
Docking and Selectivity Studies of Covalently Bound Janus Kinase 3 Inhibitors

Abstract
:1. Introduction
2. Results and Discussion
2.1. Multiple Sequence Alignment
2.2. Molecular Docking
2.2.1. Validation of Method

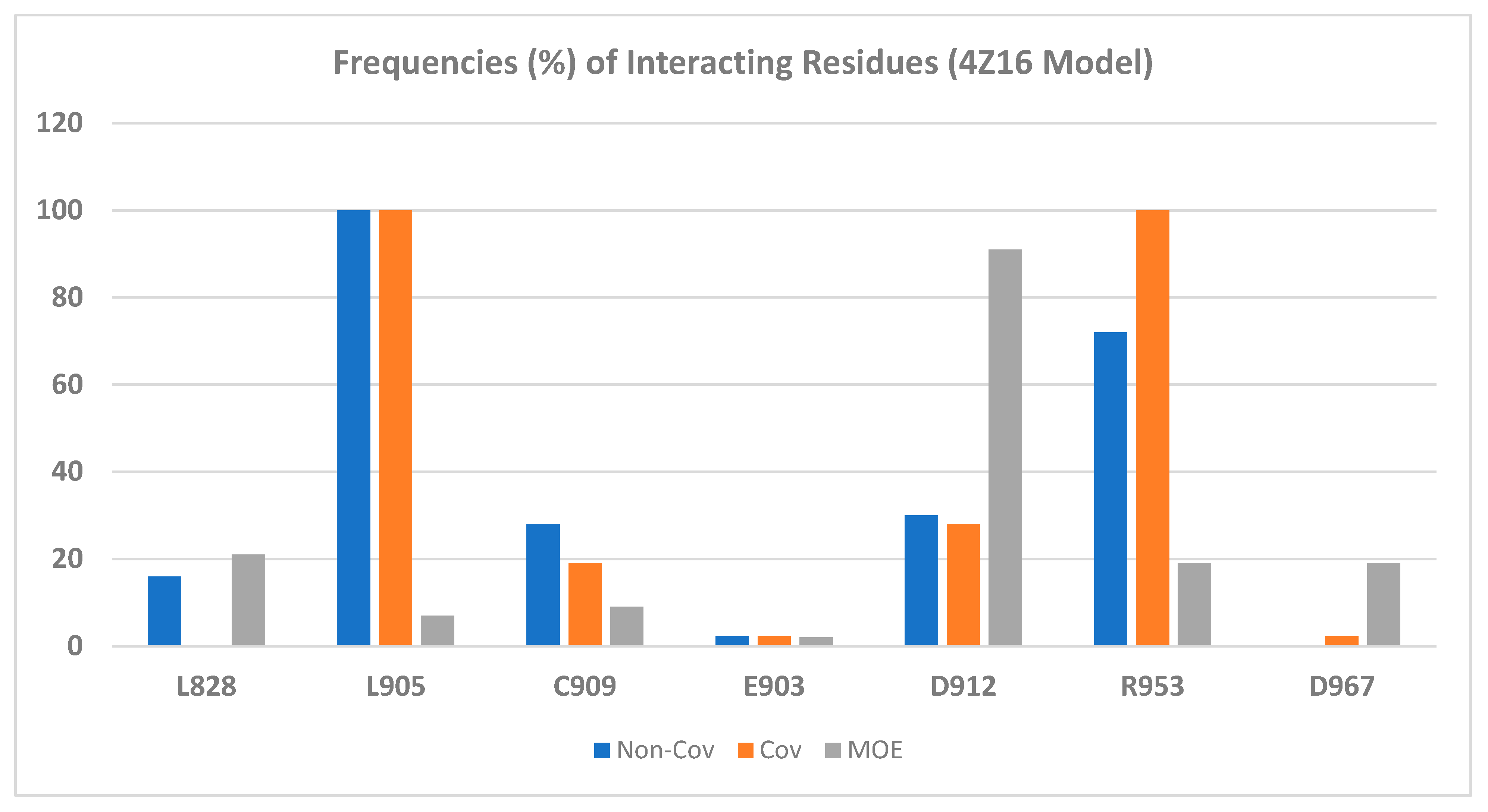
2.2.2. Binding Mode of Wild-Type JAK3 Model (4Z16)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | IC50 (nM) | ΔGExp (kcal/mol) | XP_NC | ΔΔG_NC | Res_NC | XP_Cov | ΔΔG_Cov | Res_Cov |
|---|---|---|---|---|---|---|---|---|
| 1 | 4.8 | −11.35 | −9.88 | 1.47 | D912, L905, R953 | −10.29 | 1.06 | L905, R953 |
| 2 | 46 | −10.01 | −9.97 | 0.04 | D912, L905, L828 | −10.15 | −0.14 | L905, R953, D912 |
| 3 | 2 | −11.87 | −10.54 | 1.32 | D912, L905, R953 | −10.45 | 1.42 | L905, R953, D912 |
| 4 | 20 | −10.50 | −10.96 | −0.46 | L905, R953 | −10.21 | 0.29 | L905, R953, D912 |
| 5 | 4.6 | −11.37 | −11.31 | 0.06 | L905, R953 | −10.51 | 0.86 | L905, R953 |
| 6 | 1.3 | −12.12 | −10.64 | 1.49 | L905, R953 | −9.74 | 2.38 | L905, R953 |
| 7 | 1.4 | −12.08 | −7.60 | 4.48 | L905, K855 | −10.11 | 1.97 | L905, R953 |
| 8 | 0.9 | −12.34 | −9.80 | 2.54 | L905, R953 | −10.12 | 2.22 | L905, R953 |
| 9 | 3.6 | −11.52 | −10.25 | 1.27 | L905, C909, L828 | −10.42 | 1.10 | L905, R953, R911 |
| 10 | 7.4 | −11.09 | −11.54 | −0.45 | D912, L905, R953 | −10.81 | 0.29 | L905, R953, D912 |
| 11 | 6.2 | −11.20 | −11.38 | −0.18 | L905, C909, R953 | −11.09 | 0.11 | L905, R953, C909 |
| 12 | 24 | −10.39 | −9.52 | 0.87 | L905, L828, R953 | −9.25 | 1.14 | L905, R953, P906 |
| 13 | 99 | −9.55 | −10.18 | −0.63 | L905, R953 | −9.67 | −0.12 | L905, R953 |
| 14 | 1600 | −7.91 | −10.37 | −2.46 | L905, R953 | −10.01 | −2.10 | L905, R953, C909 |
| 15 | 0.6 | −12.58 | −10.21 | 2.37 | L905, R953 | −9.88 | 2.70 | L905, R953 |
| 16 | 1.7 | −11.96 | −10.46 | 1.50 | L905, R953 | −10.02 | 1.94 | L905, R953 |
| 17 | 2.9 | −11.65 | −10.18 | 1.47 | L905, R953 | −9.68 | 1.96 | L905, R953 |
| 18 | 1.4 | −12.08 | −9.95 | 2.13 | L905, C909, L828 | −9.94 | 2.14 | L905, R953 |
| 19 | 0.7 | −12.49 | −9.85 | 2.64 | L905, R953 | −9.71 | 2.78 | L905, R953 |
| 20 | 0.5 | −12.69 | −11.60 | 1.08 | L905, C909, R953 | −10.89 | 1.80 | L905, R953, C909 |
| 21 | 1.1 | −12.22 | −11.43 | 0.79 | L905, C909, R953 | −10.88 | 1.35 | L905, R953, C909 |
| 22 | 0.6 | −12.58 | −10.52 | 2.06 | L905, C909, R953 | −10.67 | 1.91 | L905, R953, C909 |
| 23 | 0.6 | −12.58 | −9.62 | 2.96 | L905, L828 | −10.71 | 1.87 | L905, R953, C909 |
| 24 | 0.6 | −12.58 | −9.79 | 2.79 | L905, C909, L828 | −10.82 | 1.76 | L905, R953, Y904 |
| 25 | 7.8 | −11.06 | −9.50 | 1.56 | L905, C909 | −10.69 | 0.37 | L905, R953, Y904 |
| 26 | 1.2 | −12.17 | −10.06 | 2.11 | L905, R953 | −9.65 | 2.52 | L905, R953 |
| 27 | 0.7 | −12.49 | −8.88 | 3.61 | L905 | −9.71 | 2.78 | L905, R953 |
| 28 | 64 | −9.81 | −10.20 | −0.39 | L905, D912, R911 | −9.27 | 0.54 | L905, R953, D912 |
| 29 | 12 | −10.80 | −10.38 | 0.43 | L905, R953 | −9.73 | 1.07 | L905, R953, D912 |
| 30 | 30 | −10.26 | −11.31 | −1.04 | D912, L905, R953 | −10.32 | −0.05 | L905, R953 |
| 31 | 2.3 | −11.78 | −10.17 | 1.61 | D912, L905, R953 | −10.19 | 1.60 | L905, R953, D912 |
| 32 | 18 | −10.56 | −11.05 | −0.48 | D912, L905, R953 | −11.26 | −0.69 | L905, R953 |
| 33 | 31 | −10.24 | −10.84 | −0.59 | D912, L905, R953 | −10.29 | −0.05 | L905, R953, D912 |
| 34 | 4.4 | −11.40 | −10.20 | 1.20 | L905, R953 | −9.94 | 1.46 | L905, R953 |
| 35 | 83 | −9.66 | −9.28 | 0.38 | D912, L905, R953 | −9.88 | −0.22 | L905, R953, D912 |
| 36 | 0.87 | −12.36 | −9.96 | 2.40 | L905, C909 | −9.98 | 2.38 | L905, R953 |
| 37 | 0.58 | −12.60 | −10.25 | 2.35 | L905, R953 | −9.33 | 3.27 | L905, R953 |
| 38 | 2 | −11.87 | −11.32 | 0.55 | L905, C909, R953 | −10.77 | 1.10 | L905, R953, C909 |
| 39 | 1.3 | −12.12 | −11.33 | 0.79 | L905, C909, R953 | −10.23 | 1.90 | L905, R953, C909 |
| 40 | 0.7 | −12.49 | −11.16 | 1.33 | D912, L905, R953 | −10.96 | 1.53 | L905, R953, D912 |
| 41 | 0.9 | −12.34 | −11.02 | 1.32 | L905, R953 | −10.55 | 1.79 | L905, R953, D912 |
| 42 | 7 | −11.12 | −10.19 | 0.93 | D912, L905 | −9.66 | 1.46 | L905, R953 |
| 43 | 2720 | −7.59 | −7.97 | −0.38 | D912, L905, L828, C909, E903 | −9.23 | −1.63 | L905, R953, D912, E903 |
| Errors | 1.08 | 1.21 | ||||||
| StdEv | 1.38 | 1.19 |
2.2.3. Binding Mode of the JAK3 C1048S Mutant Model (5TTV)
2.2.4. Binding Selectivity of JAK3 Ligands towards Different Subtypes
3. Computational Methods
3.1. Multiple Sequence Alignment
3.2. Preparation of Protein Structures
3.3. Preparation of Ligand Structures
3.4. Glide Docking
3.5. Schrödinger Covalent Docking
3.6. MOE Covalent Docking
3.7. Binding Affinity and Protein-Ligand Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [Green Version]
- Leonard, W.J.; O′Shea, J.J. Jaks and STATs: Biological implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef] [Green Version]
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O′Shea, J.J. Mechanisms of Jak/STAT signaling in immunity and disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, S. Tofacitinib: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1987–2001. [Google Scholar] [CrossRef]
- Al-Salama, Z.T.; Scott, L.J. Baricitinib: A Review in Rheumatoid Arthritis. Drugs 2018, 78, 761–772. [Google Scholar] [CrossRef]
- Berekmeri, A.; Mahmood, F.; Wittmann, M.; Helliwell, P. Tofacitinib for the treatment of psoriasis and psoriatic arthritis. Expert Rev. Clin. Immunol. 2018, 14, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Clotet, A.; Castro-Poceiro, J.; Panés, J. Tofacitinib for the treatment of ulcerative colitis. Expert Rev. Clin. Immunol. 2018, 14, 881–892. [Google Scholar] [CrossRef]
- Mesa, R.A. Ruxolitinib, a selective JAK1 and JAK2 inhibitor for the treatment of myeloproliferative neoplasms and psoriasis. IDrugs Investig. Drugs J. 2010, 13, 394–403. [Google Scholar]
- Taylor, P.C.; Keystone, E.C.; van der Heijde, D.; Weinblatt, M.E.; Del Carmen Morales, L.; Reyes Gonzaga, J.; Yakushin, S.; Ishii, T.; Emoto, K.; Beattie, S.; et al. Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis. N. Engl. J. Med. 2017, 376, 652–662. [Google Scholar] [CrossRef]
- Rubin, R. Baricitinib Is First Approved COVID-19 Immunomodulatory Treatment. JAMA 2022, 327, 2281. [Google Scholar] [CrossRef]
- Richez, C.; Truchetet, M.E.; Kostine, M.; Schaeverbeke, T.; Bannwarth, B. Efficacy of baricitinib in the treatment of rheumatoid arthritis. Expert Opin. Pharmacother. 2017, 18, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, F.; Silva, H.T.; Busque, S.; O’Connell, P.J.; Russ, G.; Budde, K.; Yoshida, A.; Tortorici, M.A.; Lamba, M.; Lawendy, N.; et al. Evaluation of the effect of tofacitinib exposure on outcomes in kidney transplant patients. Am. J. Transpl. 2015, 15, 1644–1653. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, A.J.; Bowman, J.W.; Fici, G.J.; Zhang, M.; Mann, D.W.; Mitton-Fry, M. Oclacitinib (APOQUEL®) is a novel Janus kinase inhibitor with activity against cytokines involved in allergy. J. Vet. Pharmacol. Ther. 2014, 37, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Wollenhaupt, J.; Silverfield, J.; Lee, E.B.; Curtis, J.R.; Wood, S.P.; Soma, K.; Nduaka, C.I.; Benda, B.; Gruben, D.; Nakamura, H.; et al. Safety and efficacy of tofacitinib, an oral janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, longterm extension studies. J. Rheumatol. 2014, 41, 837–852. [Google Scholar] [CrossRef]
- Serhal, L.; Edwards, C.J. Upadacitinib for the treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 2019, 15, 13–25. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Anderson, J.K.; Magrey, M.; Merola, J.F.; Liu, Y.; Kishimoto, M.; Jeka, S.; Pacheco-Tena, C.; Wang, X.; Chen, L.; et al. Trial of Upadacitinib and Adalimumab for Psoriatic Arthritis. N. Engl. J. Med. 2021, 384, 1227–1239. [Google Scholar] [CrossRef]
- Forster, M.; Gehringer, M.; Laufer, S.A. Recent advances in JAK3 inhibition: Isoform selectivity by covalent cysteine targeting. Bioorg. Med. Chem. Lett. 2017, 27, 4229–4237. [Google Scholar] [CrossRef]
- Pesu, M.; Laurence, A.; Kishore, N.; Zwillich, S.H.; Chan, G.; O’Shea, J.J. Therapeutic targeting of Janus kinases. Immunol. Rev. 2008, 223, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Akahane, K.; McNally, R.; Reyskens, K.M.; Ficarro, S.B.; Liu, S.; Herter-Sprie, G.S.; Koyama, S.; Pattison, M.J.; Labella, K.; et al. Development of Selective Covalent Janus Kinase 3 Inhibitors. J. Med. Chem. 2015, 58, 6589–6606. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Chen, Z.; Liu, M.; He, R.; Liu, C.; Li, R.; Gao, M.; Zheng, M.; Tu, Z.; Zhang, Z.; et al. Design, synthesis and structure-activity relationship studies of pyrido[2,3-d]pyrimidin-7-ones as potent Janus Kinase 3 (JAK3) covalent inhibitors. Bioorg. Med. Chem. Lett. 2022, 64, 128680. [Google Scholar] [CrossRef]
- Shu, L.; Chen, C.; Huan, X.; Huang, H.; Wang, M.; Zhang, J.; Yan, Y.; Liu, J.; Zhang, T.; Zhang, D. Design, synthesis, and pharmacological evaluation of 4- or 6-phenyl-pyrimidine derivatives as novel and selective Janus kinase 3 inhibitors. Eur. J. Med. Chem. 2020, 191, 112148. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Chen, C.J.; Yu, R.N.; Wang, Z.J.; Zhang, T.T.; Zhang, D.Y. Structure-based design and synthesis of 1H-pyrazolo[3,4-d]pyrimidin-4-amino derivatives as Janus kinase 3 inhibitors. Bioorg. Med. Chem. 2018, 26, 4774–4786. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhong, Z.; Li, X.; Zhou, Y.; Pan, Z. Discovery of an Orally Available Janus Kinase 3 Selective Covalent Inhibitor. J. Med. Chem. 2019, 62, 1054–1066. [Google Scholar] [CrossRef]
- Xu, H.; Jesson, M.I.; Seneviratne, U.I.; Lin, T.H.; Sharif, M.N.; Xue, L.; Nguyen, C.; Everley, R.A.; Trujillo, J.I.; Johnson, D.S.; et al. PF-06651600, a Dual JAK3/TEC Family Kinase Inhibitor. ACS Chem. Biol. 2019, 14, 1235–1242. [Google Scholar] [CrossRef]
- Bao, Q.; Zhang, L.; Wang, N.; Gabet, B.; Yang, W.; Gao, X.; You, Q.; Jiang, Z. Hydrogen Peroxide Inducible JAK3 Covalent Inhibitor: Prodrug for the Treatment of RA with Enhanced Safety Profile. ACS Med. Chem. Lett. 2020, 11, 2182–2189. [Google Scholar] [CrossRef]
- Spergel, S.H.; Mertzman, M.E.; Kempson, J.; Guo, J.; Stachura, S.; Haque, L.; Lippy, J.S.; Zhang, R.F.; Galella, M.; Pitt, S.; et al. Discovery of a JAK1/3 Inhibitor and Use of a Prodrug To Demonstrate Efficacy in a Model of Rheumatoid Arthritis. ACS Med. Chem. Lett. 2019, 10, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Laux, J.; Forster, M.; Riexinger, L.; Schwamborn, A.; Guezguez, J.; Pokoj, C.; Kudolo, M.; Berger, L.M.; Knapp, S.; Schollmeyer, D.; et al. Pharmacokinetic Optimization of Small Molecule Janus Kinase 3 Inhibitors to Target Immune Cells. ACS Pharmacol. Transl. Sci. 2022, 5, 573–602. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meng, D.; Xie, J.; Li, R.; Wang, Z.; Li, J.; Mou, L.; Deng, X.; Deng, P. Design of Rational JAK3 Inhibitors Based on the Parent Core Structure of 1,7-Dihydro-Dipyrrolo [2,3-b:3′,2′-e] Pyridine. Int. J. Mol. Sci. 2022, 23, 5437. [Google Scholar] [CrossRef]
- Remenyi, J.; Naik, R.J.; Wang, J.; Razsolkov, M.; Verano, A.; Cai, Q.; Tan, L.; Toth, R.; Raggett, S.; Baillie, C.; et al. Generation of a chemical genetic model for JAK3. Sci. Rep. 2021, 11, 10093. [Google Scholar] [CrossRef]
- Vavalà, T. Role of afatinib in the treatment of advanced lung squamous cell carcinoma. Clin. Pharmacol. 2017, 9, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Remon, J.; Steuer, C.E.; Ramalingam, S.S.; Felip, E. Osimertinib and other third-generation EGFR TKI in EGFR-mutant NSCLC patients. Ann. Oncol. 2018, 29, i20–i27. [Google Scholar] [CrossRef]
- Timofeeva, N.; Gandhi, V. Ibrutinib combinations in CLL therapy: Scientific rationale and clinical results. Blood Cancer J. 2021, 11, 79. [Google Scholar] [CrossRef]
- Kulagowski, J.J.; Blair, W.; Bull, R.J.; Chang, C.; Deshmukh, G.; Dyke, H.J.; Eigenbrot, C.; Ghilardi, N.; Gibbons, P.; Harrison, T.K.; et al. Identification of imidazo-pyrrolopyridines as novel and potent JAK1 inhibitors. J. Med. Chem. 2012, 55, 5901–5921. [Google Scholar] [CrossRef] [PubMed]
- Brasca, M.G.; Nesi, M.; Avanzi, N.; Ballinari, D.; Bandiera, T.; Bertrand, J.; Bindi, S.; Canevari, G.; Carenzi, D.; Casero, D.; et al. Pyrrole-3-carboxamides as potent and selective JAK2 inhibitors. Bioorg. Med. Chem. 2014, 22, 4998–5012. [Google Scholar] [CrossRef]
- Thorarensen, A.; Dowty, M.E.; Banker, M.E.; Juba, B.; Jussif, J.; Lin, T.; Vincent, F.; Czerwinski, R.M.; Casimiro-Garcia, A.; Unwalla, R.; et al. Design of a Janus Kinase 3 (JAK3) Specific Inhibitor 1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one (PF-06651600) Allowing for the Interrogation of JAK3 Signaling in Humans. J. Med. Chem. 2017, 60, 1971–1993. [Google Scholar] [CrossRef]
- Leit, S.; Greenwood, J.R.; Mondal, S.; Carriero, S.; Dahlgren, M.; Harriman, G.C.; Kennedy-Smith, J.J.; Kapeller, R.; Lawson, J.P.; Romero, D.L.; et al. Potent and selective TYK2-JH1 inhibitors highly efficacious in rodent model of psoriasis. Bioorg. Med. Chem. Lett. 2022, 73, 128891. [Google Scholar] [CrossRef]
- The Clustal Omega. Available online: https://www.ebi.ac.uk/Tools/msa/clustalo/ (accessed on 30 June 2022).
- MOE. The Molecular Operating EnVironment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2021. [Google Scholar]
- Schrödinger Suite 2019-1 Protein Preparation Wizard, Maestro, Protein Grid Generation, Glide, Macromodel, Epik, and Covalent Dock; Schrödinger, LLC.: New York, NY, USA, 2019.
- Hynes, J., Jr.; Wu, H.; Kempson, J.; Duan, J.J.; Lu, Z.; Jiang, B.; Stachura, S.; Tokarski, J.S.; Sack, J.S.; Khan, J.A.; et al. Discovery of potent and efficacious pyrrolopyridazines as dual JAK1/3 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3101–3106. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, T.T.; Betz, R.M.; Cai, Q.W.; Cerutti, D.S.; Cheatham, T.; Darden, T.; Duke, R.E.; Gohllke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [Green Version]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Koch, P.; Laufer, S.A.; Knapp, S. The Cysteinome of Protein Kinases as a Target in Drug Development. Angew Chem. Int. Ed. Engl. 2018, 57, 4372–4385. [Google Scholar] [CrossRef] [PubMed]








| Proteins | 826 | 828 | 833 | 855 | 905 | 909 | 911 | 912 | 953 | 967 | 985 | 988 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4E4N (JAK1) | R879 | L881 | F886 | K908 | L959 | S963 | K965 | E966 | R1007 | D1021 | D1039 | D1042 |
| 4D1S (JAK2) | Q853 | L855 | F860 | K882 | L932 | S936 | R938 | D939 | R980 | D994 | E1012 | E1015 |
| 4Z16 (JAK3) | S826 | L828 | F833 | K855 | L905 | C909 | R911 | D912 | R953 | D967 | E985 | Q988 |
| 7UYT (TYK2) | R901 | L903 | F908 | K930 | V981 | S985 | R987 | D988 | R1027 | D1041 | E1059 | D1062 |
| Compd. | IC50 (nM) | ΔGExp (kcal/mol) | XP_NC | ΔΔG_NC | Res_NC | XP_Cov | ΔΔG_Cov | Res_Cov |
|---|---|---|---|---|---|---|---|---|
| 1 | 4.8 | −11.35 | −8.90 | 2.44 | L905, D912, R911 | −7.31 | 4.04 | R911 |
| 2 | 46 | −10.01 | −8.28 | 1.73 | L905, D912 | −7.80 | 2.21 | NA |
| 3 | 2 | −11.87 | −9.49 | 2.38 | L905, R911, L828 | −9.07 | 2.79 | L905, R911, L828 |
| 4 | 20 | −10.50 | −9.59 | 0.91 | L905 | −8.99 | 1.51 | L905, D912 |
| 5 | 4.6 | −11.37 | −8.50 | 2.87 | L905 | −7.22 | 4.15 | N954 |
| 6 | 1.3 | −12.12 | −6.98 | 5.15 | D967, N954 | −8.50 | 3.62 | L905, R911, L828 |
| 7 | 1.4 | −12.08 | −7.52 | 4.56 | D967, N954 | −7.42 | 4.66 | R911, R953 |
| 8 | 0.9 | −12.34 | −9.08 | 3.26 | L905 | −8.43 | 3.91 | L905, L828 |
| 9 | 3.6 | −11.52 | −6.99 | 4.52 | L905, R911, K830 | −8.59 | 2.93 | L905 |
| 10 | 7.4 | −11.09 | −7.66 | 3.44 | Q988, R911, K830 | −8.53 | 2.56 | L905, R911, D912 |
| 11 | 6.2 | −11.20 | −10.45 | 0.74 | L905, R916, K830 | −8.70 | 2.50 | L905 |
| 12 | 24 | −10.39 | −7.40 | 2.99 | L905, K830 | −8.17 | 2.22 | L905, R911 |
| 13 | 99 | −9.55 | −9.59 | −0.04 | L905, R911, L828 | −8.49 | 1.06 | L905, L828, R911 |
| 14 | 1600 | −7.91 | −9.13 | −1.22 | L905, R911 | −7.63 | 0.27 | C909 |
| 15 | 0.6 | −12.58 | −7.34 | 5.23 | L905 | −8.36 | 4.22 | L905, R911 |
| 16 | 1.7 | −11.96 | −8.97 | 2.99 | L905, K830 | −8.72 | 3.24 | L905, R911 |
| 17 | 2.9 | −11.65 | −8.49 | 3.15 | L905, R911 | −8.30 | 3.35 | L905, L828, R911 |
| 18 | 1.4 | −12.08 | −8.93 | 3.15 | L905 | −7.46 | 4.62 | L905, C909 |
| 19 | 0.7 | −12.49 | −8.37 | 4.12 | L905, R911 | −8.48 | 4.01 | L905 |
| 20 | 0.5 | −12.69 | −9.16 | 3.52 | L905, R911, Y904 | −8.38 | 4.31 | L905, R911, C909 |
| 21 | 1.1 | −12.22 | −7.50 | 4.72 | N954, R911, K830 | −9.47 | 2.75 | L905, R911, C909 |
| 22 | 0.6 | −12.58 | −8.85 | 3.73 | L905, R911, R916 | −9.39 | 3.19 | L905, L828, C909, R911 |
| 23 | 0.6 | −12.58 | −7.61 | 4.97 | L905, R911, K830 | −9.70 | 2.88 | L905, C909, R911 |
| 24 | 0.6 | −12.58 | −9.80 | 2.78 | L905, L828 | −9.05 | 3.53 | L905, R911 |
| 25 | 7.8 | −11.06 | −9.52 | 1.54 | L905, R953 | −9.69 | 1.37 | R911, C909, L905 |
| 26 | 1.2 | −12.17 | −8.75 | 3.42 | L905 | −8.28 | 3.88 | L905, R911 |
| 27 | 0.7 | −12.49 | −6.22 | 6.26 | D967 | −8.64 | 3.85 | L905, R911 |
| 28 | 64 | −9.81 | −8.41 | 1.41 | L905, N954 | −7.08 | 2.74 | N954 |
| 29 | 12 | −10.80 | −6.91 | 3.90 | D912 | −8.67 | 2.14 | C909 |
| 30 | 30 | −10.26 | −8.16 | 2.11 | L905, R911, D912 | −7.71 | 2.55 | NA |
| 31 | 2.3 | −11.78 | −8.18 | 3.60 | L905, N954 | −8.50 | 3.28 | C909 |
| 32 | 18 | −10.56 | −5.96 | 4.61 | Y904, D967 | −7.98 | 2.58 | R911 |
| 33 | 31 | −10.24 | −6.56 | 3.69 | R911 | −7.65 | 2.59 | na |
| 34 | 4.4 | −11.40 | −8.02 | 3.38 | L905 | −9.40 | 2.00 | L905, R911 |
| 35 | 83 | −9.66 | −8.80 | 0.86 | L905 | −7.81 | 1.85 | R911, D912 |
| 36 | 0.87 | −12.36 | −9.20 | 3.16 | L905 | −7.90 | 4.46 | R911, L905 |
| 37 | 0.58 | −12.60 | −9.66 | 2.94 | L905 | −7.80 | 4.80 | L828, D967 |
| 38 | 2 | −11.87 | −10.07 | 1.79 | L905, L828 | −8.72 | 3.14 | Y904, L905 |
| 39 | 1.3 | −12.12 | −9.87 | 2.25 | L905, R911 | −9.67 | 2.45 | L905, C909 |
| 40 | 0.7 | −12.49 | −9.43 | 3.06 | L905, D912 | −9.19 | 3.30 | L905, R911 |
| 41 | 0.9 | −12.34 | −6.67 | 5.67 | L905, D912 | −8.81 | 3.53 | L905, R911, E903, S989, L828 |
| 42 | 7 | −11.12 | −8.52 | 2.60 | L905, D912 | −9.23 | 1.90 | L905, R911 |
| 43 | 2720 | −7.59 | −7.73 | −0.14 | E903, D912 | −7.95 | −0.35 | D912, R911, E903 |
| Errors | 2.98 | 2.94 | ||||||
| StdEv | 1.59 | 1.15 |
| JAK1 | R879 | F886 | K908 | L959 | K965 | E966 | R1007 | D1021 | D1039 | D1042 |
| frequency (%) | 2 | 30 | 28 | 44 | 0 | 0 | 21 | 35 | 21 | 21 |
| JAK2 | Q853 | F860 | K882 | L932 | R938 | D939 | R980 | D994 | E1012 | E1015 |
| frequency (%) | 0 | 7 | 0 | 67 | 49 | 23 | 42 | 0 | 0 | 0 |
| TYK2 | R901 | F908 | K930 | V981 | R987 | D988 | R1027 | D1041 | E1059 | D1062 |
| frequency (%) | 53 | 0 | 0 | 67 | 5 | 37 | 5 | 23 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, H.A.; Almahmoud, S. Docking and Selectivity Studies of Covalently Bound Janus Kinase 3 Inhibitors. Int. J. Mol. Sci. 2023, 24, 6023. https://doi.org/10.3390/ijms24076023
Zhong HA, Almahmoud S. Docking and Selectivity Studies of Covalently Bound Janus Kinase 3 Inhibitors. International Journal of Molecular Sciences. 2023; 24(7):6023. https://doi.org/10.3390/ijms24076023
Chicago/Turabian StyleZhong, Haizhen A., and Suliman Almahmoud. 2023. "Docking and Selectivity Studies of Covalently Bound Janus Kinase 3 Inhibitors" International Journal of Molecular Sciences 24, no. 7: 6023. https://doi.org/10.3390/ijms24076023