
Transcriptome Profiling Reveals Differential Gene Expression during the Process of Microtuber Formation in Pinellia ternata

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
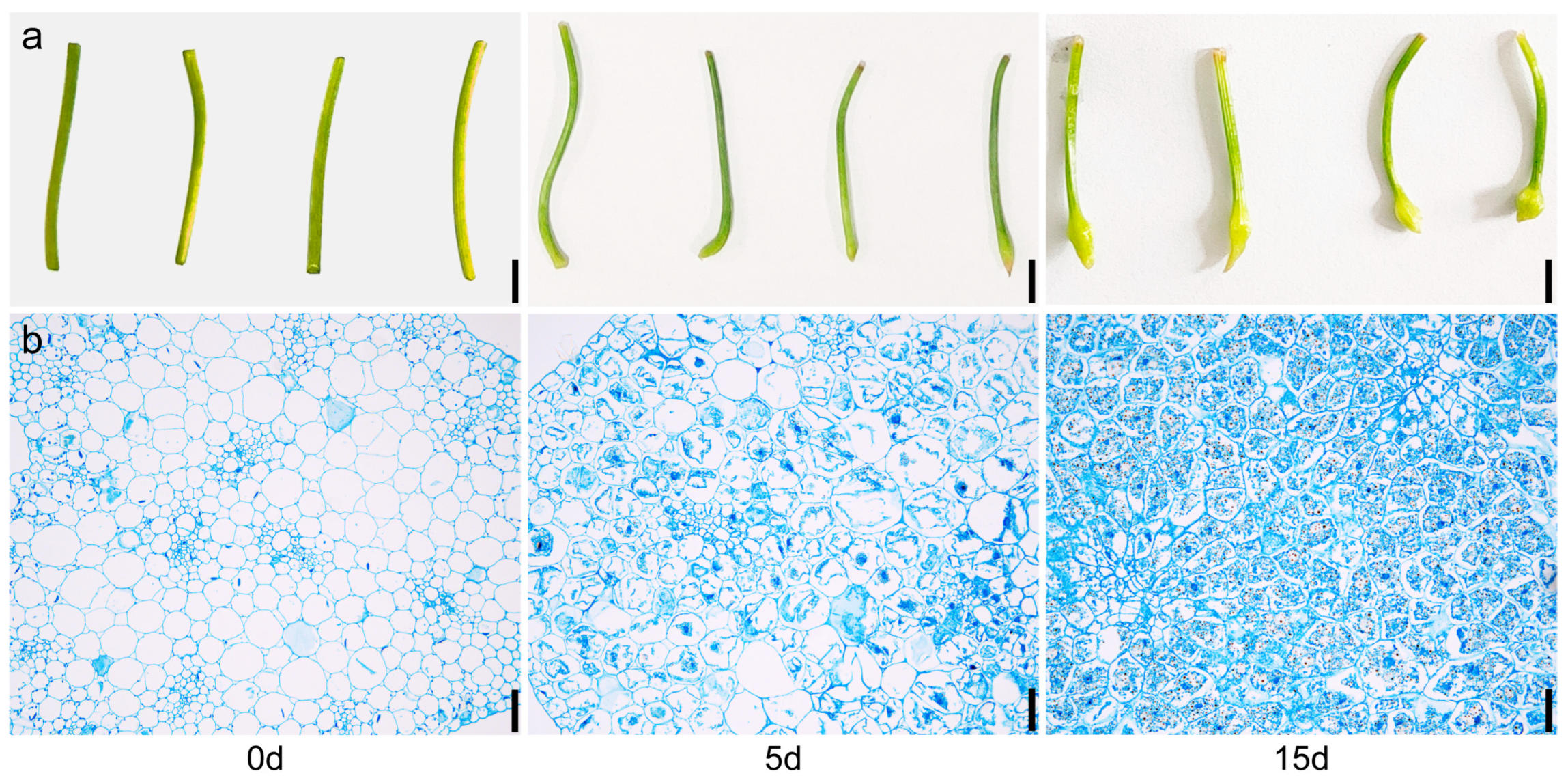
2.1. Phenotypic Comparison of P. ternata Microtubers with Different Cultivation Times
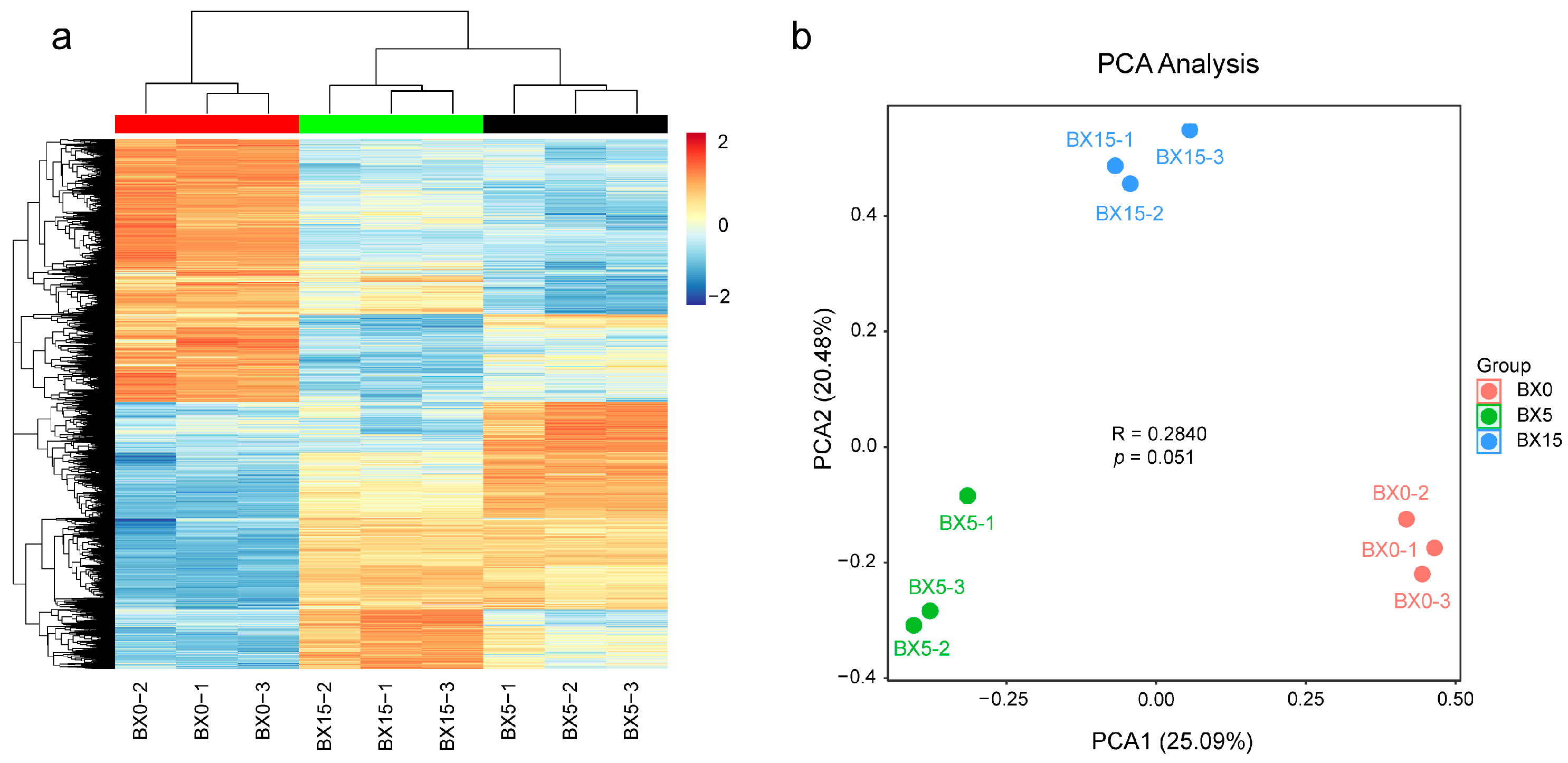
2.2. Transcriptome Sequencing of P. ternata Microtubers with Different Cultivation Times
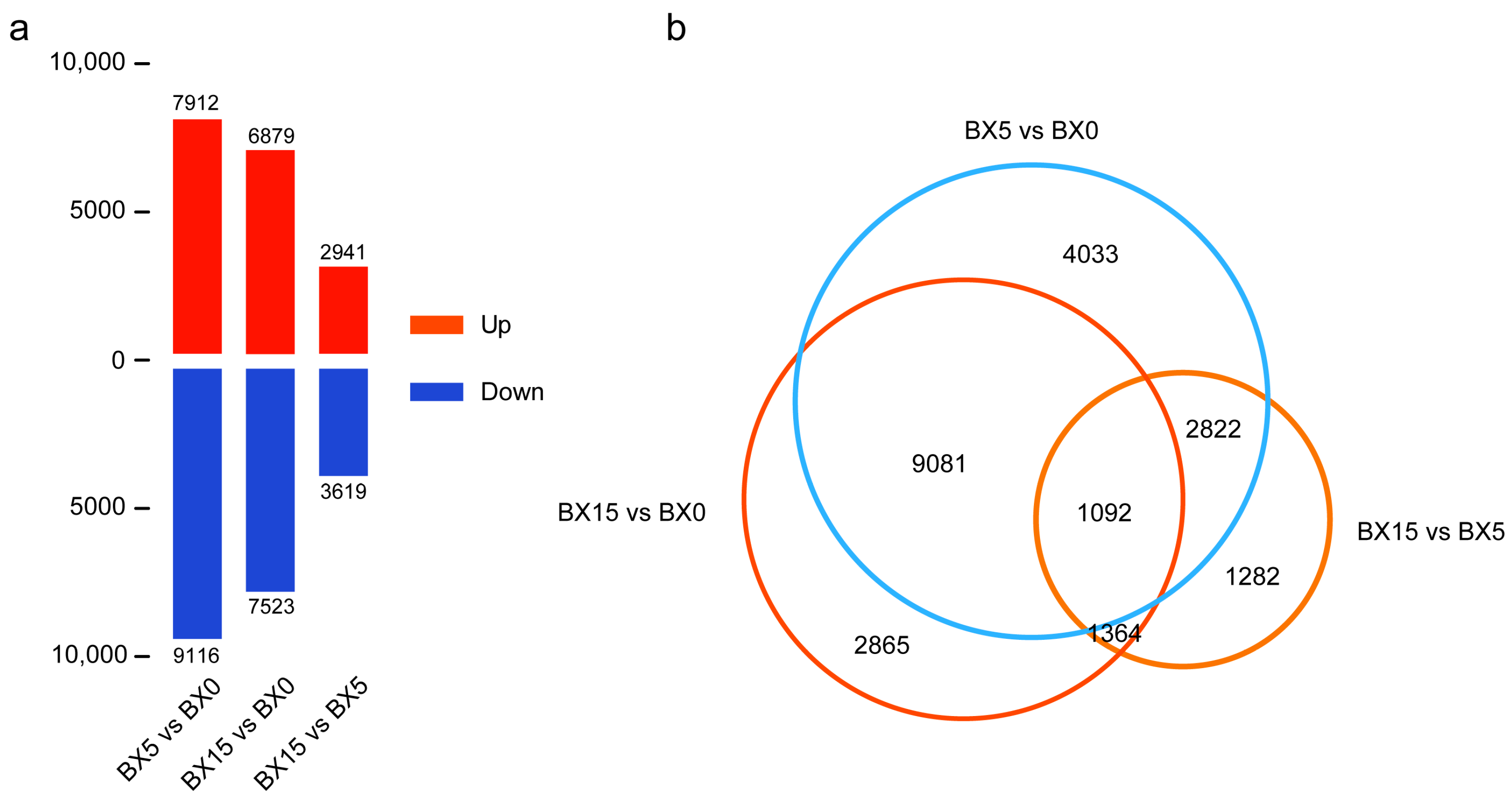
2.3. Analysis of DEGs in BX5 vs. BX0, BX15 vs. BX0, and BX15 vs. BX5 Comparisons
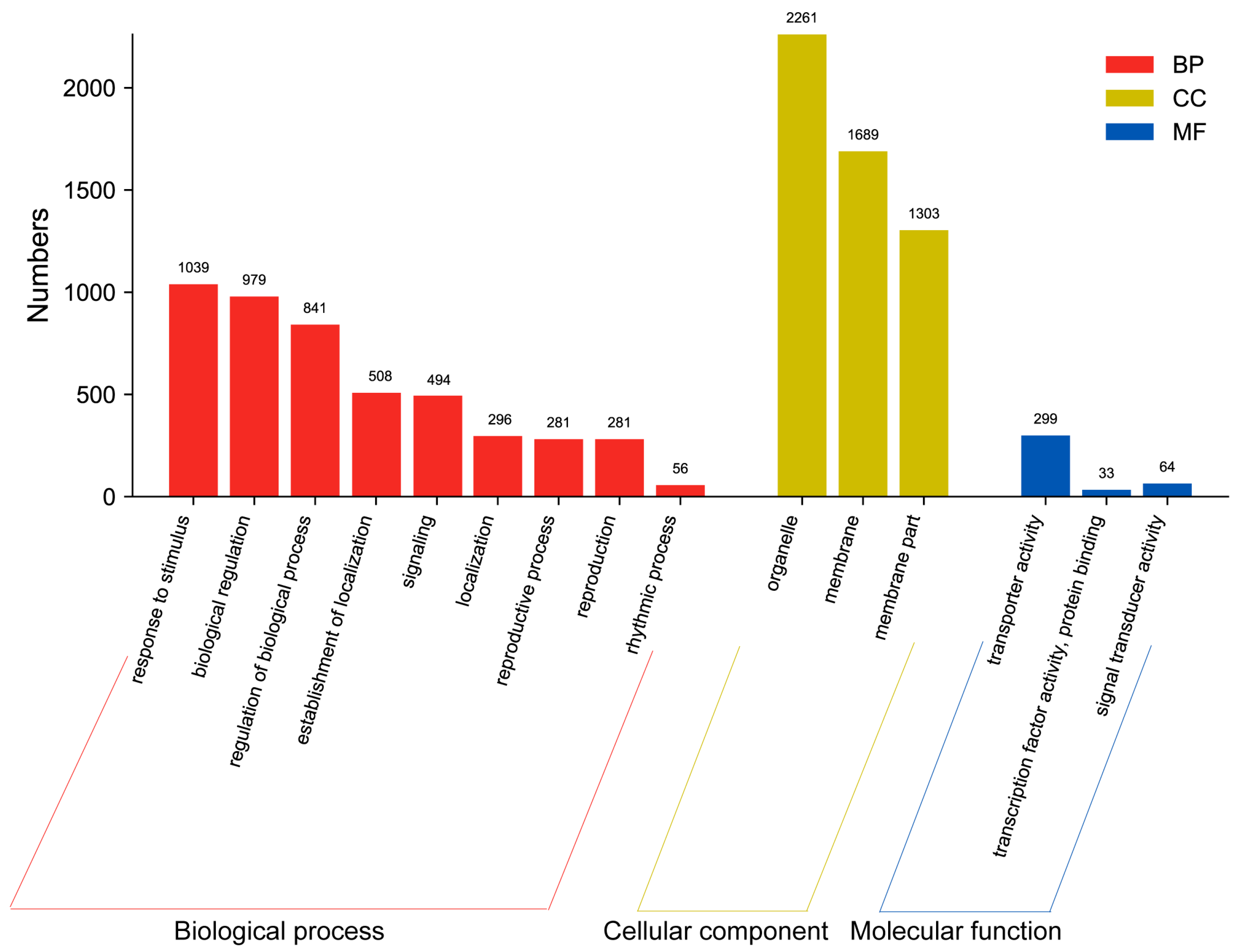
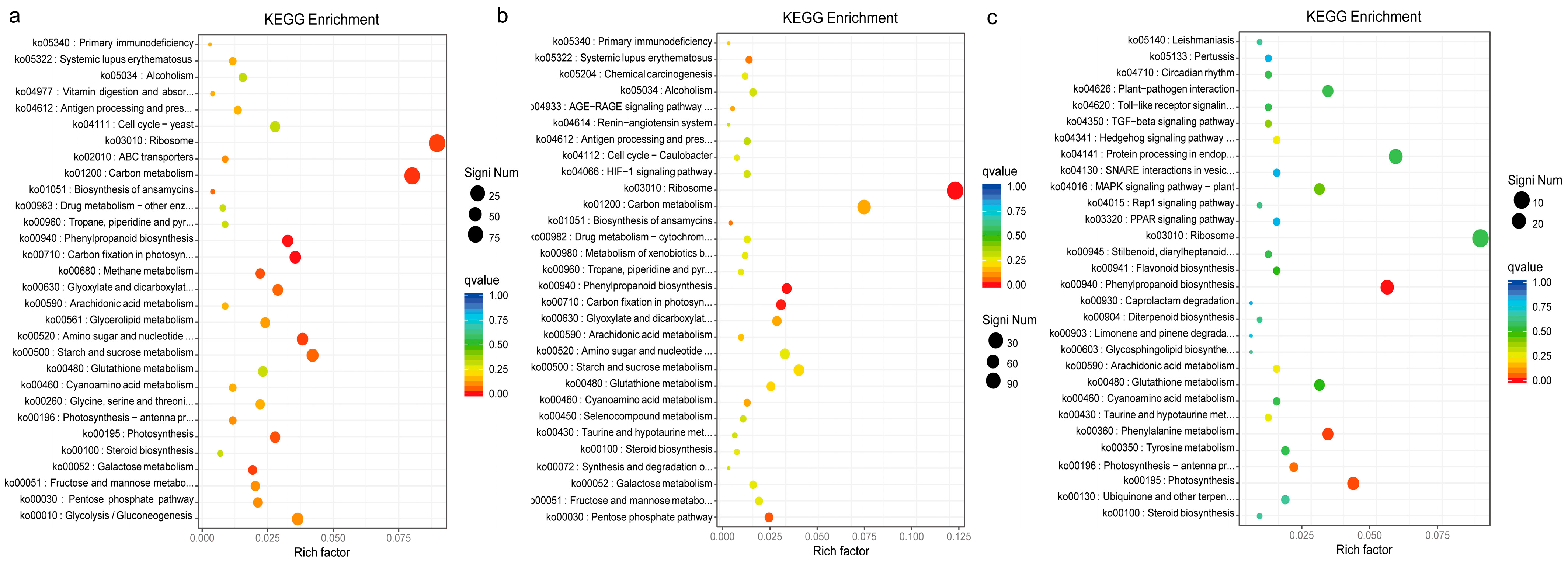
2.4. Functional Classification of DEGs
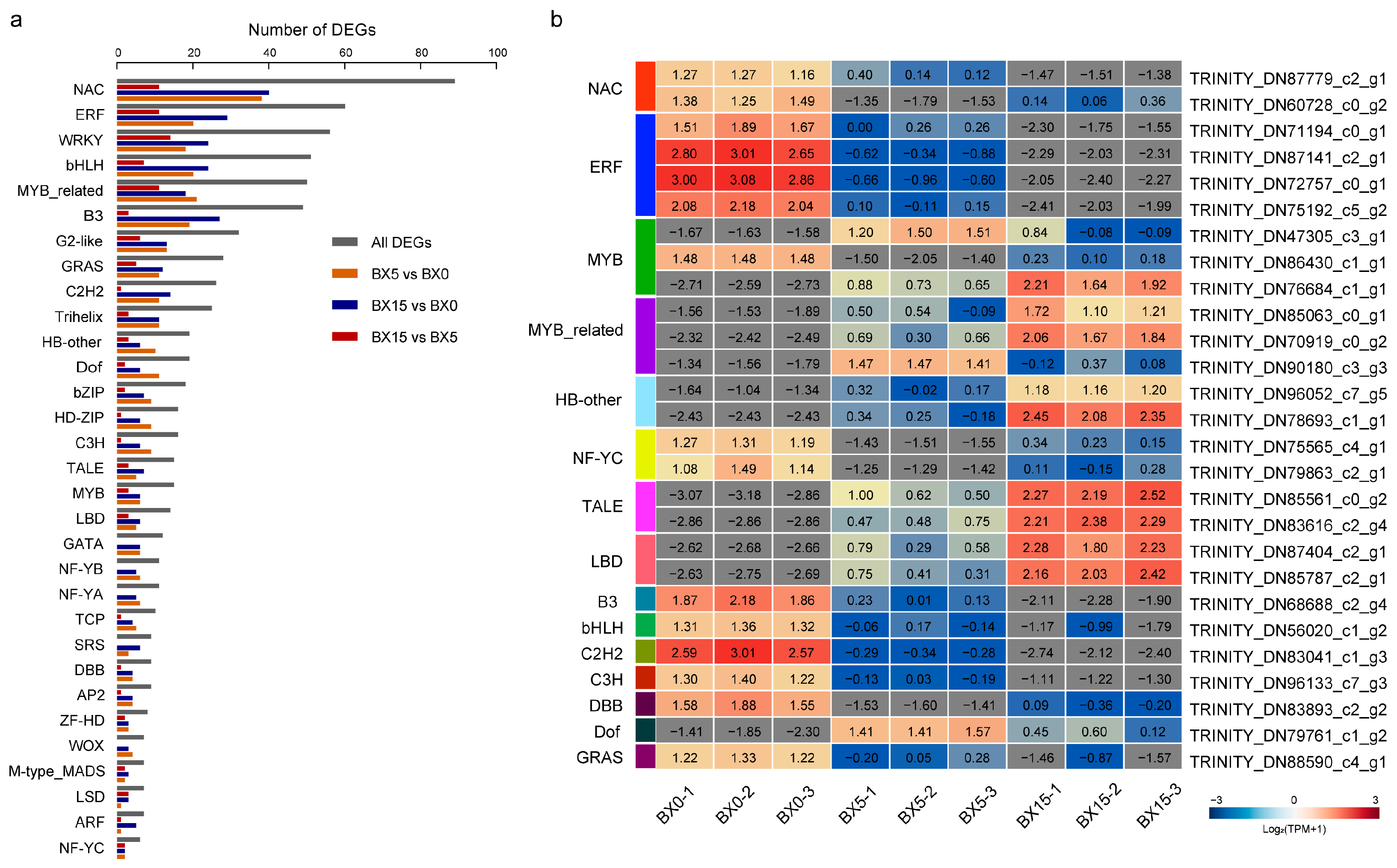
2.5. Analysis of DEGs Encoding Transcription Factors (TFs)
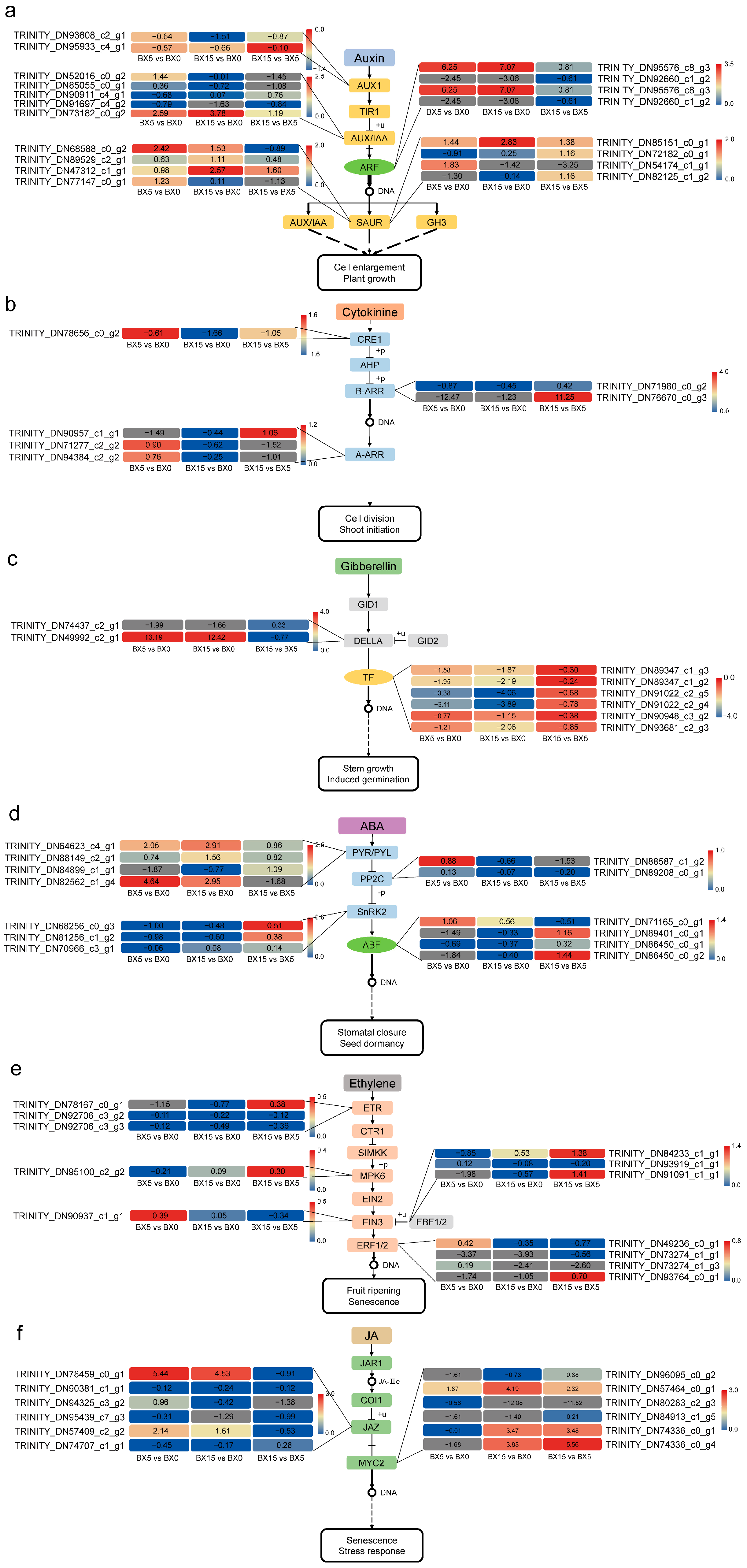
2.6. Analysis of DEGs Associated with Plant Hormones
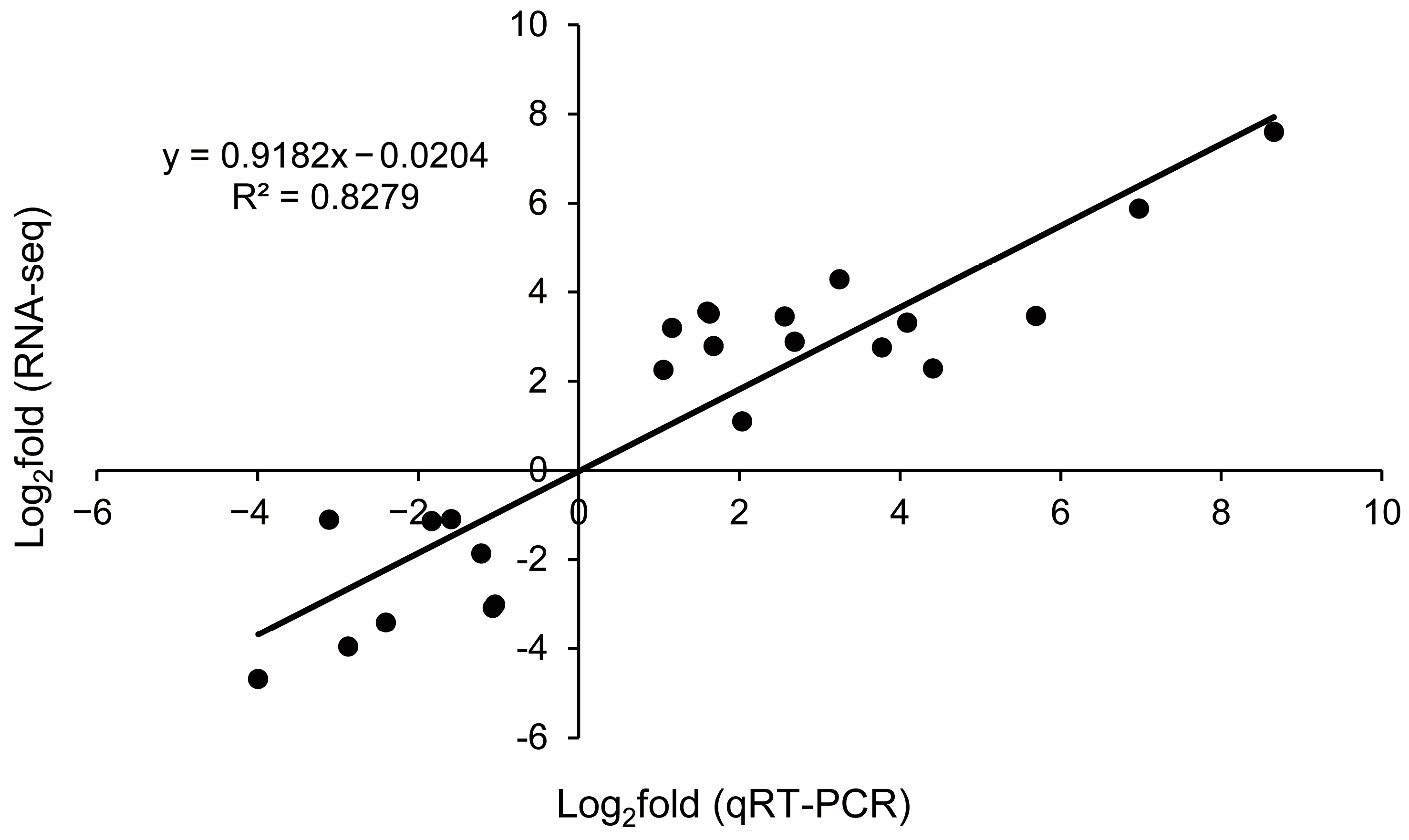
2.7. Validation of RNA-Seq Analysis by Quantitative Real-Time PCR (qRT-PCR)
3. Discussion
3.1. Transcriptomic Analysis Yields 1092 DEGs Associated with Microtuber Formation
3.2. DEGs Encoding Transcription Factors
3.3. Effect of Plant Hormones on Microtuber Formation
4. Materials and Methods
4.1. Plant Materials and Growth
4.2. Microscopic Structure Analysis of P. ternata Microtubers
4.3. cDNA Library Construction and Sequencing
4.4. Data Processing and De Novo Assembly
4.5. Functional Annotation of Unigenes and Differential Expression Genes Analysis
4.6. Validation of RNA-seq Data by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moon, B.; Kim, W.; Ji, Y.; Lee, Y.; Kang, Y.; Choi, G. Molecular identification of the traditional herbal medicines, Arisaematis Rhizoma and Pinelliae Tuber, and common adulterants via universal DNA barcode sequences. Gen. Mol. Res. 2016, 15, gmr-7064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Guo, Q.S.; Zheng, D.S. Genetic diversity analysis of Pinellia ternata based on SRAP and TRAP markers. Biochem. Syst. Ecol. 2013, 50, 258–265. [Google Scholar] [CrossRef]
- Mao, Z.C.; Peng, Z.S. Progress on research of rapid propagation system of Pinellia ternata. China J. Chin. Mater. Med. 2003, 28, 193–195. [Google Scholar]
- Zeng, X.Q.; Peng, Z.S. Growth and propagation of wild Pinellia ternata in cultivation. China J. Chin. Mater. Med. 2008, 33, 878–883. [Google Scholar]
- Iwasa, M.; Iwasaki, T.; Ono, T.; Miyazawa, M. Chemical composition and major odor-active compounds of essential oil from PINELLIA TUBER (dried rhizome of Pinellia ternata) as crude drug. J. Oleo Sci. 2014, 63, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Yang, M.; Abbas, H.M.K.; Wu, J.; Li, M.; Dong, W. Antimicrobial genes from Allium sativum and Pinellia ternata revealed by a Bacillus subtilis expression system. Sci. Rep. 2018, 8, 14514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.H.; Zhao, Y.Y.; Cheng, X.l.; Dai, Z.; Zhou, C.; Bai, X.; Lin, R.C. General toxicity of Pinellia ternata (Thunb.) Berit. in rat: A metabonomic method for profiling of serum metabolic changes. J. Ethnopharmacol. 2013, 149, 303–310. [Google Scholar] [CrossRef]
- Duan, Y.B.; Lu, F.; Cui, T.T.; Zhao, F.L.; Teng, J.T.; Sheng, W.; Zhang, A.M.; Xue, J.P. Effects of abiotic elicitors MeJA and SA on alkaloids accumulation and related enzymes metabolism in Pinellia ternata suspension cell cultures. Chin. J. Inf. TCM 2017, 24, 87–90. [Google Scholar]
- Liu, Y.; Hui, R.; Deng, R.; Wang, J.; Wang, M.; Li, Z. Abnormal male meiosis explains pollen sterility in the polyploid medicinal plant Pinellia ternata (Araceae). Genet. Mol. Res. 2012, 11, 112–120. [Google Scholar] [CrossRef]
- Eguchi, T.; Tanaka, H.; Yoshida, S.; Matsuoka, K. Temperature effects on the yield and quality of the medicinal plant Pinellia ternata Breit. Environ. Conv. Biol. 2019, 57, 83–85. [Google Scholar] [CrossRef] [Green Version]
- Tsay, H.; Gau, T.; Chen, C. Rapid clonal propagation of Pinellia ternata by tissue culture. Plant Cell Rep. 1989, 8, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Q.; Wang, J.; Lu, Y.; Xiao, X.; Gong, W.; Liu, J. Effect of different plant growth regulators on micro-tuber induction and plant regeneration of Pinellia ternate (Thunb) Briet. Physiol. Mol. Biol. Plants 2009, 15, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akita, M.; Ohta, Y. A simple method for mass propagation of potato (Solanum tuberosum L.) using a bioreactor without forced aeration. Plant Cell Rep. 1998, 18, 284–287. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, Z.; Zhang, Y. Induction and in vitro alkaloid yield of calluses and protocorm-like bodies (PLBs) from Pinellia ternata. Vitr. Cell. Dev. Biol.-Plant 2010, 46, 239–245. [Google Scholar] [CrossRef]
- Duan, Y.B.; Zhang, H.; Meng, X.; Huang, M.M.; Zhang, Z.Y.; Huang, C.H.; Zhao, F.L.; Xue, T.; Xue, J.P. Accumulation of salicylic acid-elicited alkaloid compounds in in vitro cultured Pinellia ternata microtubers and expression profiling of genes associated with benzoic acid-derived alkaloid biosynthesis. Plant Cell Tissue Organ Cult. 2009, 139, 317–325. [Google Scholar] [CrossRef]
- Yikun, H.; Changfu, Z.; Mengyuan, H.; Alin, H.; Shui, H. Morphogenesis of tubercles and production of artificial seeds in Pinellia ternata. Acta Agron. Sin. 1997, 23, 482–486. [Google Scholar]
- Lu, H.; Xue, T.; Zhang, A.; Sheng, W.; Zhu, Y.; Chang, L.; Song, Y.; Xue, J. Construction of an SSH library of Pinellia ternata under heat stress, and expression analysis of four transcripts. Plant Mol. Biol. Rep. 2013, 31, 185–194. [Google Scholar] [CrossRef]
- Xue, T.; Zhang, H.; Zhang, Y.; Wei, S.; Chao, Q.; Zhu, Y.; Teng, J.; Zhang, A.; Sheng, W.; Duan, Y. Full-length transcriptome analysis of shade-induced promotion of tuber production in Pinellia ternata. BMC Plant Biol. 2019, 19, 565. [Google Scholar] [CrossRef] [Green Version]
- Gombodorj, S.; Yang, M.H.; Shang, Z.C.; Liu, R.H.; Li, T.X.; Yin, G.P.; Kong, L.Y. New phenalenone derivatives from Pinellia ternata tubers derived Aspergillus sp. Fitoterapia 2017, 120, 72–78. [Google Scholar] [CrossRef]
- Jian, X. Research on Direct Formation of Microtubers from Pinellia ternata. Acta Agron. Sin. 2004, 30, 1060–1064. [Google Scholar]
- Li, C. Formation of Microtubers from the Petiole of Pinellia ternata (Thunb.) Berit. in vitro and Change of Endogenous Hormones Content. J. Huazhong Agric. Univ. 2007, 26, 612–615. [Google Scholar]
- Li, J.; Zhao, X.; Dong, Y.; Li, S.; Yuan, J.; Li, C.; Zhang, X.; Li, M. Transcriptome analysis reveals key pathways and hormone activities involved in early microtuber formation of Dioscorea opposita. BioMed Res. Int. 2020, 2020, 8057929. [Google Scholar] [CrossRef] [Green Version]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Front. Plant Sci. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Qi, Y.; Xu, J.; Dai, X.; Chen, J.; Dong, C.H.; Xiang, F. Arabidopsis WRKY71 regulates ethylene-mediated leaf senescence by directly activating EIN2, ORE1 and ACS2 genes. Plant J. 2021, 107, 1819–1836. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, K.; Yang, C. BpNAC012 positively regulates abiotic stress responses and secondary wall biosynthesis. Plant Physiol. 2019, 179, 700–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohto, M.A.; Fischer, R.L.; Goldberg, R.B.; Nakamura, K.; Harada, J.J. Control of seed mass by APETALA2. Proc. Natl. Acad. Sci. USA 2005, 102, 3123–3128. [Google Scholar] [CrossRef]
- Fukazawa, J.; Sakai, T.; Ishida, S.; Yamaguchi, I.; Kamiya, Y.; Takahashi, Y. Repression of shoot growth, a bZIP transcriptional activator, regulates cell elongation by controlling the level of gibberellins. Plant Cell 2000, 12, 901–915. [Google Scholar] [CrossRef] [Green Version]
- Haga, N.; Kobayashi, K.; Suzuki, T.; Maeo, K.; Kubo, M.; Ohtani, M.; Mitsuda, N.; Demura, T.; Nakamura, K.; Jürgens, G. Mutations in MYB3R1 and MYB3R4 cause pleiotropic developmental defects and preferential down-regulation of multiple G2/M-specific genes in Arabidopsis. Plant Physiol. 2011, 157, 706–717. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Koyama, H.; Bhati, K.K.; Alok, A. The biotechnological importance of the plant-specific NAC transcription factor family in crop improvement. J. Plant Res. 2021, 134, 475–495. [Google Scholar] [CrossRef]
- Feng, K.; Hou, X.L.; Xing, G.M.; Liu, J.X.; Duan, A.Q.; Xu, Z.S.; Li, M.Y.; Zhuang, J.; Xiong, A.S. Advances in AP2/ERF super-family transcription factors in plant. Crit. Rev. Biotechnol. 2020, 40, 750–776. [Google Scholar] [CrossRef]
- Lasserre, E.; Jobet, E.; Llauro, C.; Delseny, M. AtERF38 (At2g35700), an AP2/ERF family transcription factor gene from Arabidopsis thaliana, is expressed in specific cell types of roots, stems and seeds that undergo suberization. Plant Physiol. Biochem. 2008, 46, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.S. TRANSPARENT TESTA GLABRA2, a Trichome and Seed Coat Development Gene of Arabidopsis, Encodes a WRKY Transcription Factor. Plant Cell 2002, 14, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Van Leene, J.; Blomme, J.; Kulkarni, S.R.; Cannoot, B.; De Winne, N.; Eeckhout, D.; Persiau, G.; Van De Slijke, E.; Vercruysse, L.; Vanden Bossche, R. Functional characterization of the Arabidopsis transcription factor bZIP29 reveals its role in leaf and root development. J. Exp. Bot. 2016, 67, 5825–5840. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Lee, E.; Lucas, J.R.; Morohashi, K.; Li, D.; Murray, J.A.; Sack, F.D.; Grotewold, E. Regulation of cell proliferation in the stomatal lineage by the Arabidopsis MYB FOUR LIPS via direct targeting of core cell cycle genes. Plant Cell 2010, 22, 2306–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, P.J. The Plant Hormones: Their Nature, Occurrence, and Functions; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Roumeliotis, E.; Kloosterman, B.; Oortwijn, M.; Kohlen, W.; Bouwmeester, H.J.; Visser, R.G.; Bachem, C.W. The effects of auxin and strigolactones on tuber initiation and stolon architecture in potato. J. Exp. Bot. 2012, 63, 4539–4547. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Liu, J.; Fan, M.; Xin, W.; Hu, Y.; Xu, C. A genome-wide transcriptome profiling reveals the early molecular events during callus initiation in Arabidopsis multiple organs. Genomics 2012, 100, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Skoog, F.; Miller, C.O. Chemical regulation of growth and organ formation in plant tissues cultured in vitro. Cheminform 1957, 11, 118–130. [Google Scholar]
- Wang, X.; Li, J.; Ban, L.; Wu, Y.; Wu, X.; Wang, Y.; Wen, H.; Chapurin, V.; Dzyubenko, N.; Li, Z. Functional characterization of a gibberellin receptor and its application in alfalfa biomass improvement. Sci. Rep. 2017, 7, 41296. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Ren, Z.; Cheng, C.; Wang, T.; Ji, H.; Zhao, Y.; Deng, Z.; Zhi, L.; Lu, J.; Wu, X. Counteraction of ABA-mediated inhibition of seed germination and seedling establishment by ABA signaling terminator in Arabidopsis. Mol. Plant 2020, 13, 1284–1297. [Google Scholar] [CrossRef]
- Kendrick, M.D.; Chang, C. Ethylene signaling: New levels of complexity and regulation. Curr. Opin. Plant Biol. 2008, 11, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Shi, L.; Wang, Y.; Li, W.; Chen, B.; Zhu, L.; Fu, Y. Arabidopsis ECAP is a new adaptor protein that connects JAZ repressors with the TPR2 co-repressor to suppress jasmonate-responsive anthocyanin accumulation. Mol. Plant 2020, 13, 246–265. [Google Scholar] [CrossRef]
- Wang, L.; Xie, X.; Xu, Y.; Li, Z.; Xu, G.; Cheng, L.; Yang, J.; Li, L.; Pu, W.; Cao, P. Comprehensive analysis of the carboxylesterase gene reveals that NtCXE22 regulates axillary bud growth through strigolactone metabolism in tobacco. Front. Plant Sci. 2022, 13, 1019538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, J.; Huang, Y.; Wu, H.; Hu, X.; Cheng, B.; Ma, Q.; Zhao, Y. Comparative transcriptomics Reveals the molecular mechanism of the parental lines of maize hybrid An’nong876 in response to salt stress. Int. J. Mol. Sci. 2022, 23, 5231. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Bo, C.; Cai, R.; Fang, X.; Wu, H.; Ma, Z.; Yuan, H.; Cheng, B.; Fan, J.; Ma, Q. Transcription factor ZmWRKY20 interacts with ZmWRKY115 to repress expression of ZmbZIP111 for salt tolerance in maize. Plant J. 2022, 111, 1660–1675. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Z.; Xiong, Y.; Shi, J.; Chen, C.; Pan, Y.; Xue, T.; Xue, J.; Duan, Y. Stearic acid desaturase gene negatively regulates the thermotolerance of Pinellia ternata by modifying the saturated levels of fatty acids. Ind. Crops Prod. 2021, 166, 113490. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bo, C.; Su, C.; Teng, J.; Sheng, W.; Xue, T.; Zhu, Y.; Xue, J. Transcriptome Profiling Reveals Differential Gene Expression during the Process of Microtuber Formation in Pinellia ternata. Int. J. Mol. Sci. 2023, 24, 11604. https://doi.org/10.3390/ijms241411604
Bo C, Su C, Teng J, Sheng W, Xue T, Zhu Y, Xue J. Transcriptome Profiling Reveals Differential Gene Expression during the Process of Microtuber Formation in Pinellia ternata. International Journal of Molecular Sciences. 2023; 24(14):11604. https://doi.org/10.3390/ijms241411604
Chicago/Turabian StyleBo, Chen, Chuandong Su, Jingtong Teng, Wei Sheng, Tao Xue, Yanfang Zhu, and Jianping Xue. 2023. "Transcriptome Profiling Reveals Differential Gene Expression during the Process of Microtuber Formation in Pinellia ternata" International Journal of Molecular Sciences 24, no. 14: 11604. https://doi.org/10.3390/ijms241411604