
Inhibition of HSP90 in Driver Oncogene-Defined Lung Adenocarcinoma Cell Lines: Key Proteins Underpinning Therapeutic Efficacy

 ,
, 
Abstract
:1. Introduction
2. Results
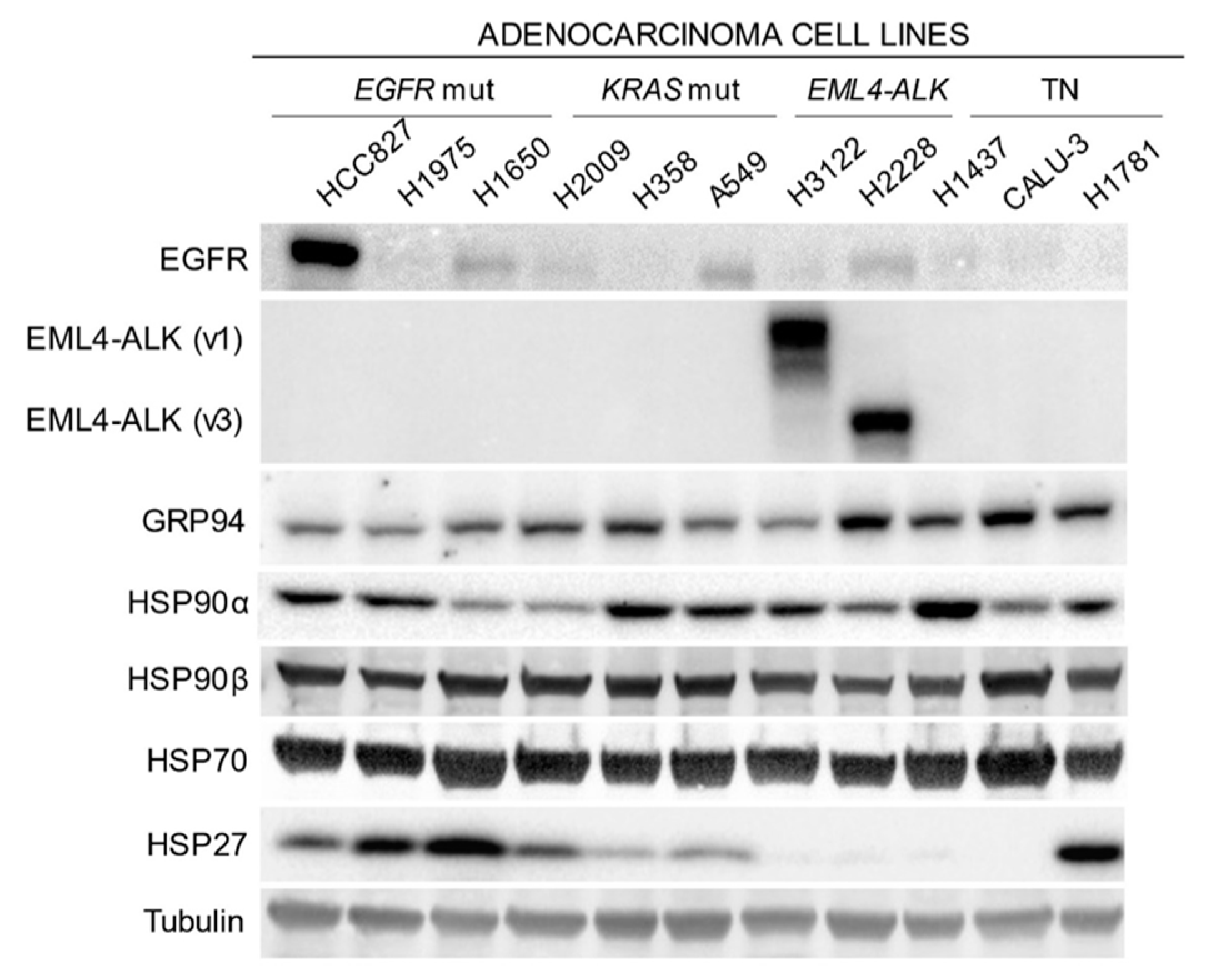
2.1. Expression of HSP90 and Related Proteins in Different Molecular Subtypes of Lung Adenocarcinoma
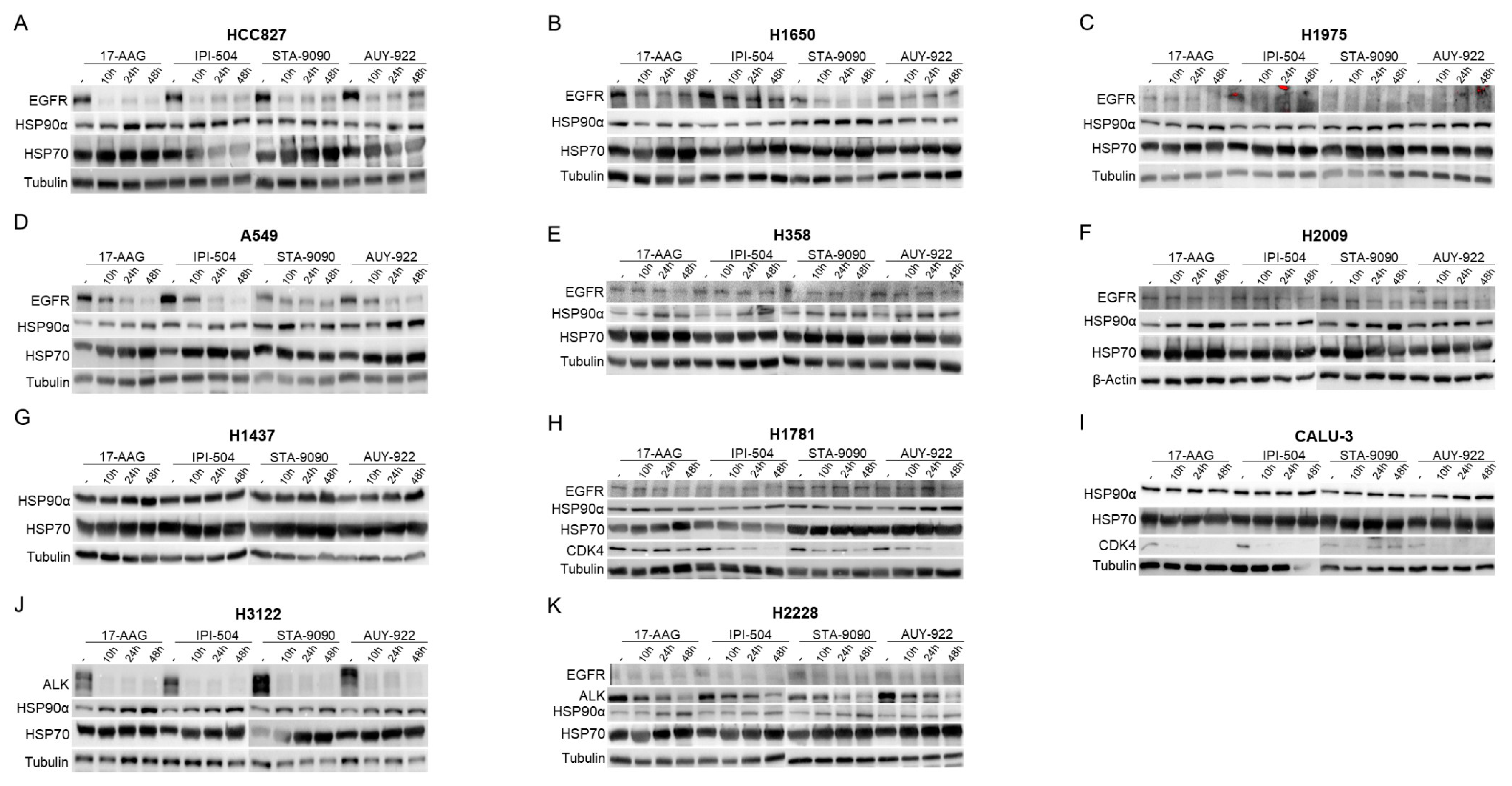
2.2. Effect of Inhibition of HSP90 According to the Lung Adenocarcinoma Molecular Subgroup
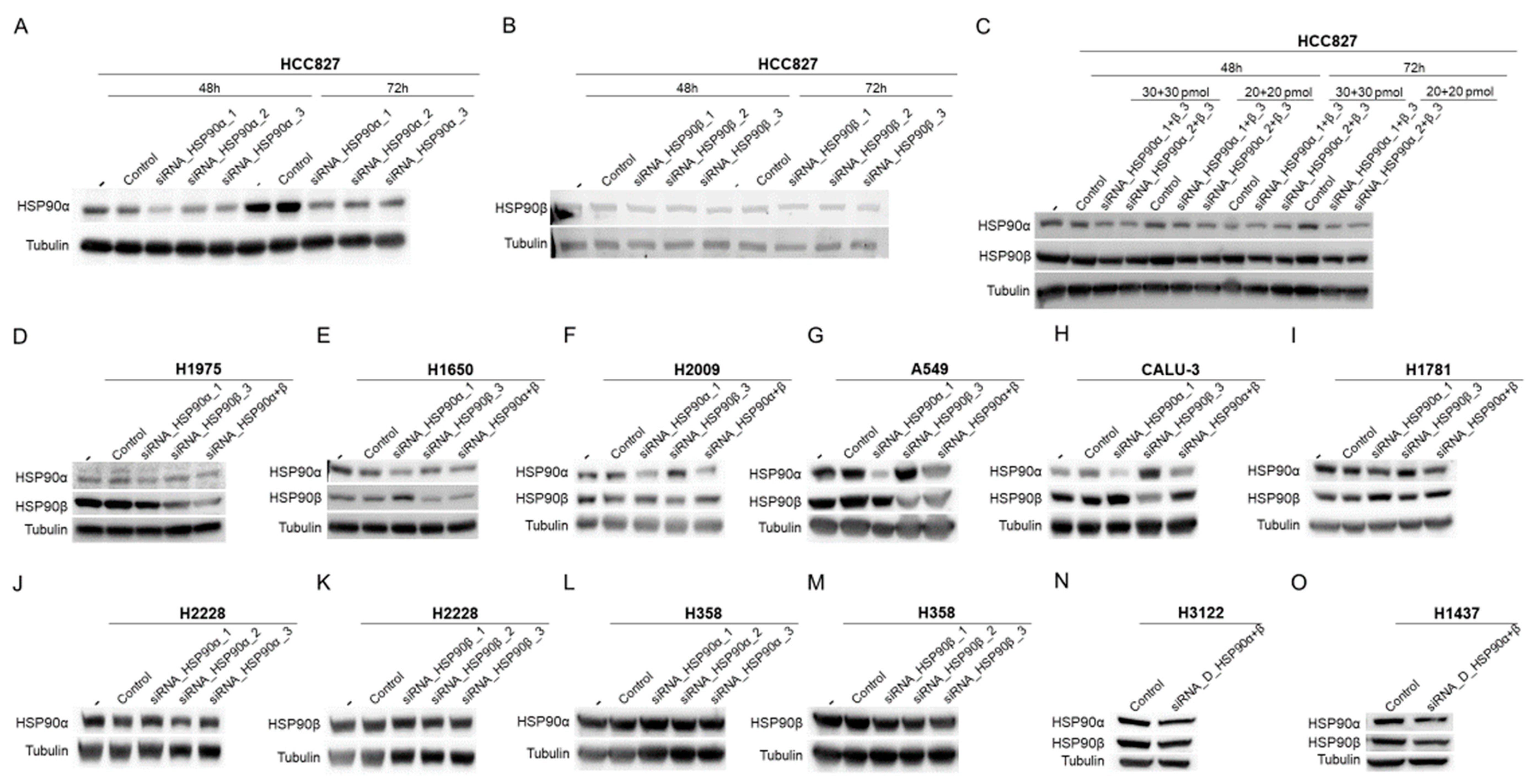
2.3. HSP90 Gene Silencing in Lung Adenocarcinoma Cell Lines

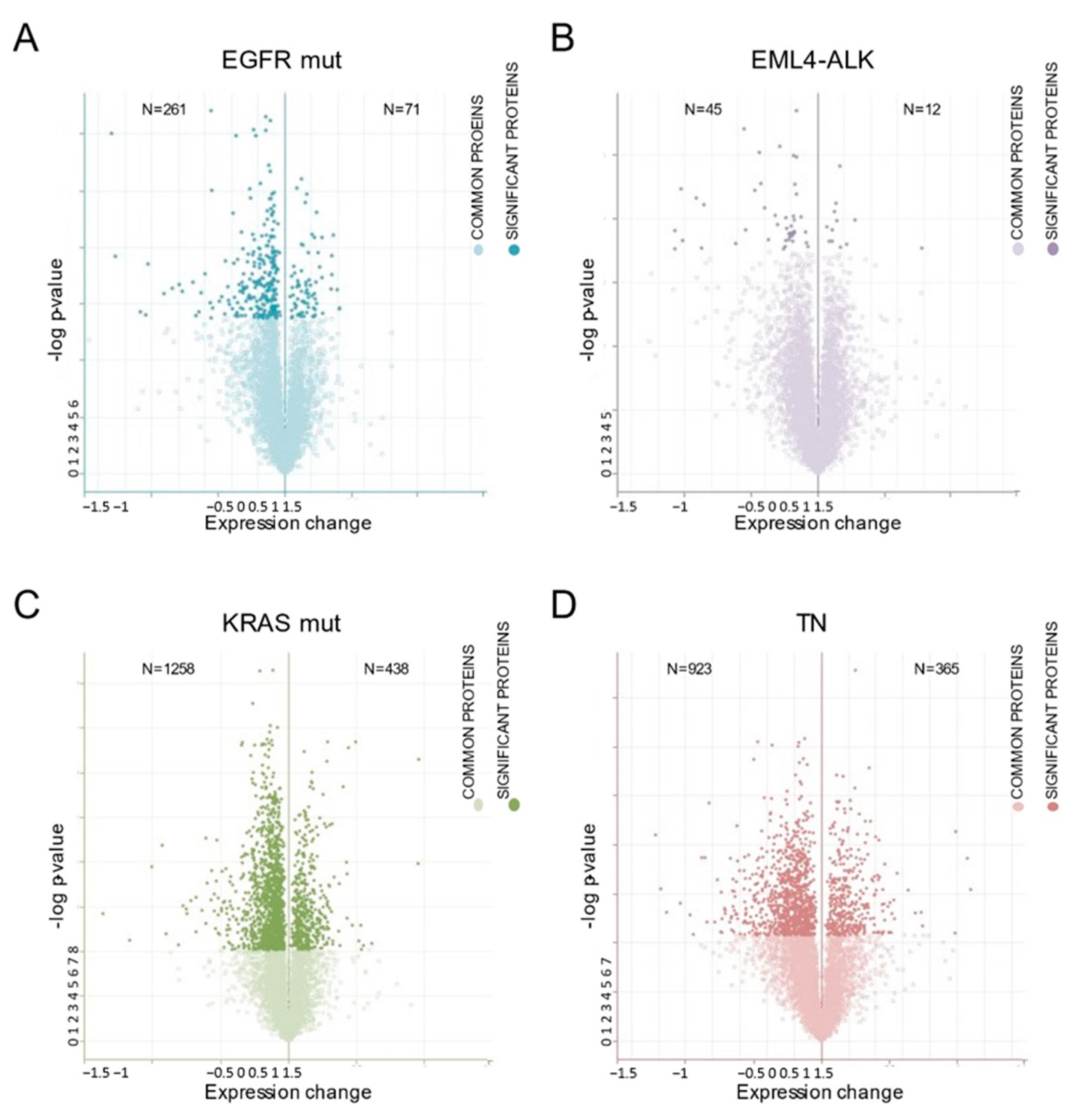
2.4. Differentially Expressed Proteins Underlying the HSP90 Inhibition Identified by Proteomic Profiling
2.5. Effect of HSP90 Inhibition on Proteomic Profiling with Respect to the Lung Adenocarcinoma Molecular Subgroup
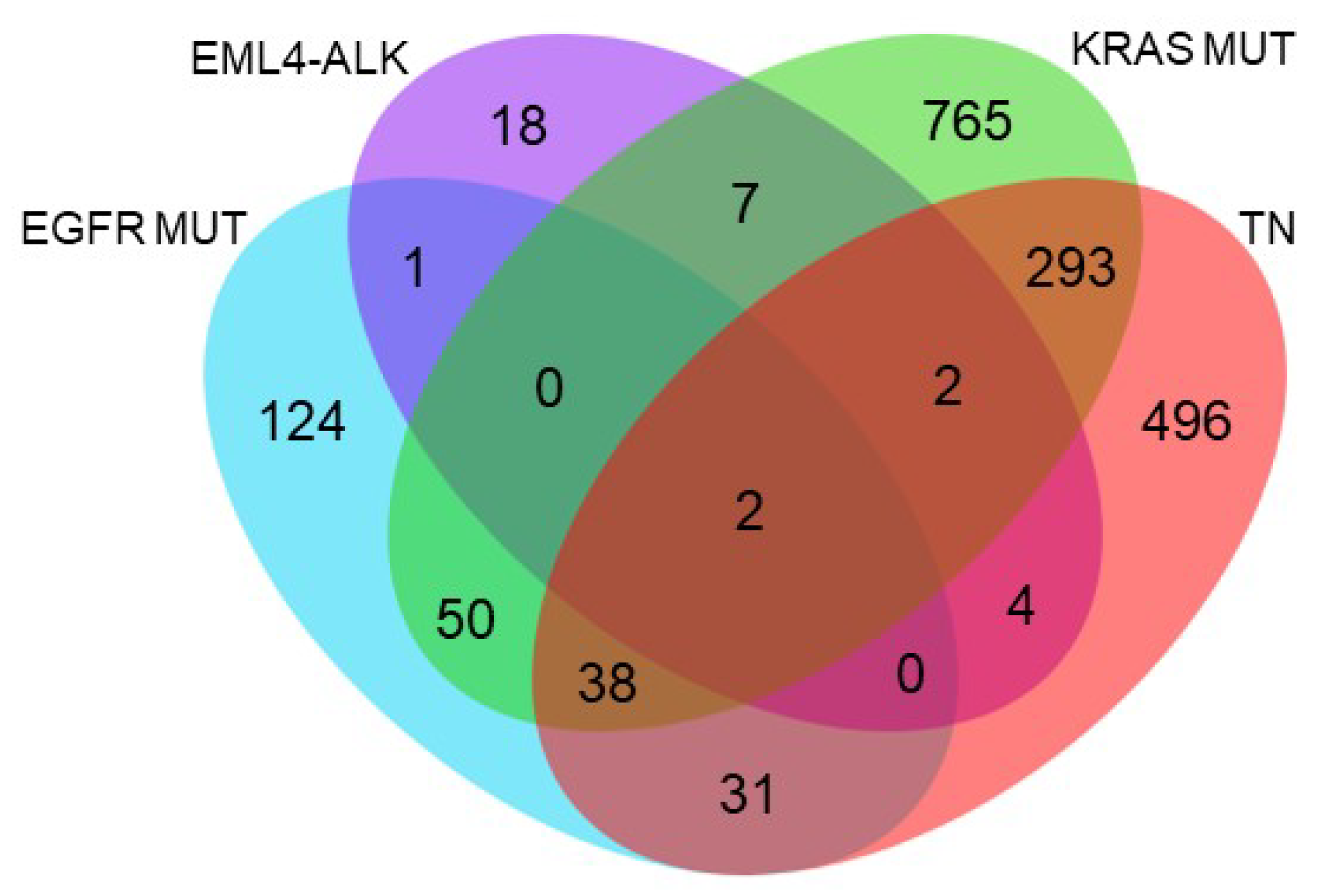
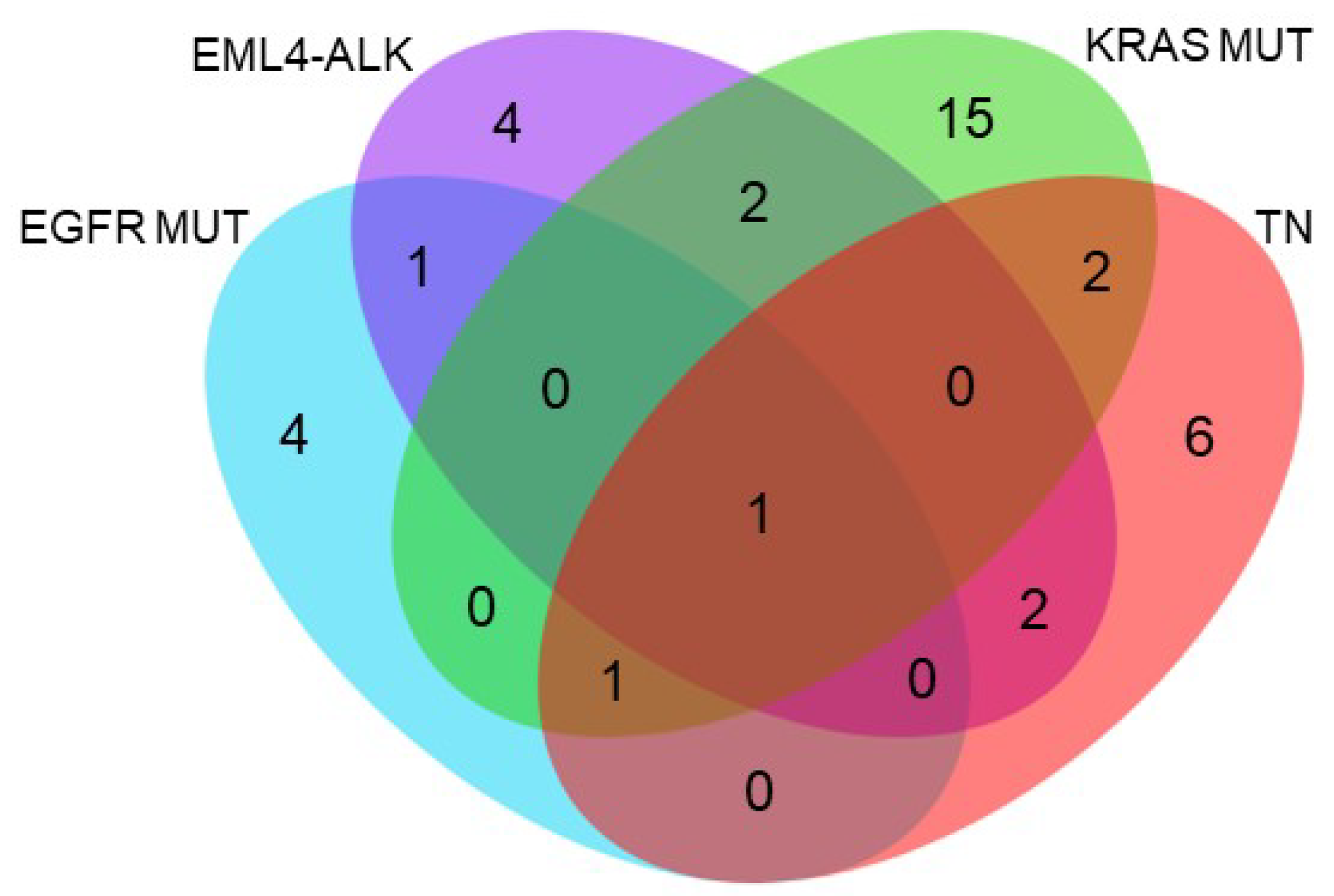
2.6. Functional Annotation of Specifically and Significantly Down-Regulated Proteins after HSP90 Inhibition in Different Molecular Subgroups of Lung Adenocarcinoma
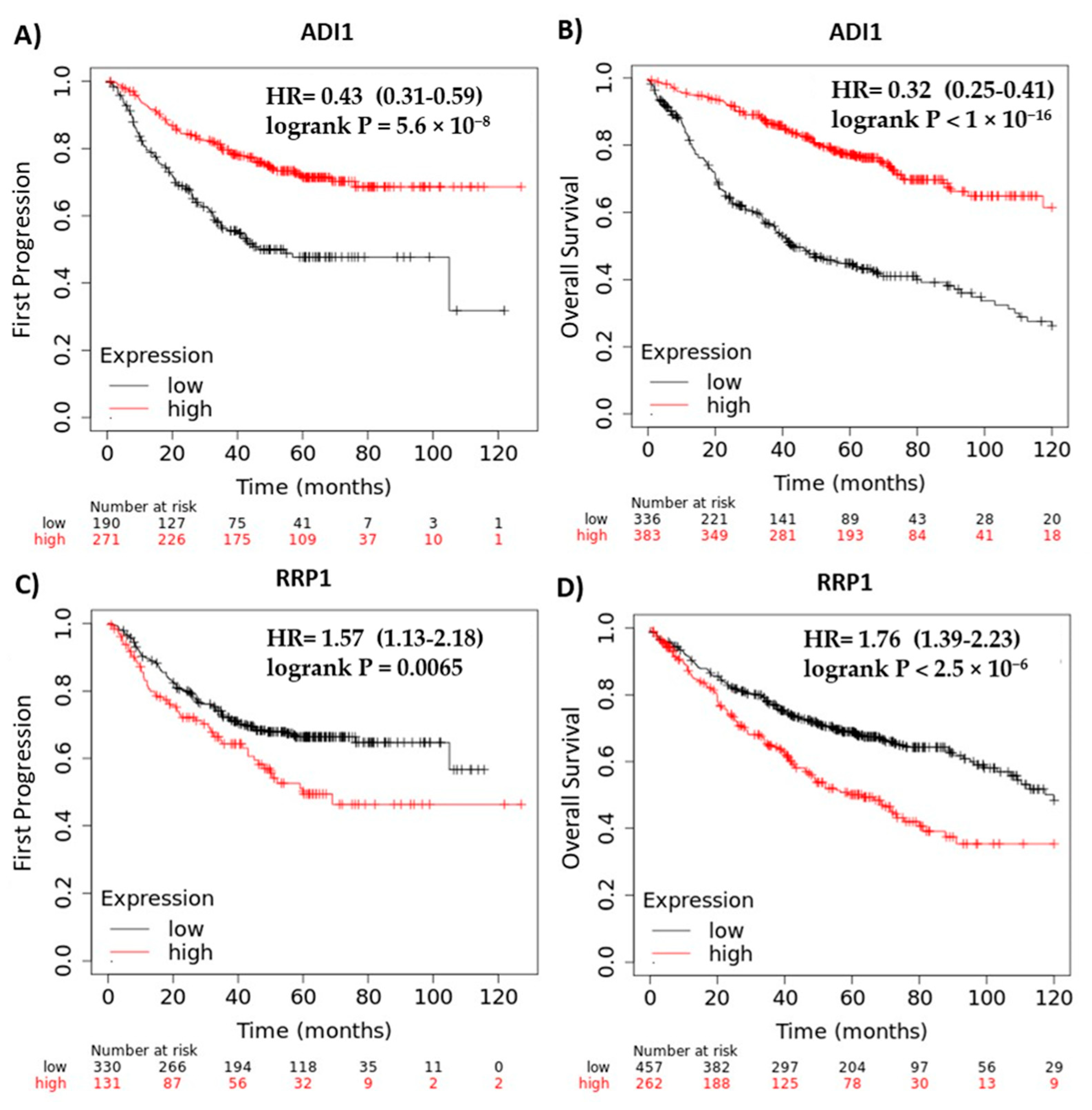
2.7. ADI1 and RRP1 mRNA Expression Are Strongly Associated with Clinical Outcome
3. Discussion
4. Materials and Methods
4.1. Cell Line Culture
4.2. Genetic Silencing of HSP90
4.3. Treatment with HSP90 Inhibitors
4.4. Western Blot (WB)
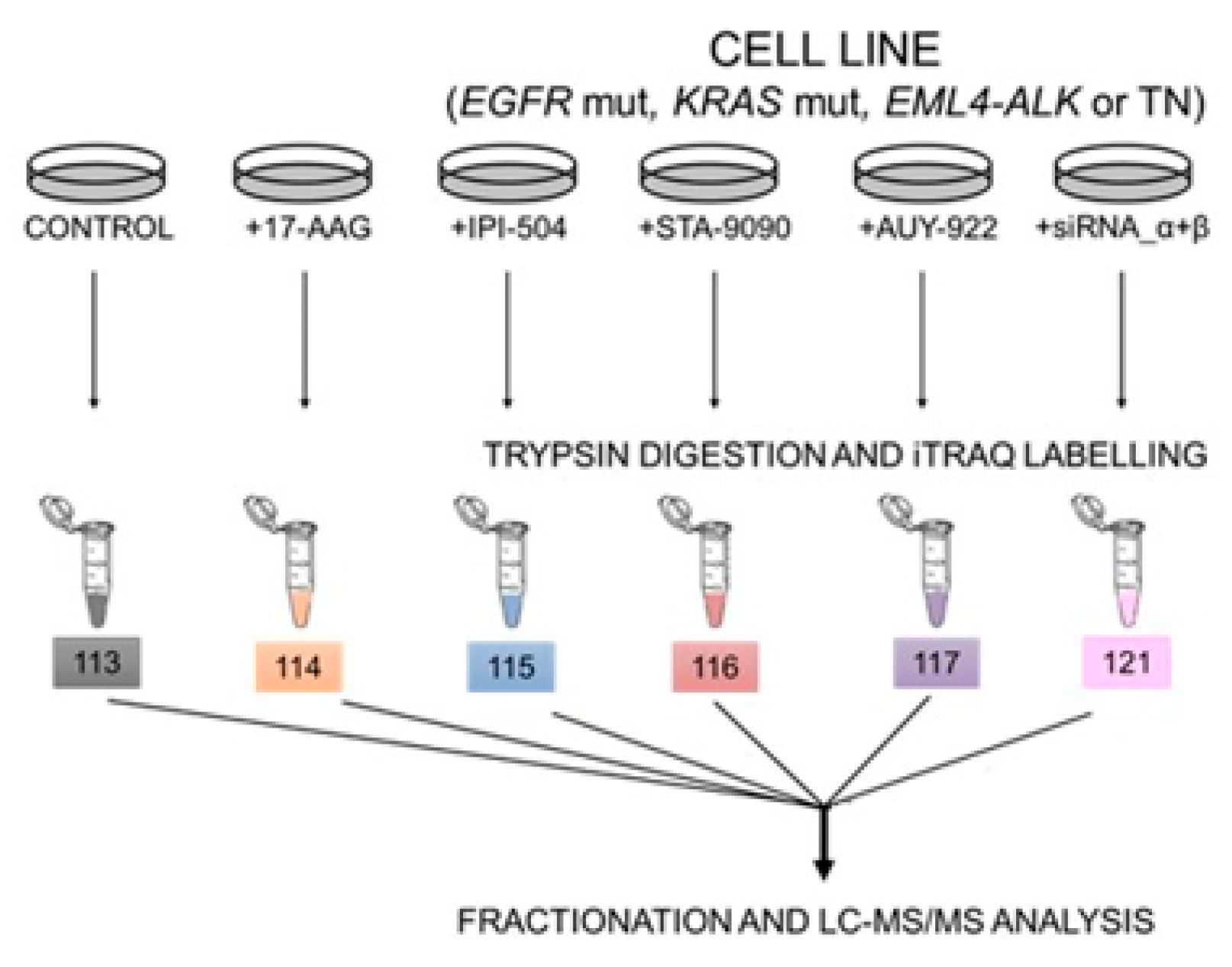
4.5. Sample Preparation for Proteomics Analysis
4.6. iTRAQ Labelling
4.7. Nano LC-MS/MS Analysis
4.8. MS Data Analysis
4.9. Bioinformatics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mathers, C.D.; Loncar, D. Projections of Global Mortality and Burden of Disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA A Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef]
- Zugazagoitia, J.; Molina-Pinelo, S.; Lopez-Rios, F.; Paz-Ares, L. Biological therapies in nonsmall cell lung cancer. Eur. Respir. J. 2017, 49, 1601520. [Google Scholar] [CrossRef]
- Somasundaram, A.; Socinski, M.A.; Burns, T.F. Personalized treatment of EGFR mutant and ALK-positive patients in NSCLC. Expert Opin. Pharmacother. 2014, 15, 2693–2708. [Google Scholar] [CrossRef] [PubMed]
- Pendharkar, D.; Ausekar, B.V.; Gupta, S. Molecular Biology of Lung Cancer—A Review. Indian J. Surg. Oncol. 2013, 4, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Chalela, R.; Curull, V.; Enríquez, C.; Pijuan, L.; Bellosillo, B.; Gea, J. Lung adenocarcinoma: From molecular basis to ge-nome-guided therapy and immunotherapy. J. Thorac. Dis. 2017, 9, 2142–2158. [Google Scholar] [CrossRef]
- Tamborero, D.; Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Kandoth, C.; Reimand, J.; Michael, S.; Getz, G.; Bader, G.D.; Lopez-Bigas, N. Comprehensive identifi-cation of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013, 3, 2650. [Google Scholar] [CrossRef]
- Gallant, J.-N.; Lovly, C.M. Established, emerging and elusive molecular targets in the treatment of lung cancer. J. Pathol. 2018, 244, 565–577. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Zhou, C.; Liam, C.-K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Bhattacharya, S.; Socinski, M.A.; Burns, T.F. HSP90 inhibitors in lung cancer: Promise still unfulfilled. Clin. Adv. Hematol. Oncol. HO 2016, 14, 346–356. [Google Scholar]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mu-tant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef]
- Spagnuolo, A.; Maione, P.; Gridelli, C. Evolution in the treatment landscape of non-small cell lung cancer with ALK gene alterations: From the first-to third-generation of ALK inhibitors. Expert Opin. Emerg. Drugs 2018, 23, 231–241. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef]
- Radli, M.; Rüdiger, S.G. Dancing with the Diva: Hsp90–Client Interactions. J. Mol. Biol. 2018, 430, 3029–3040. [Google Scholar] [CrossRef]
- Butler, L.M.; Ferraldeschi, R.; Armstrong, H.K.; Centenera, M.M.; Workman, P. Maximizing the Therapeutic Potential of HSP90 Inhibitors. Mol. Cancer Res. 2015, 13, 1445–1451. [Google Scholar] [CrossRef]
- Workman, P.; Burrows, F.; Neckers, L.; Rosen, N. Drugging the cancer chaperone HSP90: Combinatorial therapeutic exploi-tation of oncogene addiction and tumor stress. Ann. N. Y. Acad. Sci. 2007, 1113, 202–216. [Google Scholar] [CrossRef]
- Prodromou, C.; Roe, S.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Identification and Structural Characterization of the ATP/ADP-Binding Site in the Hsp90 Molecular Chaperone. Cell 1997, 90, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, E.S.; Wang, T.; Luo, K.; Xiao, Z.; Niewiadomska, A.M.; Martinez, T.; Xu, W.; Neckers, L.; Yu, X.-F. Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2009, 106, 20330–20335. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Lowell, A.M.; Engelman, J.A.; Shapiro, G.I. Epidermal Growth Factor Receptors Harboring Kinase Domain Mutations Associate with the Heat Shock Protein 90 Chaperone and Are Destabilized following Exposure to Geldanamycins. Cancer Res. 2005, 65, 6401–6408. [Google Scholar] [CrossRef]
- Xu, W.; Soga, S.; Beebe, K.; Lee, M.-J.; Kim, Y.S.; Trepel, J.; Neckers, L. Sensitivity of epidermal growth factor receptor and ErbB2 exon 20 insertion mutants to Hsp90 inhibition. Br. J. Cancer 2007, 97, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Pashtan, I.; Tsutsumi, S.; Xu, W.; Neckers, L. Cancer cells harboring MET gene amplification activate alternative signaling pathways to escape MET inhibition but remain sensitive to Hsp90 inhibitors. Cell Cycle 2009, 8, 2050–2056. [Google Scholar] [CrossRef] [PubMed]
- Dias, S.d.R.; Friedlos, F.; Light, Y.; Springer, C.; Workman, P.; Marais, R. Activated B-RAF Is an Hsp90 Client Protein That Is Targeted by the Anticancer Drug 17-Allylamino-17-Demethoxygeldanamycin. Cancer Res. 2005, 65, 10686–10691. [Google Scholar] [CrossRef] [PubMed]
- Normant, E.; Paez, G.; West, K.A.; Lim, A.R.; Slocum, K.L.; Tunkey, C.; McDougall, J.; Wylie, A.A.; Robison, K.; Caliri, K.; et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4–ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene 2011, 30, 2581–2586. [Google Scholar] [CrossRef]
- Cohen, V.; Esfahani, K. HSP90 as a novel molecular target in non-small-cell lung cancer. Lung Cancer Targets Ther. 2016, 7, 11–17. [Google Scholar] [CrossRef]
- Ruiz, M.I.G.; Floor, K.; Roepman, P.; Rodriguez, J.A.; Meijer, G.A.; Mooi, W.J.; Jassem, E.; Niklinski, J.; Muley, T.; van Zandwijk, N.; et al. Integration of Gene Dosage and Gene Expression in Non-Small Cell Lung Cancer, Identification of HSP90 as Potential Target. PLoS ONE 2008, 3, e0001722. [Google Scholar] [CrossRef]
- Park, K.-S.; Oh, B.; Lee, M.-H.; Nam, K.-Y.; Jin, H.R.; Yang, H.; Choi, J.; Kim, S.-W.; Lee, D.H. The HSP90 inhibitor, NVP-AUY922, sensitizes KRAS-mutant non-small cell lung cancer with intrinsic resistance to MEK inhibitor, trametinib. Cancer Lett. 2016, 372, 75–81. [Google Scholar] [CrossRef]
- Hirakawa, H.; Fujisawa, H.; Masaoka, A.; Noguchi, M.; Hirayama, R.; Takahashi, M.; Fujimori, A.; Okayasu, R. The combination of Hsp90 inhibitor 17 AAG and heavy-ion irradiation provides effective tumor control in human lung cancer cells. Cancer Med. 2015, 4, 426–436. [Google Scholar] [CrossRef]
- Hendriks, L.E.L.; Dingemans, A.-M.C. Heat shock protein antagonists in early stage clinical trials for NSCLC. Expert Opin. Investig. Drugs 2017, 26, 541–550. [Google Scholar] [CrossRef]
- Akram, A.; Khalil, S.; Halim, S.A.; Younas, H.; Iqbal, S.; Mehar, S. Therapeutic Uses of HSP90 Inhibitors in Non-Small Cell Lung Carcinoma (NSCLC). Curr. Drug Metab. 2018, 19, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Gettinger, S.; Senzer, N.N.; Martins, R.G.; Jänne, P.A.; Lilenbaum, R.; Gray, J.E.; Iafrate, A.J.; Katayama, R.; Hafeez, N.; et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 4953–4960. [Google Scholar] [CrossRef] [PubMed]
- Felip, E.; Barlesi, F.; Besse, B.; Chu, Q.; Gandhi, L.; Kim, S.-W.; Carcereny, E.; Sequist, L.V.; Brunsvig, P.; Chouaid, C.; et al. Phase 2 Study of the HSP-90 Inhibitor AUY922 in Previously Treated and Molecularly Defined Patients with Advanced Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 576–584. [Google Scholar] [CrossRef]
- Socinski, M.A.; Goldman, J.; El-Hariry, I.; Koczywas, M.; Vukovic, V.; Horn, L.; Paschold, E.; Salgia, R.; West, H.; Sequist, L.V.; et al. A Multicenter Phase II Study of Ganetespib Monotherapy in Patients with Genotypically Defined Advanced Non–Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 3068–3077. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Carnero, A.; Paz-Ares, L. Inhibition of HSP90 molecular chaperones: Moving into the clinic. Lancet Oncol. 2013, 14, e358–e369. [Google Scholar] [CrossRef] [PubMed]
- Rong, B.; Yang, S. Molecular mechanism and targeted therapy of Hsp90 involved in lung cancer: New discoveries and de-velopments (Review). Int. J. Oncol. 2018, 52, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Marrugal, Á.; Ferrer, I.; Pastor, M.D.; Ojeda, L.; Quintanal-Villalonga, Á.; Carnero, A.; Molina-Pinelo, S.; Paz-Ares, L. Impact of Heat Shock Protein 90 Inhibition on the Proteomic Profile of Lung Adenocarcinoma as Measured by Two-Dimensional Electrophoresis Coupled with Mass Spectrometry. Cells 2019, 8, 806. [Google Scholar] [CrossRef]
- Marrugal, Á.; Ferrer, I.; Gómez-Sánchez, D.; Quintanal-Villalonga, Á.; Pastor, M.D.; Ojeda, L.; Paz-Ares, L.; Molina-Pinelo, S. Identification of Predictive Biomarkers of Response to HSP90 Inhibitors in Lung Adenocarcinoma. Int. J. Mol. Sci. 2021, 22, 2538. [Google Scholar] [CrossRef] [PubMed]
- Yuno, A.; Lee, M.-J.; Lee, S.; Tomita, Y.; Rekhtman, D.; Moore, B.; Trepel, J.B. Clinical Evaluation and Biomarker Profiling of Hsp90 Inhibitors. Methods Mol. Biol. 2018, 1709, 423–441. [Google Scholar] [CrossRef]
- Li, F.; Zhao, D.; Yang, S.; Wang, J.; Liu, Q.; Jin, X.; Wang, W. ITRAQ-Based Proteomics Analysis of Triptolide On Human A549 Lung Adenocarcinoma Cells. Cell Physiol. Biochem. 2018, 45, 917–934. [Google Scholar] [CrossRef]
- Hsu, C.-H.; Hsu, C.-W.; Hsueh, C.; Wang, C.-L.; Wu, Y.-C.; Wu, C.-C.; Liu, C.-C.; Yu, J.-S.; Chang, Y.-S.; Yu, C.-J. Identification and Characterization of Potential Biomarkers by Quantitative Tissue Proteomics of Primary Lung Adenocarcinoma. Mol. Cell Proteom. 2016, 15, 2396–2410. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, X.; Wang, W.; Sun, R.; Liu, B.; Ma, Y.; Zhang, W.; Ma, L.; Jin, Y.; Yang, S. Quantitative proteomics analysis of mitochondrial proteins in lung adenocarcinomas and normal lung tissue using iTRAQ and tandem mass spectrometry. Am. J. Transl. Res. 2017, 9, 3918–3934. [Google Scholar]
- Richards, M.W.; Law, E.W.; Rennalls, L.V.P.; Busacca, S.; O’Regan, L.; Fry, A.M.; Fennell, D.A.; Bayliss, R. Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical beta-propeller domain. Proc. Natl. Acad. Sci. USA 2014, 111, 5195–5200. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Marcu, M.; Yuan, X.; Mimnaugh, E.; Patterson, C.; Neckers, L. Chaperone-dependent E3 ubiquitin ligase CHIP me-diates a degradative pathway for c-ErbB2/Neu. Proc. Natl. Acad. Sci. USA 2002, 99, 12847–12852. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.R.; Pochapsky, T.C.; Petsko, G.A.; Ringe, D. Dual chemistry catalyzed by human acireductone dioxygenase. Protein Eng. Des. Sel. 2017, 30, 109–206. [Google Scholar] [CrossRef]
- Uekita, T.; Gotoh, I.; Kinoshita, T.; Itoh, Y.; Sato, H.; Shiomi, T.; Okada, Y.; Seiki, M. Membrane-type 1 Matrix Metalloproteinase Cytoplasmic Tail-binding Protein-1 Is a New Member of the Cupin Superfamily. Pediatrics 2004, 279, 12734–12743. [Google Scholar] [CrossRef] [PubMed]
- Pratt, J.; Iddir, M.; Bourgault, S.; Annabi, B. Evidence of MTCBP-1 interaction with the cytoplasmic domain of MT1-MMP: Implications in the autophagy cell index of high-grade glioblastoma. Mol. Carcinog. 2015, 55, 148–160. [Google Scholar] [CrossRef]
- Su, L.; Yang, K.; Li, S.; Liu, C.; Han, J.; Zhang, Y.; Xu, G. Enolase-phosphatase 1 as a novel potential malignant glioma indicator promotes cell proliferation and migration. Oncol. Rep. 2018, 40, 2233–2241. [Google Scholar] [CrossRef]
- Oram, S.W.; Ai, J.; Pagani, G.M.; Hitchens, M.R.; Stern, J.A.; Eggener, S.; Pins, M.; Xiao, W.; Cai, X.; Haleem, R.; et al. Expression and function of the human andro-gen-responsive gene ADI1 in prostate cancer. Neoplasia 2007, 9, 643–651. [Google Scholar] [CrossRef]
- Chu, Y.-D.; Lai, H.-Y.; Pai, L.-M.; Huang, Y.-H.; Lin, Y.-H.; Liang, K.-H.; Yeh, C.-T. The methionine salvage pathway-involving ADI1 inhibits hepatoma growth by epigenetically altering genes expression via elevating S-adenosylmethionine. Cell Death Dis. 2019, 10, 240. [Google Scholar] [CrossRef] [PubMed]
- Savino, T.M.; Bastos, R.; Jansen, E.; Hernandez-Verdun, D. The nucleolar antigen Nop52, the human homologue of the yeast ribosomal RNA processing RRP1, is recruited at late stages of nucleologenesis. J. Cell Sci. 1999, 112, 1889–1900. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Ishikawa, H.; Izumikawa, K.; Miura, Y.; Hayano, T.; Isobe, T.; Simpson, R.J.; Takahashi, N. Human nucleolar protein Nop52 (RRP1/NNP-1) is involved in site 2 cleavage in internal transcribed spacer 1 of pre-rRNAs at early stages of ribosome biogenesis. Nucleic Acids Res. 2015, 43, 5524–5536. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Komatsu, W.; Hayano, T.; Miura, Y.; Homma, K.; Izumikawa, K.; Ishikawa, H.; Miyazawa, N.; Tachikawa, H.; Yamauchi, Y.; et al. Splicing Factor 2-Associated Protein p32 Participates in Ribosome Biogenesis by Regulating the Binding of Nop52 and Fibrillarin to Preribosome Particles. Mol. Cell Proteom. 2011, 10, M110.006148. [Google Scholar] [CrossRef] [PubMed]
- Senju, M.; Sueoka, N.; Sato, A.; Iwanaga, K.; Sakao, Y.; Tomimitsu, S.; Tominaga, M.; Irie, K.; Hayashi, S.; Sueoka, E. Hsp90 inhibitors cause G2/M arrest associated with the reduction of Cdc25C and Cdc2 in lung cancer cell lines. J. Cancer Res. Clin. Oncol. 2006, 132, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.N.; Ramalingam, S.S. Heat shock protein 90 inhibitors in non-small-cell lung cancer. Curr. Opin. Oncol. 2014, 26, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Jahani, M.; Noroznezhad, F.; Mansouri, K. Arginine: Challenges and opportunities of this two-faced molecule in cancer therapy. BioMedicine 2018, 102, 594–601. [Google Scholar] [CrossRef]
- Delage, B.; Fennell, D.A.; Nicholson, L.; McNeish, I.; Lemoine, N.R.; Crook, T.; Szlosarek, P.W. Arginine deprivation and argininosuc-cinate synthetase expression in the treatment of cancer. Int. J. Cancer 2010, 126, 2762–2772. [Google Scholar] [PubMed]
- Keshet, R.; Erez, A. Arginine and the metabolic regulation of nitric oxide synthesis in cancer. Dis. Model. Mech. 2018, 11, dmm03332. [Google Scholar] [CrossRef]
- Napoli, C.; Paolisso, G.; Casamassimi, A.; Al-Omran, M.; Barbieri, M.; Sommese, L.; Infante, T.; Ignarro, L.J. Effects of nitric oxide on cell prolif-eration: Novel insights. J. Am. Coll. Cardiol. 2013, 62, 89–95. [Google Scholar] [CrossRef]
- Ning, S.; Bednarski, M.; Oronsky, B.; Scicinski, J.; Knox, S.J. Novel nitric oxide generating compound glycidyl nitrate enhances the therapeutic efficacy of chemotherapy and radiotherapy. Biochem. Biophys. Res. Commun. 2014, 447, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B.; Garban, H. Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics. Redox Biol. 2015, 6, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Dingemans, A.M.; Groen, H.J.; Herder, G.J.; Stigt, J.A.; Smit, E.F.; Bahce, I.; Burgers, J.A.; van den Borne, B.E.; Biesma, B.; Vincent, A.; et al. A randomized phase II study comparing paclitaxel-carboplatin-bevacizumab with or without nitroglycerin patches in patients with stage IV nonsquamous nonsmall-cell lung cancer: NVALT12 (NCT01171170) dagger. Ann. Oncol. 2015, 26, 2286–2293. [Google Scholar] [CrossRef]
- Carter, C.A.; Oronsky, B.; Caroen, S.; Scicinski, J.; Cabrales, P.; Degesys, A.; Brzezniak, C. Partial response to carboplatin in an RRx-001 pretreated patient with EGFR-inhibitor-resistance and T790M-negative NSCLC. Respir. Med. Case Rep. 2016, 18, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Huang, J.; Sui, M. Targeting arginine metabolism pathway to treat arginine-dependent cancers. Cancer Lett. 2015, 364, 1–7. [Google Scholar] [CrossRef]
- Phillips, M.M.; Sheaff, M.T.; Szlosarek, P.W. Targeting arginine-dependent cancers with arginine-degrading enzymes: Op-portunities and challenges. Cancer Res. Treat 2013, 45, 251–262. [Google Scholar] [CrossRef]
- Patil, M.D.; Bhaumik, J.; Babykutty, S.; Banerjee, U.C.; Fukumura, D. Arginine dependence of tumor cells: Targeting a chink in cancer’s armor. Oncogene 2016, 35, 4957–4972. [Google Scholar] [CrossRef]
- Rabinovich, S.; Adler, L.; Yizhak, K.; Sarver, A.; Silberman, A.; Agron, S.; Stettner, N.; Sun, Q.; Brandis, A.; Helbling, D.; et al. Diversion of aspartate in ASS1-deficient tu-mours fosters de novo pyrimidine synthesis. Nature 2015, 527, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Li, M.; Wang, J.; Nie, F.; Li, L. The E3 Ubiquitin Ligase ITCH Negatively Regulates Canonical Wnt Signaling by Targeting Dishevelled Protein. Mol. Cell Biol. 2012, 32, 3903–3912. [Google Scholar] [CrossRef]
- Serman, L.; Martic, T.N.; Serman, A.; Vranic, S. Epigenetic alterations of the Wnt signaling pathway in cancer: A mini review. Bosn. J. Basic Med. Sci. 2014, 14, 191–194. [Google Scholar] [CrossRef]
- Chen, X.; Song, X.; Yue, W.; Chen, D.; Yu, J.; Yao, Z.; Zhang, L. Fibulin-5 inhibits Wnt/beta-catenin signaling in lung cancer. Oncotarget 2015, 6, 15022–15034. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Zhang, L.; Fan, K.; Cheng, Z.X.; Sun, Q.C.; Wang, J.J. Circular RNA-ITCH Suppresses Lung Cancer Proliferation via Inhibiting the Wnt/beta-Catenin Pathway. Biomed Res. Int. 2016, 2016, 1579490. [Google Scholar] [CrossRef]
- Han, X.; Zhang, L.; Chung, J.; Pozo, F.M.; Tran, A.; Seachrist, D.D.; Jacobberger, J.W.; Keri, R.A.; Gilmore, H.; Zhang, Y. UbcH7 regulates 53BP1 stability and DSB repair. Proc. Natl. Acad. Sci. USA 2014, 111, 17456–17461. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.J.; Vyse, S.; Shields, A.M.; Boeltz, S.; Gordon, P.A.; Spector, T.D.; Lehner, P.J.; Walczak, H.; Vyse, T.J. UBE2L3 polymorphism amplifies NF-kappa B activation and promotes plasma cell development, linking linear ubiquitination to multiple autoimmune diseases. Am. J. Hum. Genet. 2015, 96, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhao, J.; Yang, F.; Liu, H.; Qi, W. Ubiquitin conjugating enzyme E2 L3 promoted tumor growth of NSCLC through accelerating p27kip1 ubiquitination and degradation. Oncotarget 2017, 8, 84193–84203. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Győrffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online survival analysis software to assess the prognostic value of bi-omarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | HSP90 Inhibitors | ||||||
|---|---|---|---|---|---|---|---|
| IC80 17-AAG nM | IC80 IPI-504 nM | IC80 STA-9090 nM | IC80 AUY-922 nM | EGFR | ALK | KRAS | |
| HCC827 | 105.02 | 68.58 | 20.55 | 16.67 | M | WT | WT |
| H1975 | 5.03 | 51.00 | 18.96 | 10.38 | M | WT | WT |
| H1650 | 26.22 | 15.06 | 22.64 | 5.89 | M | WT | WT |
| H3122 | 104.66 | 113.45 | 31.96 | 36.44 | WT | T | WT |
| H2228 | 43.55 | 185.36 | 16.52 | 17.95 | WT | T | WT |
| H2009 | 172.79 | 135.33 | 18.63 | 9.91 | WT | WT | M |
| H358 | 52.26 | 18.65 | 30.96 | 32.42 | WT | WT | M |
| A549 | 65.18 | 77.97 | 25.24 | 122.93 | WT | WT | M |
| H1437 | 14.83 | 13.89 | 27.18 | 11.26 | WT | WT | WT |
| CALU-3 | 350.93 | 173.18 | 73.78 | 6963.64 | WT | WT | WT |
| H1781 | 49.38 | 123.9 | 39.82 | 95.15 | WT | WT | WT |
| Treatment | Expression Change | HCC827 | H1975 | H1650 | H3122 | H2228 | H2009 | H358 | A549 | H1437 | CALU3 | H1781 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EGFR mut | EML4-ALK | KRAS mut | TN | ||||||||||
| HSP90 inhibition | 17-AAG | + | 96 | 133 | 29 | 142 | 122 | 126 | 69 | 165 | 31 | 172 | 89 |
| − | 238 | 170 | 109 | 389 | 385 | 269 | 337 | 202 | 28 | 438 | 445 | ||
| IPI-504 | + | 107 | 169 | 13 | 168 | 181 | 106 | 103 | 194 | 13 | 195 | 120 | |
| − | 154 | 301 | 40 | 287 | 370 | 258 | 227 | 204 | 16 | 391 | 473 | ||
| STA-9090 | + | 94 | 143 | 7 | 167 | 135 | 78 | 188 | 70 | 22 | 288 | 58 | |
| − | 164 | 230 | 65 | 119 | 147 | 177 | 149 | 113 | 23 | 276 | 244 | ||
| AUY-922 | + | 140 | 128 | 31 | 269 | 142 | 79 | 116 | 189 | 56 | 197 | 138 | |
| − | 167 | 159 | 118 | 202 | 199 | 146 | 161 | 182 | 23 | 483 | 318 | ||
| Genetic silencing | siRNA HSP90α + β | + | 471 | 134 | 69 | 321 | 304 | 269 | 249 | 219 | 92 | 316 | 198 |
| − | 243 | 272 | 69 | 309 | 434 | 108 | 289 | 252 | 217 | 270 | 294 | ||
| Molecular Background | Significant Proteins | Final Proteins * | Biological Pathways |
|---|---|---|---|
| EGFR MUT | 332 | 246 | 7 |
| EML4-ALK | 57 | 34 | 10 |
| KRAS MUT | 1696 | 1157 | 21 |
| TN | 1288 | 866 | 12 |
| EGFR Mut | EML4-ALK | KRAS Mut | Triple Negative |
|---|---|---|---|
| Apoptosis | Apoptosis | Apoptosis | Apoptosis |
| Arginine biosynthesis | Arginine biosynthesis | Cholesterol biosynthesis | Coenzyme A biosynthesis |
| Asparagine and aspartate biosynthesis | Formyltetrahydroformate biosynthesis | Purine biosynthesis | Proline biosynthesis |
| Serine glycine biosynthesis | Succinate to propionate conversion | Pyrimidine ribonucleotides byosinthesis | Huntington’s disease |
| Methylcitrate cycle | EGFR signaling pathway | Heme biosynthesis | Pyrimidine metabolism |
| Huntington’s disease | FGF signaling pathway | Insulin/IGF-PKB pathway | DNA replication |
| p53 pathway | PDGF signaling pathway | Cell cycle | Ubiquitin proteasome pathway |
| AR-α signaling pathway | Huntington’s disease | Pentose phosphate pathway | |
| GnRHR pathway | Parkinson’s disease | L signaling pathway | |
| Methylmalonil pathway | Axon guidance mediated by semaphorins | PDGF signaling pathway | |
| CCKR signaling map | Endothelin signaling pathway | ||
| Salvage pyrimidine ribonucleotides | GnRHR pathway | ||
| Cytoskeletal regulation by Rho GTPase | |||
| DNA replication | |||
| Ubiquitin proteasome pathway | |||
| p53 pathway by glucose deprivation | |||
| EGFR signaling pathway | |||
| FGF signaling pathway | |||
| VEGF signaling pathway | |||
| Cadherin signaling pathway | |||
| Integrin signaling pathway | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marrugal, Á.; Ferrer, I.; Quintanal-Villalonga, Á.; Ojeda, L.; Pastor, M.D.; García-Luján, R.; Carnero, A.; Paz-Ares, L.; Molina-Pinelo, S. Inhibition of HSP90 in Driver Oncogene-Defined Lung Adenocarcinoma Cell Lines: Key Proteins Underpinning Therapeutic Efficacy. Int. J. Mol. Sci. 2023, 24, 13830. https://doi.org/10.3390/ijms241813830
Marrugal Á, Ferrer I, Quintanal-Villalonga Á, Ojeda L, Pastor MD, García-Luján R, Carnero A, Paz-Ares L, Molina-Pinelo S. Inhibition of HSP90 in Driver Oncogene-Defined Lung Adenocarcinoma Cell Lines: Key Proteins Underpinning Therapeutic Efficacy. International Journal of Molecular Sciences. 2023; 24(18):13830. https://doi.org/10.3390/ijms241813830
Chicago/Turabian StyleMarrugal, Ángela, Irene Ferrer, Álvaro Quintanal-Villalonga, Laura Ojeda, María Dolores Pastor, Ricardo García-Luján, Amancio Carnero, Luis Paz-Ares, and Sonia Molina-Pinelo. 2023. "Inhibition of HSP90 in Driver Oncogene-Defined Lung Adenocarcinoma Cell Lines: Key Proteins Underpinning Therapeutic Efficacy" International Journal of Molecular Sciences 24, no. 18: 13830. https://doi.org/10.3390/ijms241813830