC73 via sponging miR-182-5p. Cancer Cell Int. 2020, 20, 407. [Google Scholar] [CrossRef]
- Dinami, R.; Pompili, L.; Petti, E.; Porru, M.; D’Angelo, C.; Di Vito, S.; Rizzo, A.; Campani, V.; De Rosa, G.; Bruna, A.; et al. MiR-182-3p targets TRF2 and impairs tumor growth of triple-negative breast cancer. EMBO Mol. Med. 2023, 15, e16033. [Google Scholar] [CrossRef]
- Soheilifar, M.H.; Vaseghi, H.; Seif, F.; Ariana, M.; Ghorbanifar, S.; Habibi, N.; Papari Barjasteh, F.; Pornour, M. Concomitant overexpression of mir-182-5p and mir-182-3p raises the possibility of IL-17-producing Treg formation in breast cancer by targeting CD3d, ITK, FOXO1, and NFATs: A meta-analysis and experimental study. Cancer Sci. 2021, 112, 589–603. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, M.J.; Ren, A.M.; Wu, H.F.; Han, W.M.; Tan, R.Y.; Tu, R.Q. A ten-microRNA signature identified from a genome-wide microRNA expression profiling in human epithelial ovarian cancer. PLoS ONE 2014, 9, e96472. [Google Scholar] [CrossRef]
- Shi, Z.; Wang, R.; Huang, L.; Chen, X.; Xu, M.; Zha, D.; Ma, Y. Integrative analysis of miRNAs-mRNAs reveals that miR-182 up-regulation contributes to proliferation and invasion of nasopharyngeal carcinoma by targeting PTEN. Aging 2020, 12, 11568–11578. [Google Scholar] [CrossRef]
- Song, W.; Hu, L.; Li, W.; Wang, G.; Li, Y.; Yan, L.; Li, A.; Cui, J. Oncogenic Fli-1 is a potential prognostic marker for the progression of epithelial ovarian cancer. BMC Cancer 2014, 14, 424. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Luo, H.; Liu, T.; Zacksenhaus, E.; Ben-David, Y. The ets transcription factor Fli-1 in development, cancer and disease. Oncogene 2015, 34, 2022–2031. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Song, W.; Yan, X.; Li, A.; Zhang, X.; Li, W.; Wen, X.; Zhou, L.; Yu, D.; Hu, J.F.; et al. Friend leukemia virus integration 1 promotes tumorigenesis of small cell lung cancer cells by activating the miR-17-92 pathway. Oncotarget 2017, 8, 41975–41987. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Yu, Y.; Li, L.; Chen, N.; Song, W.; He, H.; Dong, J.; Liu, X.; Cui, J. Friend leukemia virus integration 1 is a predictor of poor prognosis of breast cancer and promotes metastasis and cancer stem cell properties of breast cancer cells. Cancer Med. 2018, 7, 3548–3560. [Google Scholar] [CrossRef]
- Del Portillo, A.; Komissarova, E.V.; Bokhari, A.; Hills, C.; de Gonzalez, A.K.; Kongkarnka, S.; Remotti, H.E.; Sepulveda, J.L.; Sepulveda, A.R. Downregulation of Friend Leukemia Integration 1 (FLI1) follows the stepwise progression to gastric adenocarcinoma. Oncotarget 2019, 10, 3852–3864. [Google Scholar] [CrossRef] [Green Version]
- Beger, M.; Butz, K.; Denk, C.; Williams, T.; Hurst, H.C.; Hoppe-Seyler, F. Expression pattern of AP-2 transcription factors in cervical cancer cells and analysis of their influence on human papillomavirus oncogene transcription. J. Mol. Med. 2001, 79, 314–320. [Google Scholar] [CrossRef]
- Wu, H.R.; Zhang, J. AP-2α expression in papillary thyroid carcinoma predicts tumor progression and poor prognosis. Cancer Manag. Res. 2018, 10, 2615–2625. [Google Scholar] [CrossRef] [Green Version]
- Scheiber, M.N.; Watson, P.M.; Rumboldt, T.; Stanley, C.; Wilson, R.C.; Findlay, V.J.; Anderson, P.E.; Watson, D.K. FLI1 expression is correlated with breast cancer cellular growth, migration, and invasion and altered gene expression. Neoplasia 2014, 16, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, Y.; Yu, C.; Cao, Y.; Yu, Y.; Pan, Y.; Su, D.; Lu, Q.; Yang, W.; Zuo, Y.; et al. Characterization of the relationship between FLI1 and immune infiltrate level in tumour immune microenvironment for breast cancer. J. Cell. Mol. Med. 2020, 24, 5501–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Zhai, Y.; Kuick, R.; Nan, B.; Ota, I.; Weiss, S.J.; Trimble, C.L.; Fearon, E.R.; Cho, K.R. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007, 67, 10163–10172. [Google Scholar] [CrossRef] [Green Version]
- Den Boon, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E3255–E3264. [Google Scholar] [CrossRef] [Green Version]
- Balacescu, O.; Balacescu, L.; Tudoran, O.; Todor, N.; Rus, M.; Buiga, R.; Susman, S.; Fetica, B.; Pop, L.; Maja, L.; et al. Gene expression profiling reveals activation of the FA/BRCA pathway in advanced squamous cervical cancer with intrinsic resistance and therapy failure. BMC Cancer 2014, 14, 246. [Google Scholar] [CrossRef] [Green Version]
- Wilting, S.M.; Snijders, P.J.; Verlaat, W.; Jaspers, A.; van de Wiel, M.A.; van Wieringen, W.N.; Meijer, G.A.; Kenter, G.G.; Yi, Y.; le Sage, C.; et al. Altered microRNA expression associated with chromosomal changes contributes to cervical carcinogenesis. Oncogene 2013, 32, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Zhang, Y.; Zhu, M.; Liu, S.; Wang, X. miRNA Expression Profiles of HPV-Infected Patients with Cervical Cancer in the Uyghur Population in China. PLoS ONE 2016, 11, e0164701. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S.N.; et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Salmerón-Bárcenas, E.G.; Zacapala-Gómez, A.E.; Lozano-Amado, D.; Castro-Muñoz, L.J.; Leyva-Vázquez, M.A.; Manzo-Merino, J.; Ávila-López, P.A. Comprehensive bioinformatic analysis reveals oncogenic role of H2A.Z isoforms in cervical cancer progression. Iran. J. Basic Med. Sci. 2021, 24, 1470. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Goksuluk, D.; Korkmaz, S.; Zararsiz, G.; Karaagaoglu, A.E. easyROC: An interactive web-tool for ROC curve analysis using R language environment. R J. 2016, 8, 213. [Google Scholar] [CrossRef] [Green Version]
- Győrffy, B.J.C.; Journal, S.B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Kehl, T.; Kern, F.; Backes, C.; Fehlmann, T.; Stöckel, D.; Meese, E.; Lenhof, H.-P.; Keller, A. miRPathDB 2.0: A novel release of the miRNA Pathway Dictionary Database. Nucleic Acids Res. 2020, 48, D142–D147. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Périer, R.C.; Junier, T.; Bucher, P. The Eukaryotic Promoter Database EPD. Nucleic Acids Res. 1998, 26, 353–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Wei, Y.; Gu, Y.; Zhang, S.; Lyu, J.; Zhang, B.; Chen, C.; Zhu, J.; Wang, Y.; Liu, H.; et al. DiseaseMeth version 2.0: A major expansion and update of the human disease methylation database. Nucleic Acids Res. 2017, 45, D888–D895. [Google Scholar] [CrossRef]
- Zhuang, J.; Jones, A.; Lee, S.H.; Ng, E.; Fiegl, H.; Zikan, M.; Cibula, D.; Sargent, A.; Salvesen, H.B.; Jacobs, I.J.; et al. The dynamics and prognostic potential of DNA methylation changes at stem cell gene loci in women’s cancer. PLoS Genet. 2012, 8, e1002517. [Google Scholar] [CrossRef]
- Farkas, S.A.; Milutin-Gašperov, N.; Grce, M.; Nilsson, T.K. Genome-wide DNA methylation assay reveals novel candidate biomarker genes in cervical cancer. Epigenetics 2013, 8, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Sandelin, A.; Wasserman, W.W.; Lenhard, B. ConSite: Web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res. 2004, 32, W249–W252. [Google Scholar] [CrossRef] [Green Version]
- Grabe, N. AliBaba2: Context specific identification of transcription factor binding sites. Silico Biol. 2002, 2, S1–S15. [Google Scholar]
- Messeguer, X.; Escudero, R.; Farré, D.; Núñez, O.; Martínez, J.; Albà, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Zheng, R.; Wan, C.; Mei, S.; Qin, Q.; Wu, Q.; Sun, H.; Chen, C.H.; Brown, M.; Zhang, X.; Meyer, C.A.; et al. Cistrome Data Browser: Expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019, 47, D729–D735. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmerón-Bárcenas, E.G.; Mendoza-Catalan, M.A.; Ramírez-Bautista, Á.U.; Lozano-Santos, R.A.; Torres-Rojas, F.I.; Ávila-López, P.A.; Zacapala-Gómez, A.E. Identification of Mir-182-3p/FLI-1 Axis as a Key Signaling in Immune Response in Cervical Cancer: A Comprehensive Bioinformatic Analysis. Int. J. Mol. Sci. 2023, 24, 6032. https://doi.org/10.3390/ijms24076032
Salmerón-Bárcenas EG, Mendoza-Catalan MA, Ramírez-Bautista ÁU, Lozano-Santos RA, Torres-Rojas FI, Ávila-López PA, Zacapala-Gómez AE. Identification of Mir-182-3p/FLI-1 Axis as a Key Signaling in Immune Response in Cervical Cancer: A Comprehensive Bioinformatic Analysis. International Journal of Molecular Sciences. 2023; 24(7):6032. https://doi.org/10.3390/ijms24076032
Chicago/Turabian StyleSalmerón-Bárcenas, Eric Genaro, Miguel Angel Mendoza-Catalan, Ángela Uray Ramírez-Bautista, Rafael Acxel Lozano-Santos, Francisco Israel Torres-Rojas, Pedro Antonio Ávila-López, and Ana Elvira Zacapala-Gómez. 2023. "Identification of Mir-182-3p/FLI-1 Axis as a Key Signaling in Immune Response in Cervical Cancer: A Comprehensive Bioinformatic Analysis" International Journal of Molecular Sciences 24, no. 7: 6032. https://doi.org/10.3390/ijms24076032