
Transcriptome-Based WGCNA Analysis Reveals the Mechanism of Drought Resistance Differences in Sweetpotato (Ipomoea batatas (L.) Lam.)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
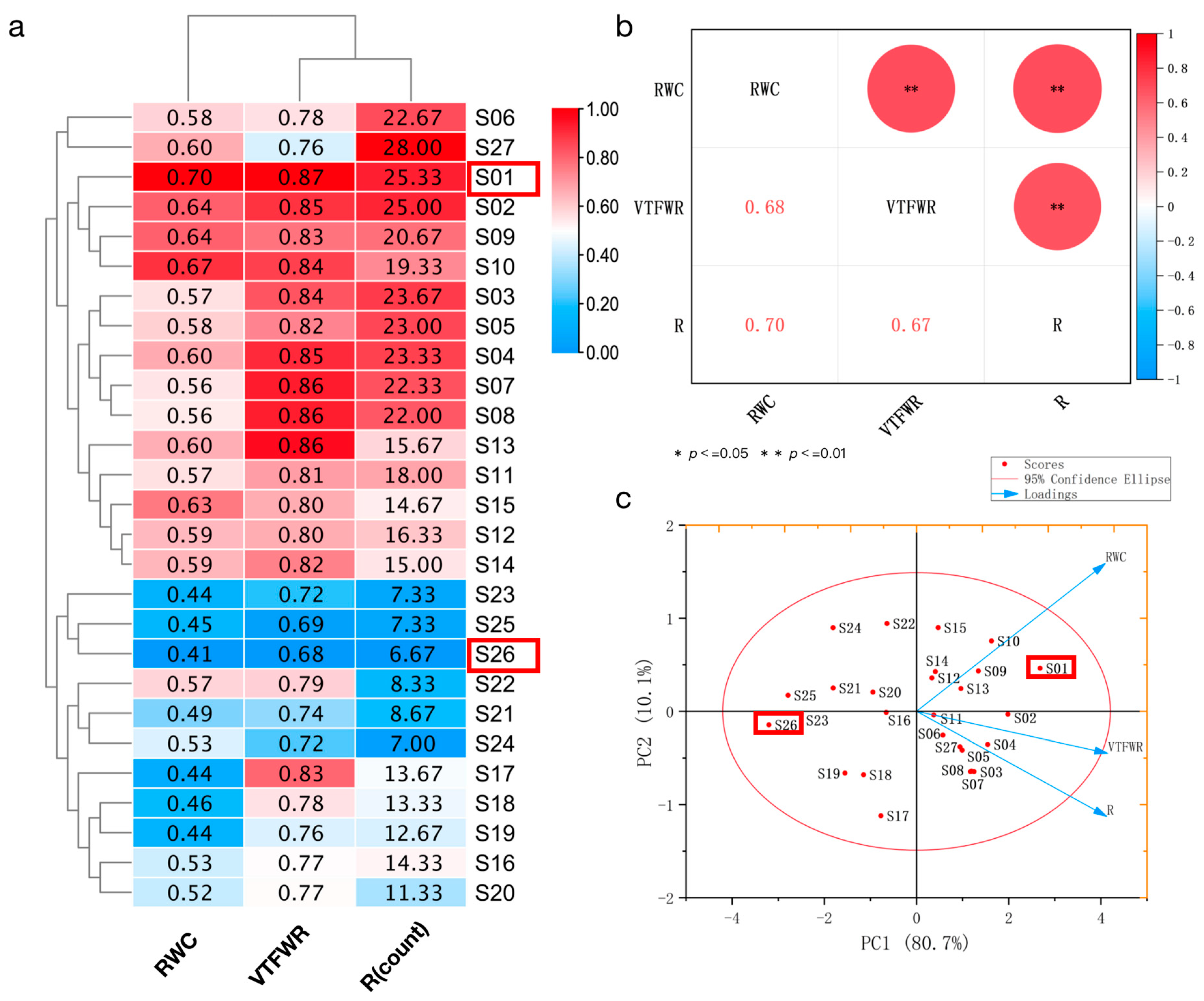
2.1. Selection of Drought-Resistant Sweetpotato Genotypes
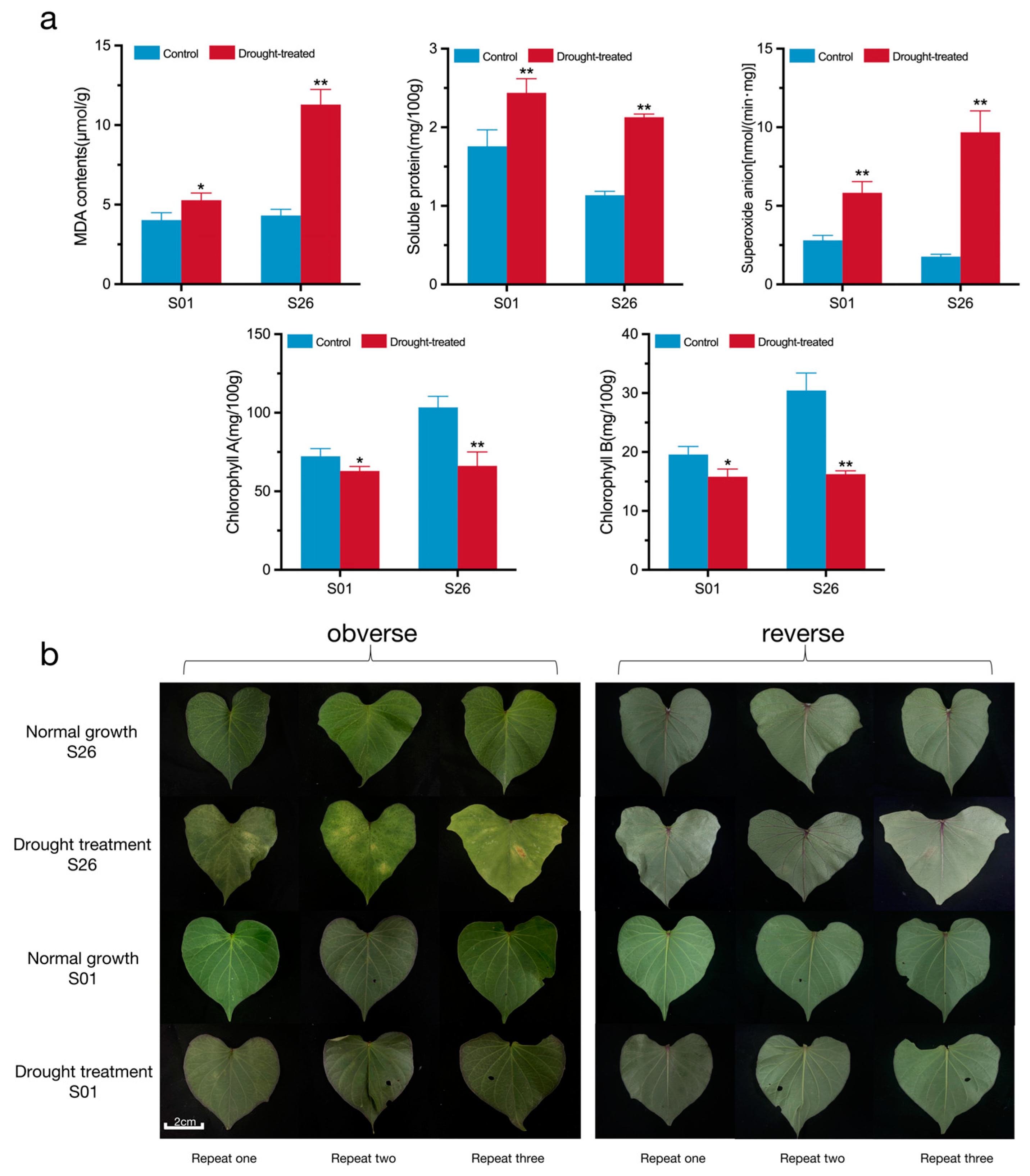
2.2. Physiological Responses of Sweetpotato Genotypes S01 and S26 to Drought Stress
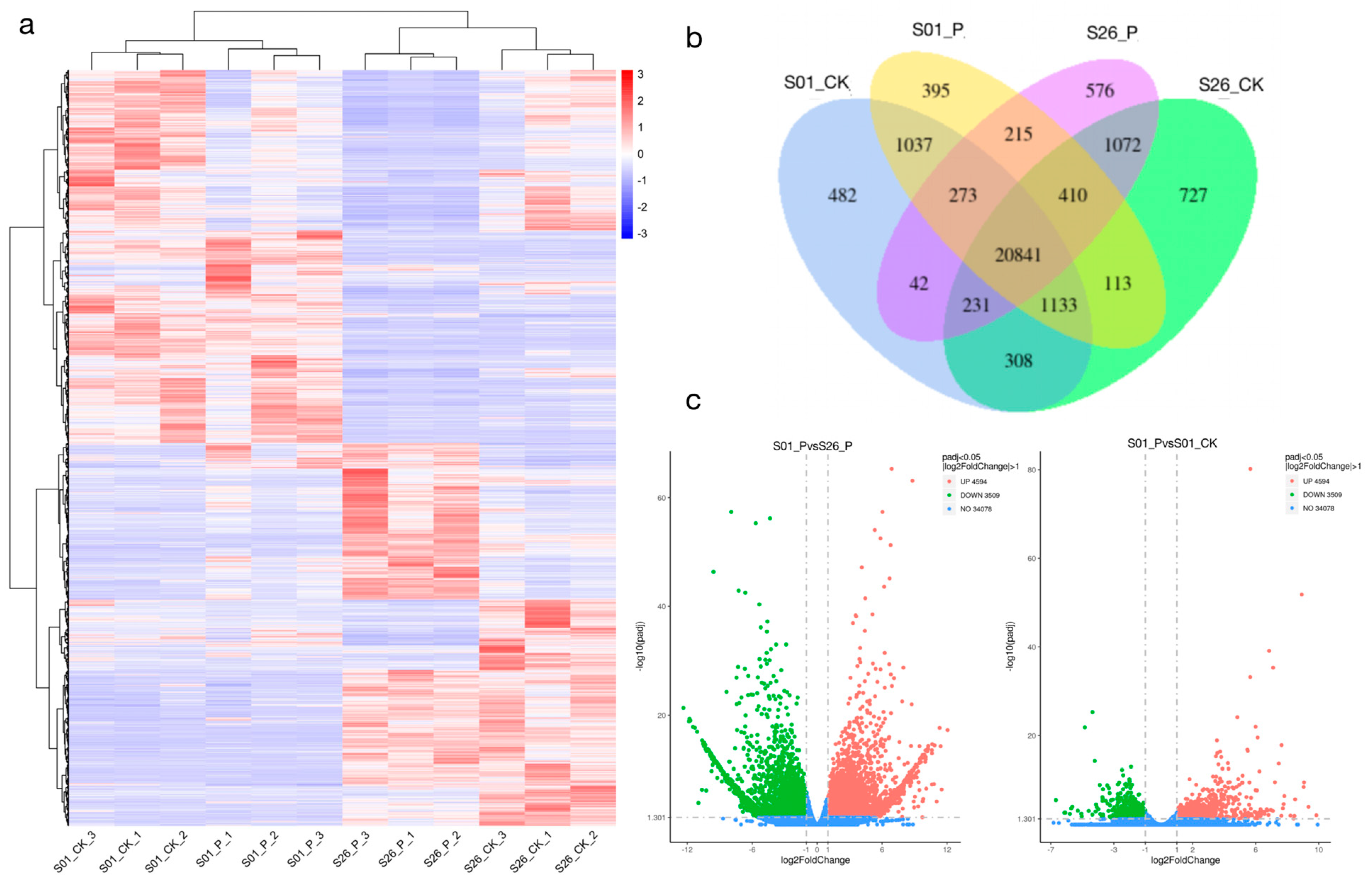
2.3. Transcriptome Sequencing Analysis
2.4. Differentially Expressed Genes (DEGs) Analysis
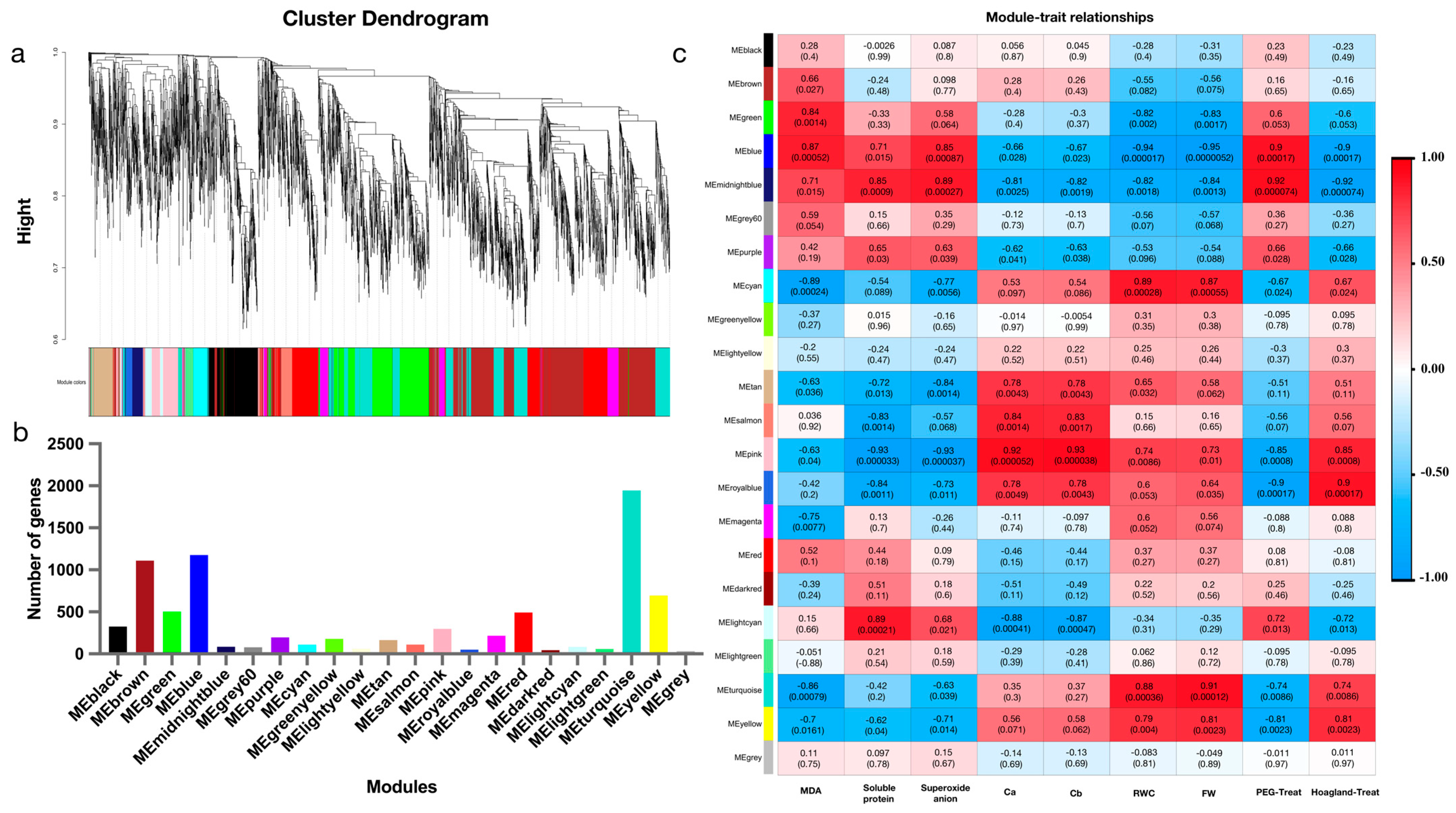
2.5. Key Gene Modules for Drought Tolerance Screened by WGCNA
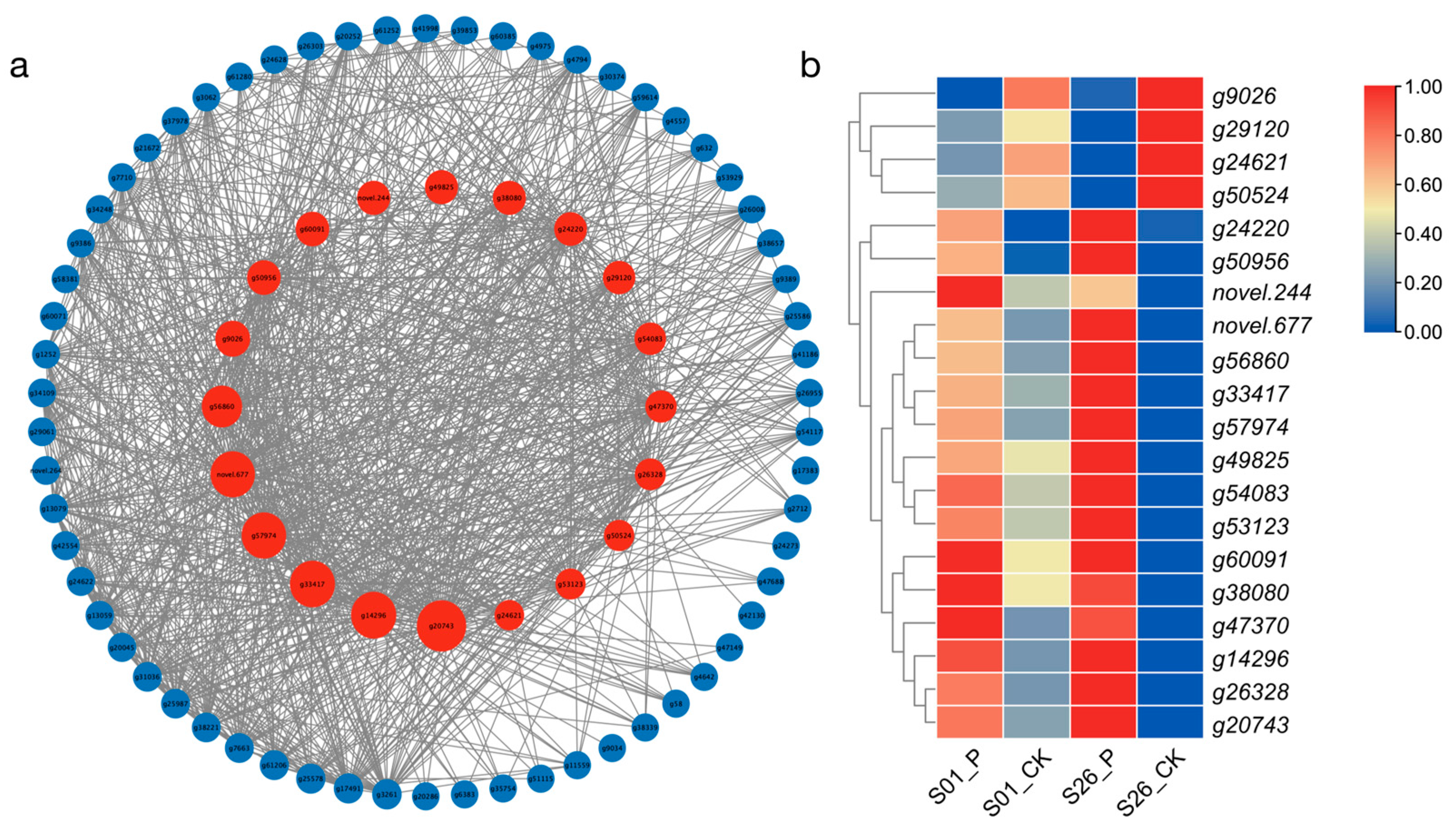
2.6. Hub Genes for Drought Tolerance Screened by WGCNA
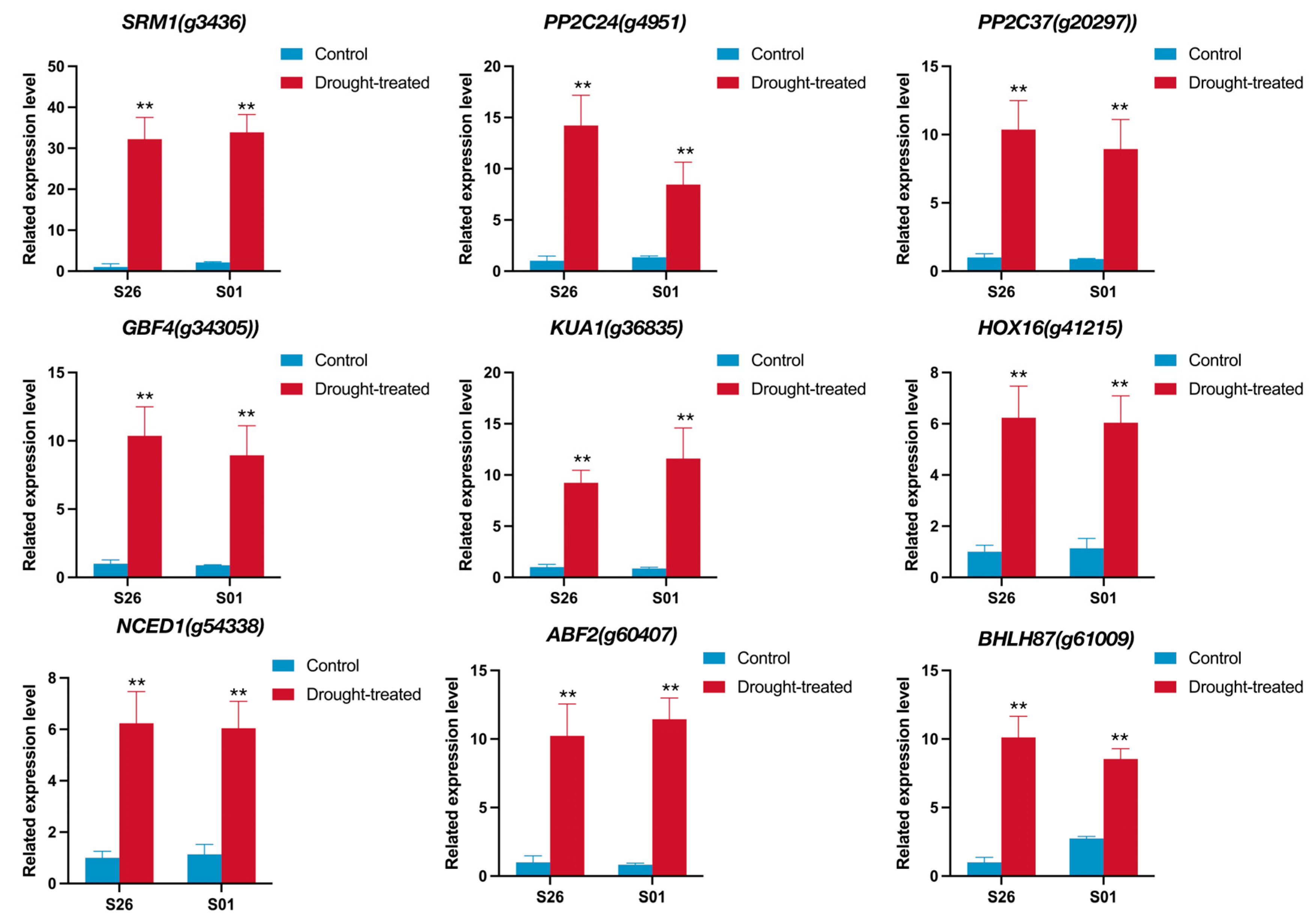
2.7. qRTPCR Validation of RNA-Seq Data
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Determination of Water Loss and Counting Rooting Number after PEG Treatment
4.3. Determination of the Contents of Malondialdehyde, Soluble Protein, and Superoxide Anion in the Blade
4.4. Chlorophyll Content Determination
4.5. cDNA Library Construction and RNA-Seq
4.6. Transcriptome Assembly and Functional Annotation
4.7. Differentially Expressed Genes (DEGs) Analysis
4.8. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.9. Quantitative Real-Time PCR (qRT-PCR) Analysis of DEGs
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- El Sheikha, A.F.; Ray, R.C. Potential impacts of bioprocessing of sweet potato: Review. Crit. Rev. Food Sci. Nutr. 2017, 57, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Vithu, P.; Dash, S.K.; Rayaguru, K. Post-harvest processing and utilization of sweet potato: A review. Food Rev. Int. 2019, 35, 726–762. [Google Scholar]
- Ramírez, D.A.; Grüneberg, W.; Andrade, M.I.; De Boeck, B.; Loayza, H.; Makunde, G.S.; Ninanya, J.; Rinza, J.; Heck, S.; Campos, H. Phenotyping of productivity and resilience in sweetpotato under water stress through UAV-based multispectral and thermal imagery in Mozambique. J. Agron. Crop Sci. 2021, 209, 41–55. [Google Scholar] [CrossRef]
- Jia, L.; Yang, Y.F.; Zhai, H.; He, S.Z.; Xin, G.S.; Zhao, N.; Zhang, H.; Gao, S.P.; Liu, Q.C. Production and characterization of a novel interspecific somatic hybrid combining drought tolerance and high quality of sweet potato and Ipomoea triloba L. Plant Cell Rep. 2022, 41, 2159–2171. [Google Scholar] [CrossRef] [PubMed]
- Shu, G.P.; Tang, Y.L.; Yuan, M.Y.; Wei, N.; Zhang, F.Y.; Yang, C.X.; Lan, X.Z.; Chen, M.; Tang, K.X.; Xiang, L.; et al. Molecular insights into AabZIP1-mediated regulation on artemisinin biosynthesis and drought tolerance in Artemisia annua. Acta. Pharm. Sin. B 2022, 12, 1500–1513. [Google Scholar] [CrossRef]
- Rong, W.; Qi, L.; Wang, A.Y.; Ye, X.G.; Du, L.P.; Liang, H.X.; Xin, Z.Y.; Zhang, Z.Y. The ERF transcription factor TaERF3 promotes tolerance to salt and drought stresses in wheat. Plant Biotechnol. J. 2014, 12, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; El-Badri, A.M.; Wang, Z.K.; Mohamed, I.a.A.; Yang, H.Y.; Ai, X.Y.; Salah, A.; Hassan, M.U.; Sami, R.; Kuai, J.; et al. Rapeseed morpho-physio-biochemical responses to drought stress induced by PEG-6000. Agronomy 2022, 12, 579. [Google Scholar] [CrossRef]
- Ayaz, M.; Ahmad, R.; Shahzad, M.; Khan, N.; Shah, M.M.; Khan, S.A. Drought stress stunt tomato plant growth and up-regulate expression of SlAREB, SlNCED3, and SlERF024 genes. Sci. Hortic. 2015, 195, 48–55. [Google Scholar] [CrossRef]
- Cui, G.P.; Zhao, Y.F.; Zhang, J.L.; Chao, M.N.; Xie, K.L.; Zhang, C.; Sun, F.L.; Liu, S.D.; Xi, Y.J. Proteomic analysis of the similarities and differences of soil drought and polyethylene glycol stress responses in wheat (Triticum aestivum L.). Plant Mol. Biol. 2019, 100, 391–410. [Google Scholar] [CrossRef]
- Ma, L.J.; Wang, L.L.; Mei, Y.X.; Zhang, S.W.; Wei, W.; Wang, J.Y.; Zhang, Y.L. Cross adaptation tolerance in rice seedlings exposed to PEG induced salinity and drought stress. Int. J. Agric. Biol. 2016, 18, 535–541. [Google Scholar] [CrossRef]
- Yooyongwech, S.; Theerawitaya, C.; Samphumphuang, T.; Cha-Um, S. Water-deficit tolerant identification in sweet potato genotypes (Ipomoea batatas (L.) Lam.) in vegetative developmental stage using multivariate physiological indices. Sci. Hortic. 2013, 162, 242–251. [Google Scholar] [CrossRef]
- Arunyanark, A.; Jogloy, S.; Akkasaeng, C.; Vorasoot, N.; Kesmala, T.; Rao, R.C.N.; Wright, G.C.; Patanothai, A. Chlorophyll stability is an indicator of drought tolerance in peanut. J. Agron. Crop Sci. 2008, 194, 113–125. [Google Scholar] [CrossRef]
- Rontein, D.; Basset, G.; Hanson, A.D. Metabolic engineering of osmoprotectant accumulation in plants. Metab. Eng. 2002, 4, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Bai, L.; Zhang, Y.; Zhang, G.T.; Mao, Y.Q.; Min, L.L.; Xiang, F.Y.; Qian, D.D.; Zhu, X.H.; Song, C.P. Modulation of guard cell turgor and drought tolerance by a peroxisomal acetate-malate shunt. Mol. Plant 2018, 11, 1278–1291. [Google Scholar] [CrossRef]
- Melo, A.S.; Yule, T.S.; Barros, V.A.; Rivas, R.; Santos, M.G. C3-species Calotropis procera increase specific leaf area and decrease stomatal pore size, alleviating gas exchange under drought and salinity. Acta Physiol. Plant. 2021, 43, 140. [Google Scholar] [CrossRef]
- Bao, X.Y.; Hou, X.Y.; Duan, W.W.; Yin, B.Z.; Ren, J.J.; Wang, Y.D.; Liu, X.J.; Gu, L.M.; Zhen, W.C. Screening and evaluation of drought resistance traits of winter wheat in the North China Plain. Front. Plant Sci. 2023, 14, 1194759. [Google Scholar] [CrossRef]
- Sharma, A.; Wang, J.F.; Xu, D.B.; Tao, S.C.; Chong, S.L.; Yan, D.L.; Li, Z.; Yuan, H.W.; Zheng, B.S. Melatonin regulates the functional components of photosynthesis, antioxidant system, gene expression, and metabolic pathways to induce drought resistance in grafted Carya cathayensis plants. Sci. Total Environ. 2020, 713, 136675. [Google Scholar] [CrossRef]
- Abid, M.; Ali, S.; Qi, L.K.; Zahoor, R.; Tian, Z.; Jiang, D.; Snider, J.L.; Dai, T. Physiological and biochemical changes during drought and recovery periods at tillering and jointing stages in wheat (Triticum aestivum L.). Sci. Rep. 2018, 8, 4615. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, V.; Shahzad, B.; Ramakrishnan, M.; Singh Sidhu, G.P.; Bali, A.S.; Handa, N.; Kapoor, D.; Yadav, P.; Khanna, K.; et al. Photosynthetic response of plants under different abiotic stresses: A review. J. Plant Growth Regul. 2019, 39, 509–531. [Google Scholar] [CrossRef]
- Zhang, K.; Wu, Z.D.; Tang, D.B.; Luo, K.; Lu, H.X.; Liu, Y.Y.; Dong, J.; Wang, X.; Lv, C.W.; Wang, J.C.; et al. Comparative Transcriptome Analysis Reveals Critical Function of Sucrose Metabolism Related-Enzymes in Starch Accumulation in the Storage Root of Sweet Potato. Front. Plant Sci. 2017, 8, 914. [Google Scholar] [CrossRef]
- Chen, P.T.; Ran, H.R.; Li, J.X.; Zong, J.K.; Luo, Q.Q.; Zhao, T.F.; Liao, Z.H.; Tang, Y.L.; Fu, Y.F. Antioxidant activity of phenolic extraction from different sweetpotato (Ipomoea batatas (L.) Lam.) blades and comparative transcriptome analysis reveals differentially expressed genes of phenolic metabolism in two genotypes. Genes 2022, 13, 1078. [Google Scholar] [CrossRef] [PubMed]
- Imran, Q.M.; Falak, N.; Hussain, A.; Mun, B.G.; Yun, B.W. Abiotic stress in plants; stress perception to molecular response and role of biotechnological tools in stress resistance. Agronomy 2021, 11, 1579. [Google Scholar] [CrossRef]
- El-Sharkawy, I.; Liang, D.; Xu, K. Transcriptome analysis of an apple (Malus x domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Bot. 2015, 66, 7359–7376. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.Q.; Cai, Z.P.; Huang, J.L.; Wang, A.Q.; Ntambiyukuri, A.; Chen, B.M.; Zheng, G.H.; Li, H.F.; Huang, Y.M.; Zhan, J.; et al. Transcriptomic analysis of tuberous root in two sweet potato varieties reveals the important genes and regulatory pathways in tuberous root development. BMC Genom. 2022, 23, 473. [Google Scholar] [CrossRef]
- Liu, E.L.; Xu, L.L.; Luo, Z.Q.; Li, Z.Q.; Zhou, G.H.; Gao, H.F.; Fang, F.R.; Tang, J.; Zhao, Y.; Zhou, Z.; et al. Transcriptomic analysis reveals mechanisms for the different drought tolerance of sweet potatoes. Front. Plant Sci. 2023, 14, 1136709. [Google Scholar] [CrossRef]
- Magwanga, R.O.; Lu, P.; Kirungu, J.N.; Cai, X.Y.; Zhou, Z.L.; Wang, X.X.; Diouf, L.; Xu, Y.C.; Hou, Y.Q.; Hu, Y.Q.; et al. Whole genome analysis of cyclin dependent kinase (CDK) gene family in cotton and functional evaluation of the role of CDKF4 gene in drought and salt stress tolerance in plants. Int. J. Mol. Sci. 2018, 19, 2625. [Google Scholar] [CrossRef]
- Zheng, Y.Y.; Wang, N.; Zhang, Z.Y.; Liu, W.H.; Xie, W.G. Identification of flowering regulatory networks and hub genes expressed in the leaves of Elymus sibiricus L. Using Comparative Transcriptome Analysis. Front. Plant Sci. 2022, 13, 877908. [Google Scholar] [CrossRef]
- Kim, K.N.; Cheong, Y.H.; Grant, J.J.; Pandey, G.K.; Luan, S. CIPK3, a calcium sensor-associated protein kinase that regulates abscisic acid and cold signal transduction in Arabidopsis. Plant Cell 2003, 15, 411–423. [Google Scholar] [CrossRef]
- Kanwar, P.; Sanyal, S.K.; Tokas, I.; Yadav, A.K.; Pandey, A.; Kapoor, S.; Pandey, G.K. Comprehensive structural, interaction and expression analysis of CBL and CIPK complement during abiotic stresses and development in rice. Cell Calcium 2014, 56, 81–95. [Google Scholar] [CrossRef]
- Lv, Q.; Zhang, L.; Zan, T.; Li, L.; Li, X.J. Wheat RING E3 ubiquitin ligase TaDIS1 degrade TaSTP via the 26S proteasome pathway. Plant Sci. 2020, 296, 110494. [Google Scholar] [CrossRef]
- Garcia, M.E.; Lynch, T.; Peeters, J.; Snowden, C.; Finkelstein, R. A small plant-specific protein family of ABI five binding proteins (AFPs) regulates stress response in germinating Arabidopsis seeds and seedlings. Plant Mol. Biol. 2008, 67, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Daryanto, S.; Wang, L.X.; Jacinthe, P.A. Drought effects on root and tuber production: A meta-analysis. Agric. Water Manag. 2016, 176, 122–131. [Google Scholar] [CrossRef]
- Laurie, S.M.; Bairu, M.W.; Laurie, R.N. Analysis of the nutritional composition and drought tolerance traits of sweet potato: Selection criteria for breeding lines. Plants 2022, 11, 1804. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.L.; Zhao, Q.; Chen, L.R.; Yao, X.D.; Zhang, W.; Zhang, B.; Xie, F.T. Effect of drought stress on sugar metabolism in leaves and roots of soybean seedlings. Plant Physiol. Biochem. 2020, 146, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.K.; Dubeaux, G.; Takahashi, Y.; Schroeder, J.I. Signaling mechanisms in abscisic acid-mediated stomatal closure. Plant J. 2021, 105, 307–321. [Google Scholar] [CrossRef]
- Kang, G.Z.; Li, G.Z.; Xu, W.; Peng, X.Q.; Han, Q.X.; Zhu, Y.J.; Guo, T.C. Proteomics reveals the effects of salicylic acid on growth and tolerance to subsequent drought stress in wheat. J. Proteome Res. 2012, 11, 6066–6079. [Google Scholar] [CrossRef]
- Salehin, M.; Li, B.H.; Tang, M.; Katz, E.; Song, L.; Ecker, J.R.; Kliebenstein, D.J.; Estelle, M. Auxin-sensitive Aux/IAA proteins mediate drought tolerance in Arabidopsis by regulating glucosinolate levels. Nat. Commun. 2019, 10, 4021. [Google Scholar] [CrossRef]
- Wang, S.K.; Bai, Y.H.; Shen, C.J.; Wu, Y.R.; Zhang, S.N.; Jiang, D.A.; Guilfoyle, T.J.; Chen, M.; Qi, Y.H. Auxin-related gene families in abiotic stress response in Sorghum bicolor. Funct. Integr. Genom. 2010, 10, 533–546. [Google Scholar] [CrossRef]
- Wang, F.B.; Niu, H.F.; Xin, D.Q.; Long, Y.; Wang, G.P.; Liu, Z.M.; Li, G.; Zhang, F.; Qi, M.Y.; Ye, Y.X.; et al. OsIAA18, an Aux/IAA transcription factor gene, is involved in Salt and drought tolerance in rice. Front. Plant Sci. 2021, 12, 738660. [Google Scholar] [CrossRef]
- Miao, J.; Li, X.F.; Li, X.B.; Tan, W.C.; You, A.Q.; Wu, S.J.; Tao, Y.J.; Chen, C.; Wang, J.; Zhang, D.P.; et al. OsPP2C09, a negative regulatory factor in abscisic acid signalling, plays an essential role in balancing plant growth and drought tolerance in rice. New Phytol. 2020, 227, 1417–1433. [Google Scholar] [CrossRef]
- Xu, T.T.; Meng, S.; Zhu, X.P.; Di, J.C.; Zhu, Y.; Yang, X.; Yan, W. Integrated GWAS and transcriptomic analysis reveal the candidate salt-responding genes regulating Na(+)/K(+) balance in barley (Hordeum vulgare L.). Front. Plant Sci. 2022, 13, 1004477. [Google Scholar] [CrossRef]
- Sairam, R.K.; Rao, K.V.; Srivastava, G.C. Differential response of wheat genotypes to long term salinity stress in relation to oxidative stress, antioxidant activity and osmolyte concentration. Plant Sci. 2002, 163, 1037–1046. [Google Scholar] [CrossRef]
- Hodges, D.M.; Toivonen, P.M.A. Quality of fresh-cut fruits and vegetables as affected by exposure to abiotic stress. Postharvest Biol. Technol. 2008, 48, 155–162. [Google Scholar] [CrossRef]
- Wang, X.Y.; Wu, Z.H.; Zhou, Q.; Wang, X.; Song, S.; Dong, S.K. Physiological response of soybean plants to water deficit. Front. Plant Sci. 2021, 12, 809692. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.B.; Zhao, X.X.; Liu, S.D.; Sun, F.L.; Zhang, C.; Xi, Y.J. Beneficial effects of melatonin in overcoming drought stress in wheat seedlings. Plant Physiol. Biochem. 2017, 118, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yu, X.Q.; Ottosen, C.O.; Rosenqvist, E.; Zhao, L.P.; Wang, Y.L.; Yu, W.G.; Zhao, T.M.; Wu, Z. Drought stress had a predominant effect over heat stress on three tomato cultivars subjected to combined stress. BMC Plant Biol. 2017, 17, 24. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zong, J.; Chen, P.; Luo, Q.; Gao, J.; Qin, R.; Wu, C.; Lv, Q.; Zhao, T.; Fu, Y. Transcriptome-Based WGCNA Analysis Reveals the Mechanism of Drought Resistance Differences in Sweetpotato (Ipomoea batatas (L.) Lam.). Int. J. Mol. Sci. 2023, 24, 14398. https://doi.org/10.3390/ijms241814398
Zong J, Chen P, Luo Q, Gao J, Qin R, Wu C, Lv Q, Zhao T, Fu Y. Transcriptome-Based WGCNA Analysis Reveals the Mechanism of Drought Resistance Differences in Sweetpotato (Ipomoea batatas (L.) Lam.). International Journal of Molecular Sciences. 2023; 24(18):14398. https://doi.org/10.3390/ijms241814398
Chicago/Turabian StyleZong, Jikai, Peitao Chen, Qingqing Luo, Jilong Gao, Ruihua Qin, Chunli Wu, Qina Lv, Tengfei Zhao, and Yufan Fu. 2023. "Transcriptome-Based WGCNA Analysis Reveals the Mechanism of Drought Resistance Differences in Sweetpotato (Ipomoea batatas (L.) Lam.)" International Journal of Molecular Sciences 24, no. 18: 14398. https://doi.org/10.3390/ijms241814398