
Construction of Histone–Protein Complex Structures by Peptide Growing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
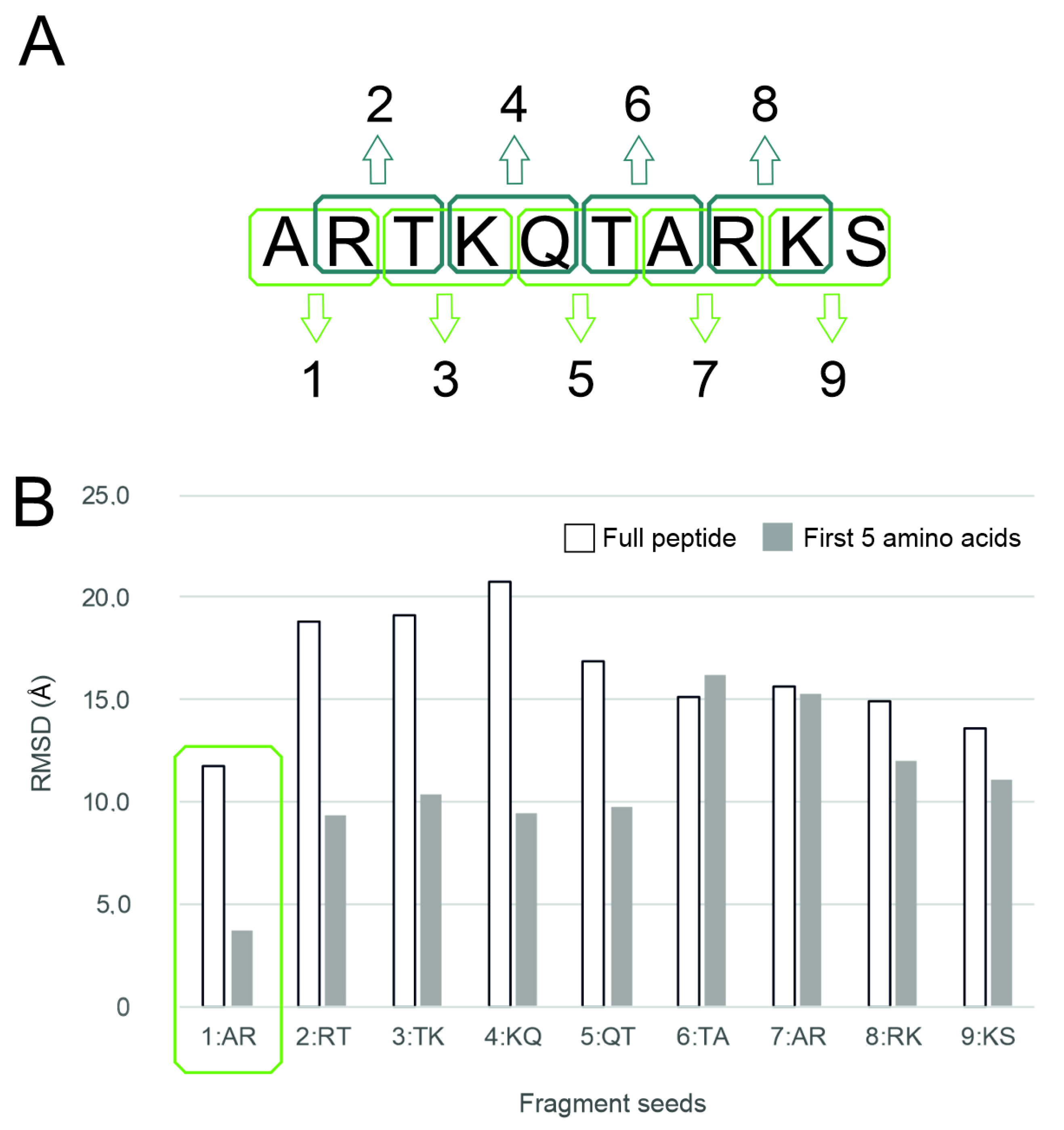
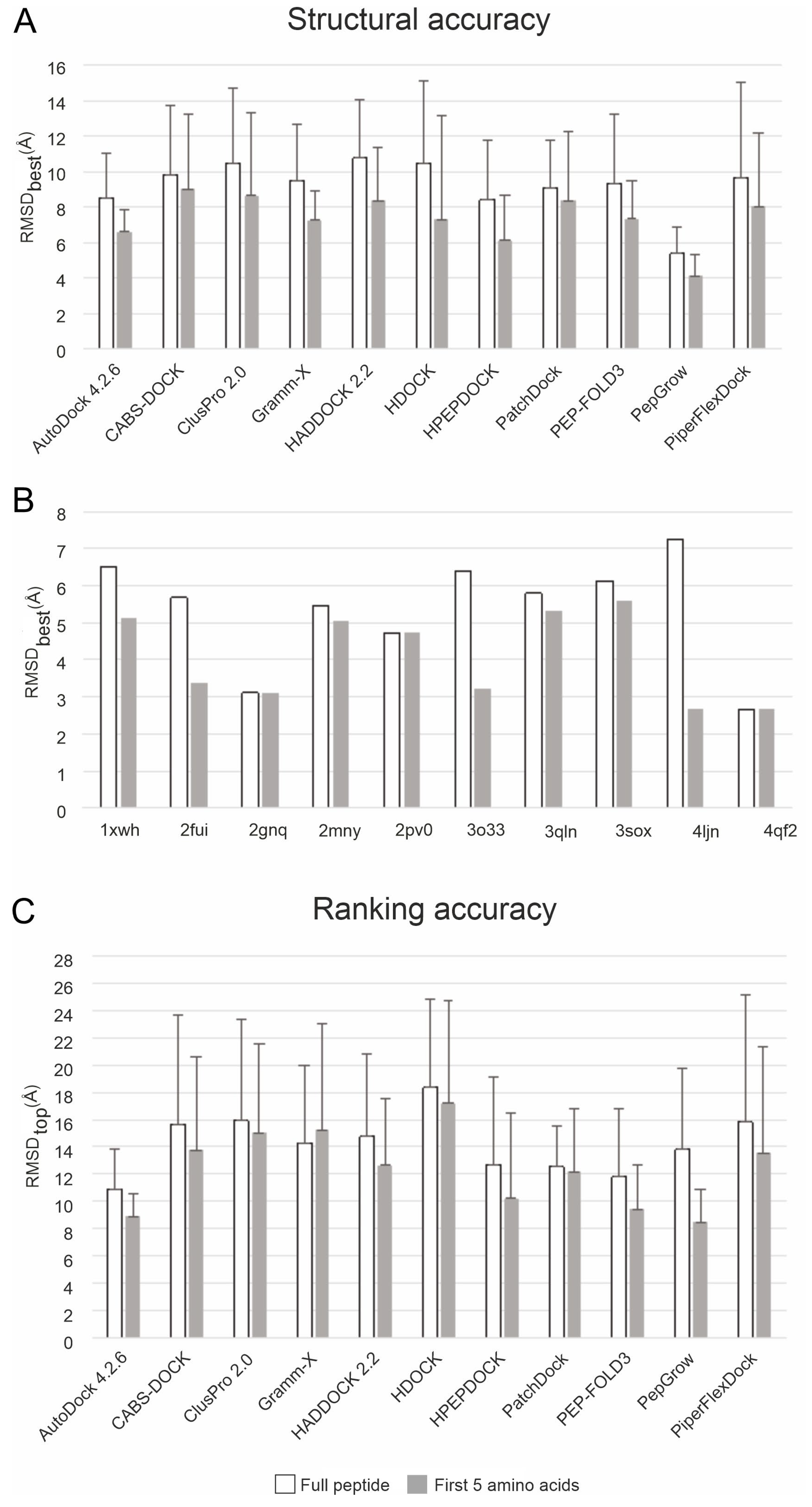
2.1. Histone Systems and Benchmark Methods
2.2. The PepGrow Protocol
2.3. Performance
3. Materials and Methods
3.1. Selection of Test Systems and Benchmark Methods
3.2. Performance Metrics
3.3. Application of Benchmark Methods
3.4. PepGrow
3.5. Calculation of Einter and Energy Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shvedunova, M.; Akhtar, A. Modulation of Cellular Processes by Histone and Non-Histone Protein Acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Enetics, E.P.I.G.; Gamblin, S.J.; Wilson, J.O.N.R. A Key to Unlock Chromatin. Nature 2019, 573, 355–356. [Google Scholar]
- Izzo, L.T.; Wellen, K.E. Histone Lactylation Links Metabolism and Gene Regulation. Nature 2019, 574, 492–493. [Google Scholar] [CrossRef] [PubMed]
- Org, T.; Chignola, F.; Hetényi, C.; Gaetani, M.; Rebane, A.; Liiv, I.; Maran, U.; Mollica, L.; Bottomley, M.J.; Musco, G.; et al. The Autoimmune Regulator PHD Finger Binds to Non-Methylated Histone H3K4 to Activate Gene Expression. EMBO Rep. 2008, 9, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Zsidó, B.Z.; Hetényi, C. Molecular Structure, Binding Affinity, and Biological Activity in the Epigenome. Int. J. Mol. Sci. 2020, 21, 4134. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Musselman, C.A.; Lalonde, M.E.; Côté, J.; Kutateladze, T.G. Perceiving the Epigenetic Landscape through Histone Readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic Protein Families: A New Frontier for Drug Discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef]
- Bortoluzzi, A.; Amato, A.; Lucas, X.; Blank, M.; Ciulli, A. Structural Basis of Molecular Recognition of Helical Histone H3 Tail by PHD Finger Domains. Biochem. J. 2017, 474, 1633–1651. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Wang, W.; Graybosch, D.M.; Li, H.; Allis, C.D.; Patel, D.J.; Verdine, G.L. Histone H3 Recognition and Presentation by the WDR5 Module of the MLL1 Complex. Nat. Struct. Mol. Biol. 2006, 13, 704–712. [Google Scholar] [CrossRef]
- Ooi, S.K.T.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L Connects Unmethylated Lysine 4 of Histone H3 to de Novo Methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Iwase, S.; Xiang, B.; Ghosh, S.; Ren, T.; Lewis, P.W.; Cochrane, J.C.; Allis, C.D.; Picketts, D.J.; Patel, D.J.; Li, H.; et al. ATRX ADD Domain Links an Atypical Histone Methylation Recognition Mechanism to Human Mental-Retardation Syndrome. Nat. Struct. Mol. Biol. 2011, 18, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Rajakumara, E.; Wang, Z.; Ma, H.; Hu, L.; Chen, H.; Lin, Y.; Guo, R.; Wu, F.; Li, H.; Lan, F.; et al. PHD Finger Recognition of Unmodified Histone H3R2 Links UHRF1 to Regulation of Euchromatic Gene Expression. Mol. Cell 2011, 43, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.Y.; Shi, X.; Schwarzer, D.; Plunkett, W.; et al. TRIM24 Links a Non-Canonical Histone Signature to Breast Cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef]
- Chignola, F.; Gaetani, M.; Rebane, A.; Org, T.; Mollica, L.; Zucchelli, C.; Spitaleri, A.; Mannella, V.; Peterson, P.; Musco, G. The Solution Structure of the First PHD Finger of Autoimmune Regulator in Complex with Non-Modified Histone H3 Tail Reveals the Antagonistic Role of H3R2 Methylation. Nucleic Acids Res. 2009, 37, 2951–2961. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, H.; Guo, X.; Rong, N.; Song, Y.; Xu, Y.; Lan, W.; Zhang, X.; Liu, M.; Xu, Y.; et al. The PHD1 Finger of KDM5B Recognizes Unmodified H3K4 during the Demethylation of Histone H3K4me2/3 by KDM5B. Protein Cell 2014, 5, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ilin, S.; Wang, W.; Duncan, E.M.; Wysocka, J.; Allis, C.D.; Patel, D.J. Molecular Basis for Site-Specific Read-out of Histone H3K4me3 by the BPTF PHD Finger of NURF. Nature 2006, 442, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Dreveny, I.; Deeves, S.E.; Fulton, J.; Yue, B.; Messmer, M.; Bhattacharya, A.; Collins, H.M.; Heery, D.M. The Double PHD Finger Domain of MOZ/MYST3 Induces α-Helical Structure of the Histone H3 Tail to Facilitate Acetylation and Methylation Sampling and Modification. Nucleic Acids Res. 2014, 42, 822–835. [Google Scholar] [CrossRef]
- Sanchez, R.; Meslamani, J.; Zhou, M.-M. The Bromodomain: From Epigenome Reader to Druggable Target. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2014, 1839, 676–685. [Google Scholar] [CrossRef]
- Li, X.; Yao, Y.; Wu, F.; Song, Y. A Proteolysis-Targeting Chimera Molecule Selectively Degrades ENL and Inhibits Malignant Gene Expression and Tumor Growth. J. Hematol. Oncol. 2022, 15, 41. [Google Scholar] [CrossRef]
- Mosca, R.; Céol, A.; Aloy, P. Interactome3D: Adding Structural Details to Protein Networks. Nat. Methods 2013, 10, 47–53. [Google Scholar] [CrossRef]
- Srivastava, A.; Nagai, T.; Srivastava, A.; Miyashita, O.; Tama, F. Role of Computational Methods in Going beyond X-Ray Crystallography to Explore Protein Structure and Dynamics. Int. J. Mol. Sci. 2018, 19, 3401. [Google Scholar] [CrossRef]
- Frank, J. Electron Microscopy Applied to Molecular Machines. Biopolymers 2013, 99, 832–836. [Google Scholar] [CrossRef]
- Verardi, R.; Traaseth, N.J.; Masterson, L.R.; Vostrikov, V.V.; Veglia, G. Isotope Labeling in Biomolecular NMR.; Springer: Dordrecht, The Netherlands, 2012; Volume 992, ISBN 978-94-007-4953-5. [Google Scholar]
- Antunes, D.A.; Devaurs, D.; Kavraki, L.E. Understanding the Challenges of Protein Flexibility in Drug Design. Expert Opin. Drug Discov. 2015, 10, 1301–1313. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Hauser, A.S.; Windshügel, B. LEADS-PEP: A Benchmark Data Set for Assessment of Peptide Docking Performance. J. Chem. Inf. Model. 2016, 56, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, K.; Felekyan, S.; Kühnemuth, R.; Dimura, M.; Tóth, K.; Seidel, C.A.M.; Langowski, J. Dynamics of the Nucleosomal Histone H3 N-Terminal Tail Revealed by High Precision Single-Molecule FRET. Nucleic Acids Res. 2020, 48, 1551–1571. [Google Scholar] [CrossRef] [PubMed]
- Morrison, E.A.; Bowerman, S.; Sylvers, K.L.; Wereszczynski, J.; Musselman, C.A. The Conformation of the Histone H3 Tail Inhibits Association of the BPTF PHD Finger with the Nucleosome. eLife 2018, 7, e31481. [Google Scholar] [CrossRef]
- Morrison, E.A.; Baweja, L.; Poirier, M.G.; Wereszczynski, J.; Musselman, C.A. Nucleosome Composition Regulates the Histone H3 Tail Conformational Ensemble and Accessibility. Nucleic Acids Res. 2021, 49, 4750–4767. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, R.; Renard, B.Y. Docking Small Peptides Remains a Great Challenge: An Assessment Using AutoDock Vina. Brief. Bioinform. 2015, 16, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef]
- Weaver, T.M.; Morrison, E.A.; Musselman, C.A. Reading More than Histones: The Prevalence of Nucleic Acid Binding among Reader Domains. Molecules 2018, 23, 2614. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.; Kurcinski, M.; Kamel, K.; Kolinski, A.; Alam, N.; Schueler-Furman, O.; Kmiecik, S. Protein–Peptide Docking: Opportunities and Challenges. Drug Discov. Today 2018, 23, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.L.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.X.; Roy, A.; Christoffer, C.; Terashi, G.; Kihara, D. Modeling Disordered Protein Interactions from Biophysical Principles. PLoS Comput. Biol. 2017, 13, e1005485. [Google Scholar] [CrossRef]
- Xiong, G.-L.; Ye, W.-L.; Shen, C.; Lu, A.-P.; Hou, T.-J.; Cao, D.-S. Improving Structure-Based Virtual Screening Performance via Learning from Scoring Function Components. Brief. Bioinform. 2021, 22, bbaa094. [Google Scholar] [CrossRef]
- Zsidó, B.Z.; Hetényi, C. The Role of Water in Ligand Binding. Curr. Opin. Struct. Biol. 2021, 67, 1–8. [Google Scholar] [CrossRef]
- Richmond, T.J.; Davey, C.A. The Structure of DNA in the Nucleosome Core. Nature 2003, 423, 145–150. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC.: New York, NY, USA, 2021. [Google Scholar]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein-Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef]
- Alam, N.; Goldstein, O.; Xia, B.; Porter, K.A.; Kozakov, D.; Schueler-Furman, O. High-Resolution Global Peptide-Protein Docking Using Fragments-Based PIPER-FlexPepDock. PLoS Comput. Biol. 2017, 13, e1005905. [Google Scholar] [CrossRef]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-Dock Web Server for the Flexible Docking of Peptides to Proteins without Prior Knowledge of the Binding Site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de Novo Structure Prediction for Linear Peptides in Solution and in Complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef]
- Castro-Alvarez, A.; Costa, A.M.; Vilarrasa, J. The Performance of Several Docking Programs at Reproducing Protein-Macrolide-like Crystal Structures. Molecules 2017, 22, 136. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; Körtvélyesi, T.; Penke, B. Mapping of Possible Binding Sequences of Two Beta-Sheet Breaker Peptides on Beta Amyloid Peptide of Alzheimer’s Disease. Bioorg. Med. Chem. 2002, 10, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Xie, X.Q. Computational Fragment-Based Drug Design: Current Trends, Strategies, and Applications. AAPS J. 2019, 20, 59. [Google Scholar] [CrossRef]
- Evans, D.J.; Yovanno, R.A.; Rahman, S.; Cao, D.W.; Beckett, M.Q.; Patel, M.H.; Bandak, A.F.; Lau, A.Y. Finding Druggable Sites in Proteins Using TACTICS. J. Chem. Inf. Model. 2021, 61, 2897–2910. [Google Scholar] [CrossRef]
- Aguayo-Ortiz, R.; Dominguez, L. Unveiling the Possible Oryzalin-Binding Site in the α-Tubulin of Toxoplasma Gondii. ACS Omega 2022, 7, 18434–18442. [Google Scholar] [CrossRef]
- Aguayo-Ortiz, R.; Guzmán-Ocampo, D.C.; Dominguez, L. Insights into the Binding of Morin to Human ΓD-Crystallin. Biophys. Chem. 2022, 282, 106750. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Portelli, S.; Rezende, P.M.; Veloso, W.N.P.; Xavier, J.S.; Karmakar, M.; Myung, Y.; Linhares, J.P.V.; Rodrigues, C.H.M.; Silk, M.; et al. A Comprehensive Computational Platform to Guide Drug Development Using Graph-Based Signature Methods. Methods Mol. Biol. 2020, 2112, 91–106. [Google Scholar]
- Lamoree, B.; Hubbard, R.E. Current Perspectives in Fragment-Based Lead Discovery (FBLD). Essays Biochem. 2017, 61, 453–464. [Google Scholar] [CrossRef]
- de Beauchene, I.C.; de Vries, S.J.; Zacharias, M. Binding Site Identification and Flexible Docking of Single Stranded RNA to Proteins Using a Fragment-Based Approach. PLoS Comput. Biol. 2016, 12, e1004697. [Google Scholar] [CrossRef]
- Liao, J.M.; Wang, Y.T.; Lin, C.L.S. A Fragment-Based Docking Simulation for Investigating Peptide-Protein Bindings. Phys. Chem. Chem. Phys. 2017, 19, 10436–10442. [Google Scholar] [CrossRef] [PubMed]
- Budin, N.; Majeux, N.; Caflisch, A. Fragment-Based Flexible Ligand Docking by Evolutionary Optimization. Biol. Chem. 2001, 382, 1365–1372. [Google Scholar] [CrossRef]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, S.B.; Johnson, A.P. EHiTS: A New Fast, Exhaustive Flexible Ligand Docking System. J. Mol. Graph. Model. 2007, 26, 198–212. [Google Scholar] [CrossRef]
- Thompson, D.C.; Denny, R.A.; Nilakantan, R.; Humblet, C.; Joseph-McCarthy, D.; Feyfant, E. CONFIRM: Connecting Fragments Found in Receptor Molecules. J. Comput. Aided Mol. Des. 2008, 22, 761–772. [Google Scholar] [CrossRef]
- Samsonov, S.A.; Zacharias, M.; de Chauvot Beauchene, I. Modeling Large Protein–Glycosaminoglycan Complexes Using a Fragment-Based Approach. J. Comput. Chem. 2019, 40, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.S.J. Improved FlexX Docking Using FlexS-Determined Base Fragment Placement. J. Chem. Inf. Model. 2005, 45, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Bálint, M.; Horváth, I.; Mészáros, N.; Hetényi, C. Towards Unraveling the Histone Code by Fragment Blind Docking. Int. J. Mol. Sci. 2019, 20, 422. [Google Scholar] [CrossRef]
- Hoffer, L.; Muller, C.; Roche, P.; Morelli, X. Chemistry-Driven Hit-to-Lead Optimization Guided by Structure-Based Approaches. Mol. Inform. 2018, 37, e1800059. [Google Scholar] [CrossRef]
- Yuan, Y.; Pei, J.; Lai, L. LigBuilder V3: A Multi-Target de Novo Drug Design Approach. Front. Chem. 2020, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Soler, D.; Soliva, R.; Guallar, V. FragPELE: Dynamic Ligand Growing within a Binding Site. A Novel Tool for Hit-To-Lead Drug Design. J. Chem. Inf. Model. 2020, 60, 1728–1736. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Bálint, M.; Jeszenoi, N.; Horváth, I.; Van Der Spoel, D.; Hetényi, C. Systematic Exploration of Multiple Drug Binding Sites. J. Cheminform. 2017, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Jeszenoi, N.; Bálint, M.; Horváth, I.; Van Der Spoel, D.; Hetényi, C. Exploration of Interfacial Hydration Networks of Target-Ligand Complexes. J. Chem. Inf. Model. 2016, 56, 148–158. [Google Scholar] [CrossRef]
- Zhao, S.; Yang, M.; Zhou, W.; Zhang, B.; Cheng, Z.; Huang, J.; Zhang, M.; Wang, Z.; Wang, R.; Chen, Z.; et al. Kinetic and High-Throughput Profiling of Epigenetic Interactions by 3D-Carbene Chip-Based Surface Plasmon Resonance Imaging Technology. Proc. Natl. Acad. Sci. USA 2017, 114, E7245–E7254. [Google Scholar] [CrossRef] [PubMed]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short Linear Motifs: Ubiquitous and Functionally Diverse Protein Interaction Modules Directing Cell Regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef]
- Davis, A.M.; Teague, S.J.; Kleywegt, G.J. Application and Limitations of X-Ray Crystallographic Data in Structure-Based Ligand and Drug Design. Angew. Chem. Int. Ed. 2003, 42, 2718–2736. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein–Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Tovchigrechko, A.; Vakser, I.A. GRAMM-X Public Web Server for Protein-Protein Docking. Nucleic Acids Res. 2006, 34, W310–W314. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.-Y. HPEPDOCK: A Web Server for Blind Peptide–Protein Docking Based on a Hierarchical Algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A Web Server for Protein–Protein and Protein–DNA/RNA Docking Based on a Hybrid Strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-Based Protein Docking Program with Pairwise Potentials. Proteins Struct. Funct. Bioinform. 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, R.; Jia, Y.; Tang, P.; Wei, B.; Zhang, Q.; Wang, V.Y.-F.; Yan, R. Inhibition and Structure-Activity Relationship of Dietary Flavones against Three Loop 1-Type Human Gut Microbial β-Glucuronidases. Int. J. Biol. Macromol. 2022, 220, 1532–1544. [Google Scholar] [CrossRef]
- Fiser, A.; Do, R.K.G.; Šali, A. Modeling of Loops in Protein Structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef]
- Hetényi, C.; van der Spoel, D. Efficient Docking of Peptides to Proteins without Prior Knowledge of the Binding Site. Protein Sci. 2009, 11, 1729–1737. [Google Scholar] [CrossRef]
- Basciu, A.; Callea, L.; Motta, S.; Bonvin, A.M.J.J.; Bonati, L.; Vargiu, A.V. No Dance, No Partner! A Tale of Receptor Flexibility in Docking and Virtual Screening. Annu. Rep. Med. Chem. 2022, 59, 43–97. [Google Scholar]
- Li, C.; Sun, J.; Li, L.-W.; Wu, X.; Palade, V. An Effective Swarm Intelligence Optimization Algorithm for Flexible Ligand Docking. IEEE ACM Trans. Comput. Biol. Bioinform. 2022, 19, 2672–2684. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed]
- Kolinski, A. Protein Modeling and Structure Prediction with a Reduced Representation. Acta Biochim. Pol. 2004, 51, 349–371. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Zou, X. MDockPP: A Hierarchical Approach for Protein-Protein Docking and Its Application to CAPRI Rounds 15–19. Proteins Struct. Funct. Bioinform. 2010, 78, 3096–3103. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Huang, S.-Y. Efficient Conformational Ensemble Generation of Protein-Bound Peptides. J. Cheminform. 2017, 9, 59. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Zou, X. Ensemble Docking of Multiple Protein Structures: Considering Protein Structural Variations in Molecular Docking. Proteins Struct. Funct. Bioinform. 2006, 66, 399–421. [Google Scholar] [CrossRef]
- Gront, D.; Kulp, D.W.; Vernon, R.M.; Strauss, C.E.M.; Baker, D. Generalized Fragment Picking in Rosetta: Design, Protocols and Applications. PLoS ONE 2011, 6, e23294. [Google Scholar] [CrossRef]
- Raveh, B.; London, N.; Schueler-Furman, O. Sub-Angstrom Modeling of Complexes between Flexible Peptides and Globular Proteins. Proteins Struct. Funct. Bioinform. 2010, 78, 2029–2040. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative Partial Equalization of Orbital Electronegativity—A Rapid Access to Atomic Charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Rackers, J.A.; Wang, Z.; Lu, C.; Laury, M.L.; Lagardère, L.; Schnieders, M.J.; Piquemal, J.-P.; Ren, P.; Ponder, J.W. Tinker 8: Software Tools for Molecular Design. J. Chem. Theory Comput. 2018, 14, 5273–5289. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Hetényi, C.; Van Der Spoel, D. Blind Docking of Drug-Sized Compounds to Proteins with up to a Thousand Residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef]
- Shen, M.; Sali, A. Statistical Potential for Assessment and Prediction of Protein Structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Mehler, E.L.; Solmajer, T. Electrostatic Effects in Proteins: Comparison of Dielectric and Charge Models. Protein Eng. Des. Sel. 1991, 4, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cieplak, P.; Li, J.; Cai, Q.; Hsieh, M.-J.; Luo, R.; Duan, Y. Development of Polarizable Models for Molecular Mechanical Calculations. 4. van Der Waals Parametrization. J. Phys. Chem. B 2012, 116, 7088–7101. [Google Scholar] [CrossRef]
- Ferreira, L.; dos Santos, R.; Oliva, G.; Andricopulo, A. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Vitoria, M.; Granich, R.; Gilks, C.F.; Gunneberg, C.; Hosseini, M.; Were, W.; Raviglione, M.; De Cock, K.M. The Global Fight Against HIV/AIDS, Tuberculosis, and Malaria. Am. J. Clin. Pathol. 2009, 131, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva, F.P., Jr. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef]
- de Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M. Molecular Docking as a Popular Tool in Drug Design, an in Silico Travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Schreiber, G.; Fleishman, S.J. Computational Design of Protein–Protein Interactions. Curr. Opin. Struct. Biol. 2013, 23, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, S.; Fernandez-Recio, J. Protein-Protein Docking and Hot-Spot Prediction for Drug Discovery. Curr. Pharm. Des. 2012, 18, 4607–4618. [Google Scholar] [CrossRef] [PubMed]
- Bienstock, R.J. Computational Drug Design Targeting Protein-Protein Interactions. Curr. Pharm. Des. 2012, 18, 1240–1254. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zsidó, B.Z.; Bayarsaikhan, B.; Börzsei, R.; Hetényi, C. Construction of Histone–Protein Complex Structures by Peptide Growing. Int. J. Mol. Sci. 2023, 24, 13831. https://doi.org/10.3390/ijms241813831
Zsidó BZ, Bayarsaikhan B, Börzsei R, Hetényi C. Construction of Histone–Protein Complex Structures by Peptide Growing. International Journal of Molecular Sciences. 2023; 24(18):13831. https://doi.org/10.3390/ijms241813831
Chicago/Turabian StyleZsidó, Balázs Zoltán, Bayartsetseg Bayarsaikhan, Rita Börzsei, and Csaba Hetényi. 2023. "Construction of Histone–Protein Complex Structures by Peptide Growing" International Journal of Molecular Sciences 24, no. 18: 13831. https://doi.org/10.3390/ijms241813831