
Conjunctive Analyses of BSA-Seq and BSR-Seq to Identify Candidate Genes Controlling the Black Lemma and Pericarp Trait in Barley
 , , and
, , and
Abstract
:1. Introduction
2. Results
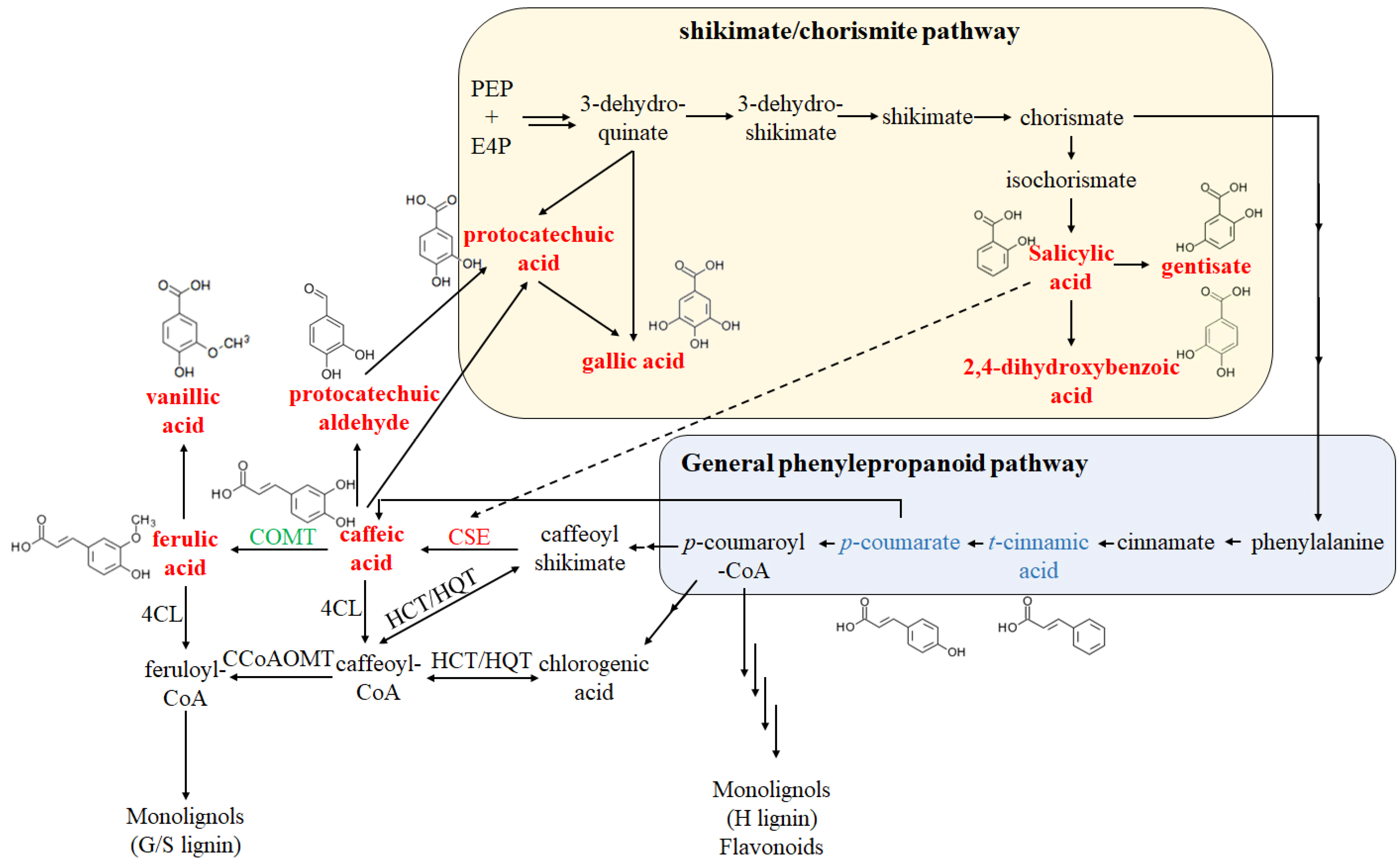
2.1. Phenolic Compounds Content Measurements
2.2. Bulked Segregant Analysis Sequencing
2.2.1. Sequencing Data Assessment and Mapping Analysis
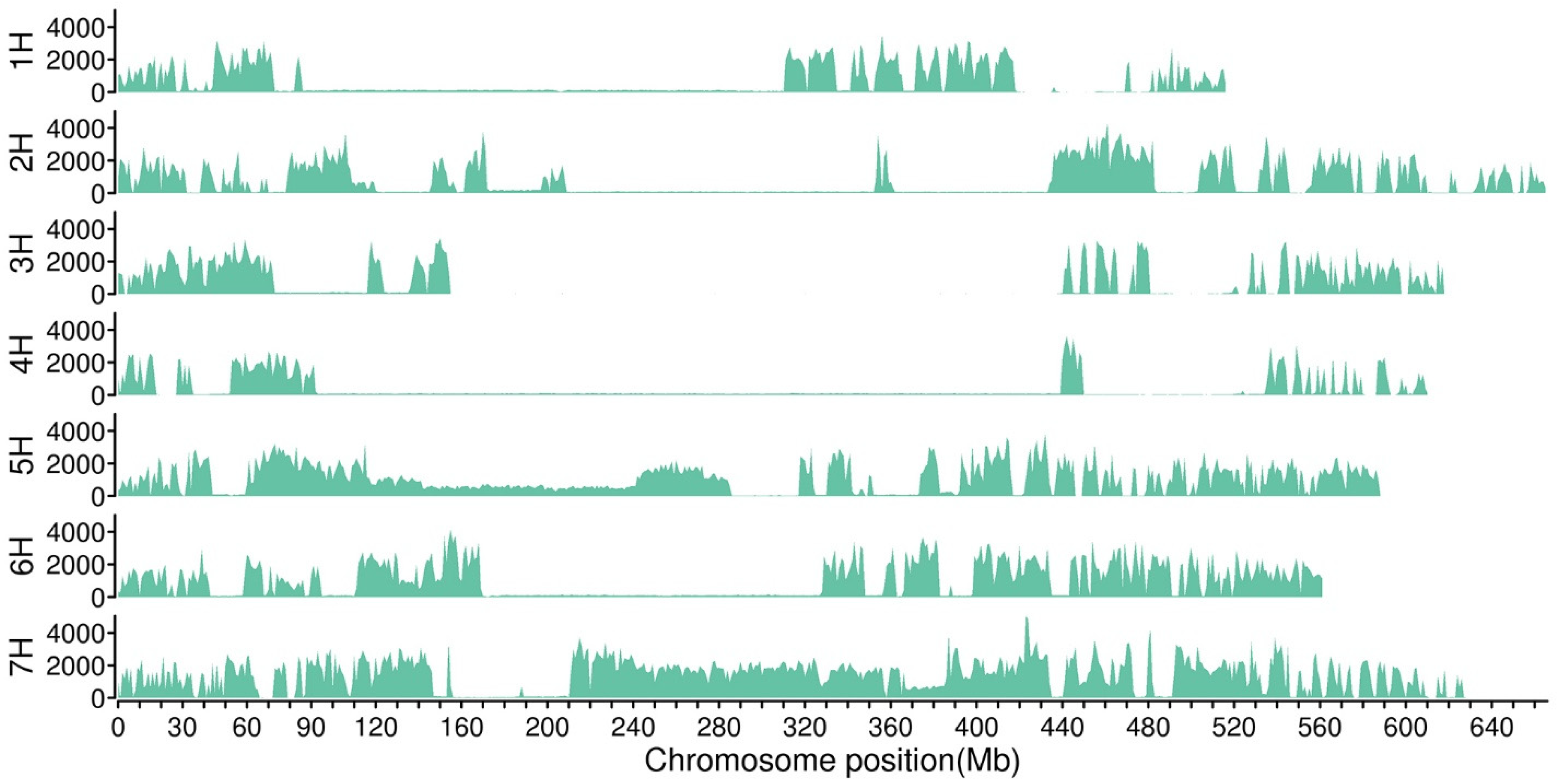
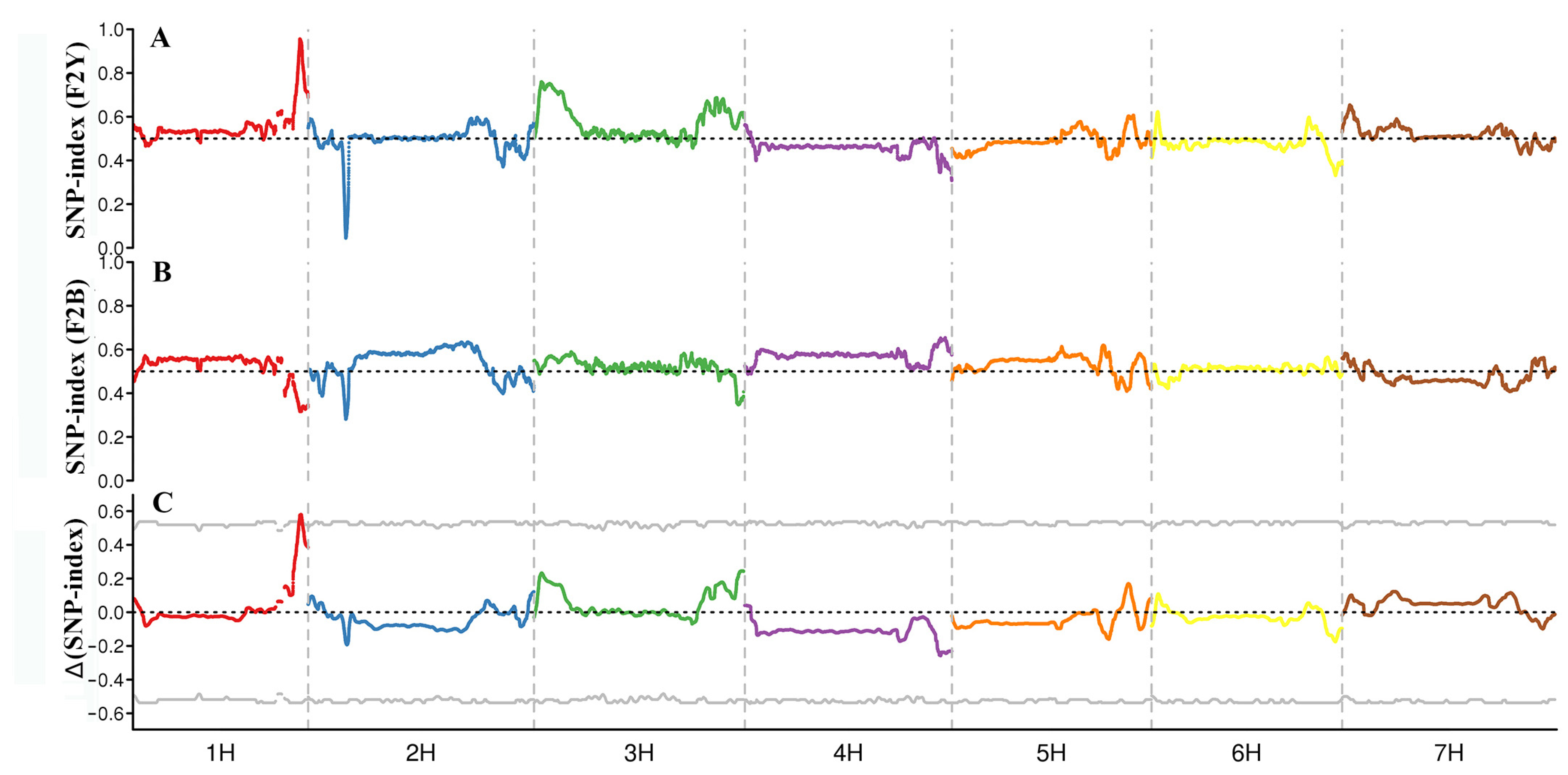
2.2.2. Location of Candidate Regions of BSA-Seq Data
2.3. Bulked Segregant RNA-Seq Analysis
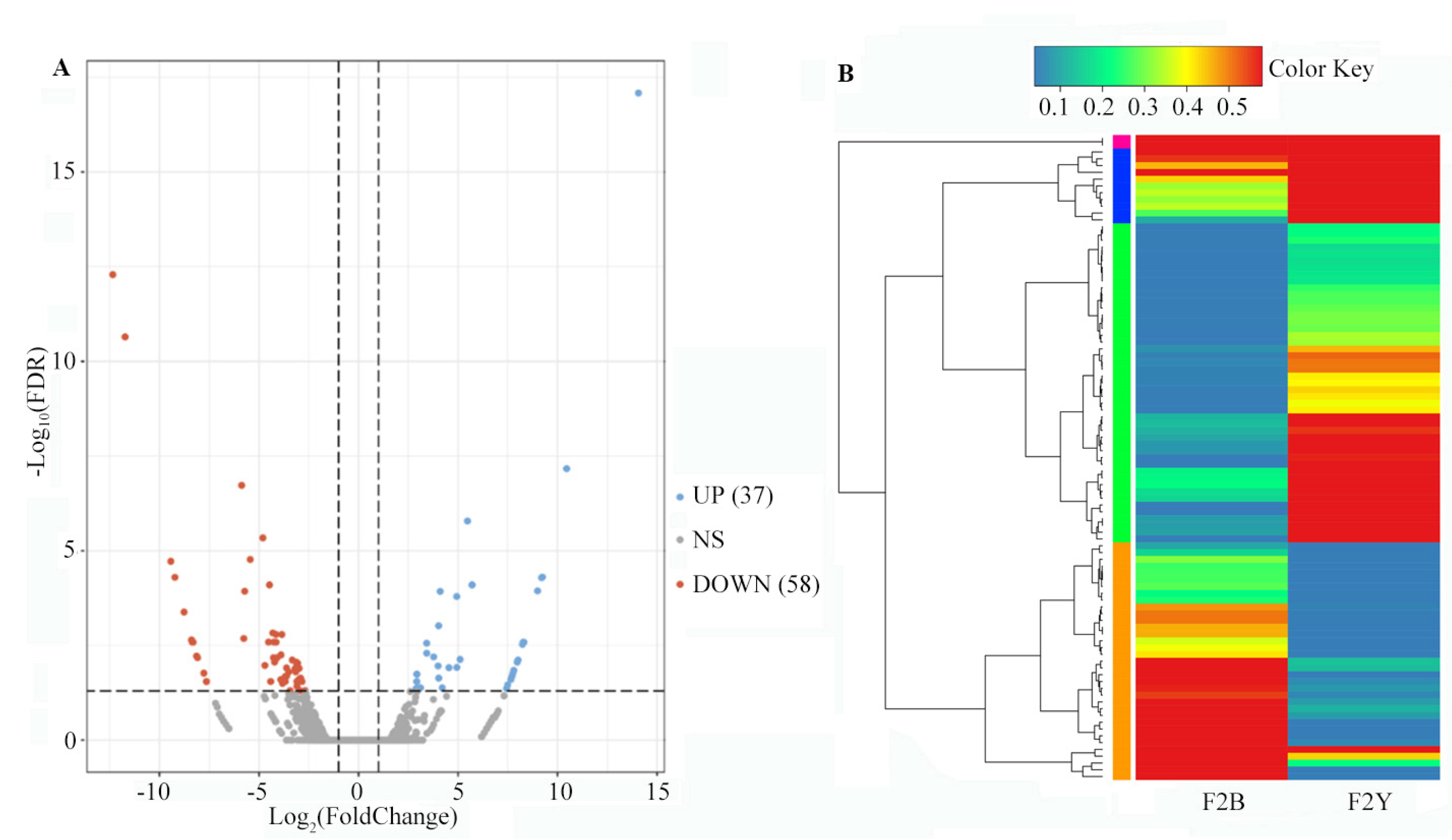
2.3.1. Sequencing Data Analysis of two cDNA Bulks
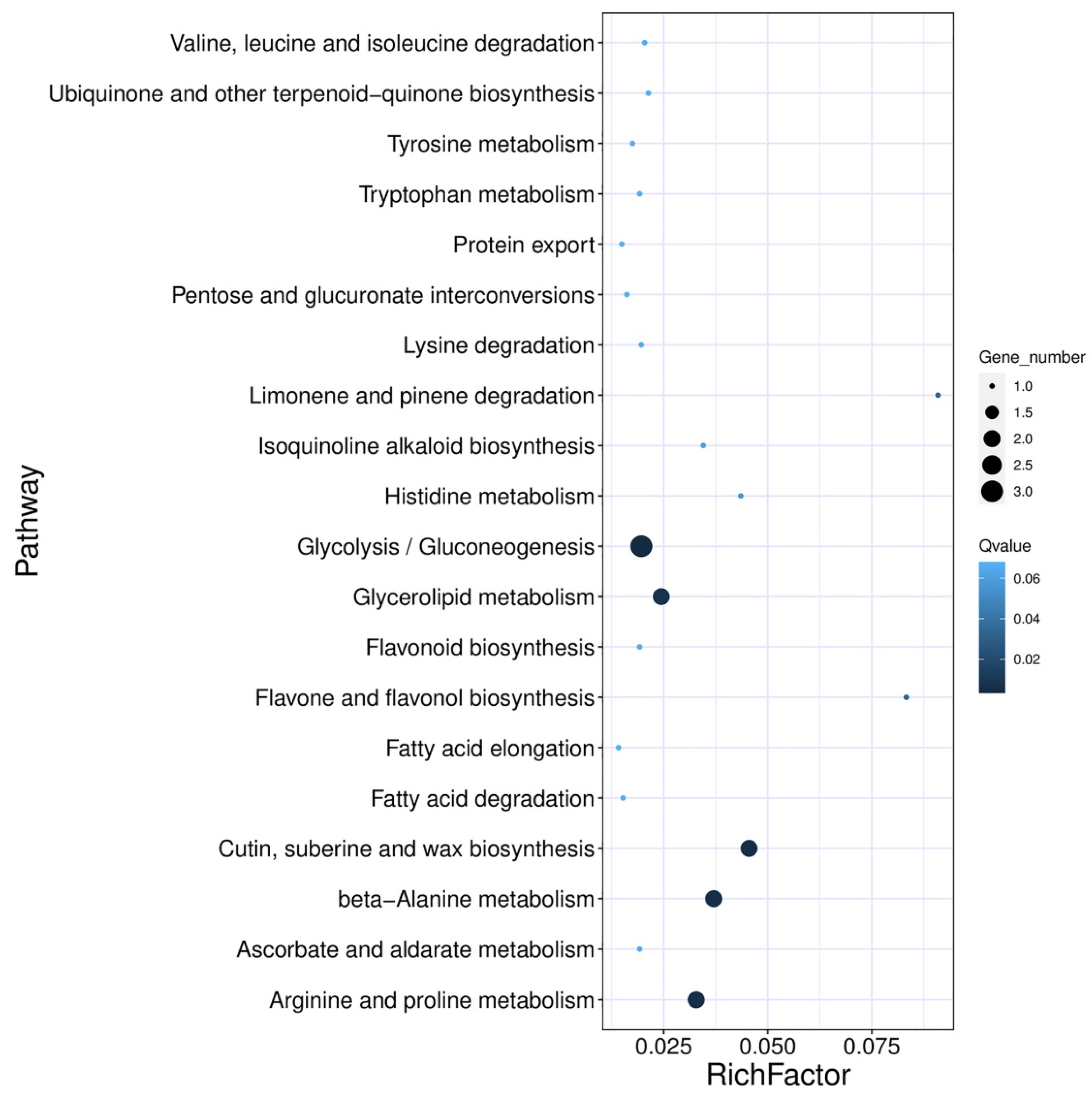
2.3.2. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes Pathway Enrichment Analysis of Differentially Expressed Genes
2.4. Combined Analysis of Bulk Segregant Analysis Sequencing and Bulk Segregant RNA Sequencing
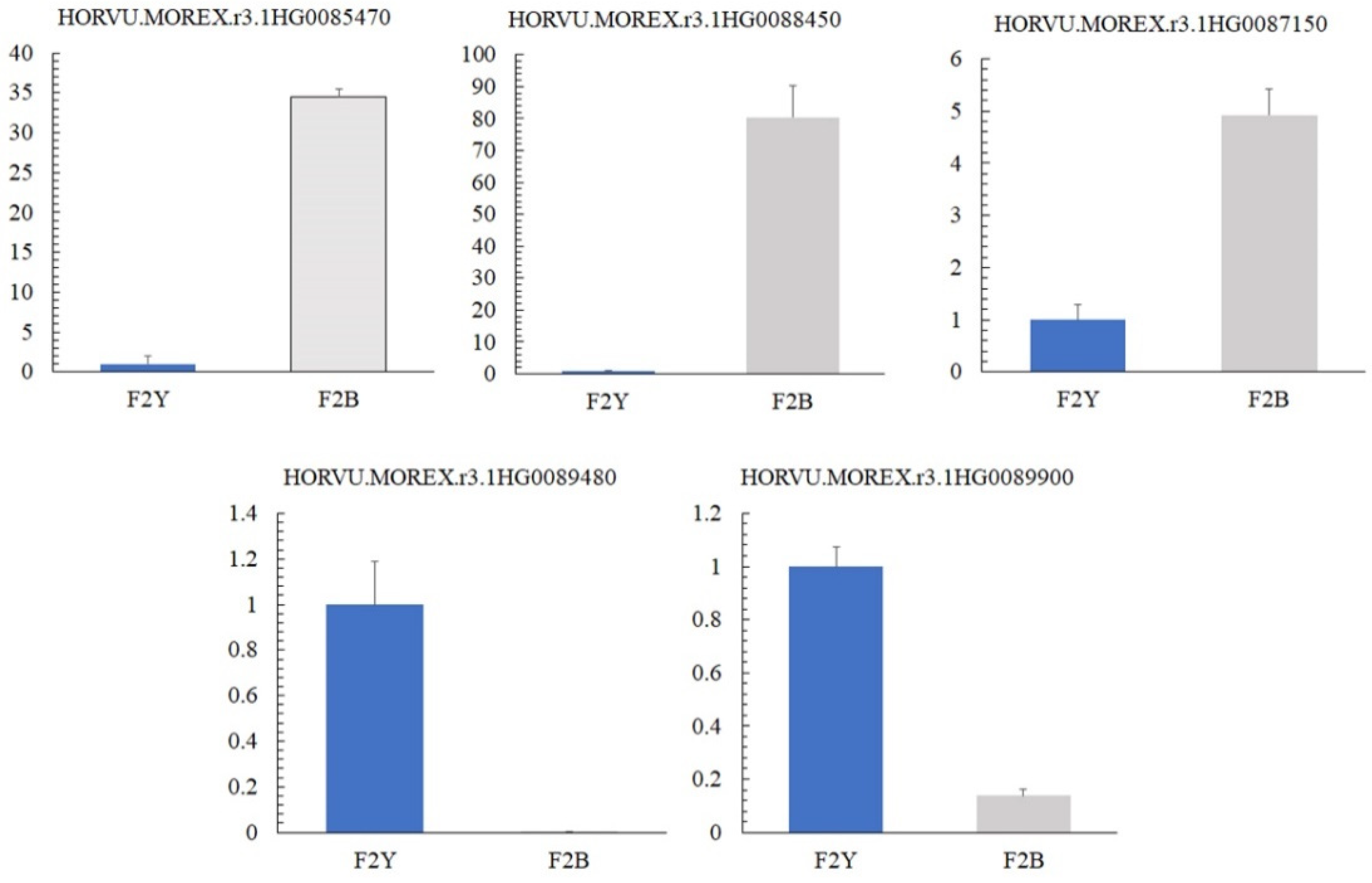
2.5. Validation of Quantitative Reverse Transcription PCR for Candidate Genes Related to Pigmentation
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Extraction and Chromatographic Analysis of Phenolic Compounds
4.3. Bulked Segregant Analysis Sequencing
4.4. Bulked Segregant RNA Sequencing
4.5. Analysis of Differential Expression Genes and its Association Analysis with BSA
4.6. Real-Time Quantitative PCR Validation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Šterna1, V.; Zute1, S.; Jansone, I.; Kantane, I. Chemical composition of covered and naked spring barley varieties and their potential for food production. Pol. J. Food Nutr. Sci. 2017, 67, 151–158. [Google Scholar] [CrossRef]
- Obadi, M.; Sun, J.; Xu, B. Highland barley: Chemical composition, bioactive compounds, health effects, and applications. Food Res. Int. 2021, 140, 110065. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, G.; Sowrirajan, S.; Joseph, B. β-Glucan, a dietary fiber in effective prevention of lifestyle diseases-an insight. Bioact. Carbohydr. Diet. Fibre 2019, 19, 2212–6198. [Google Scholar] [CrossRef]
- Tosh, S.M.; Bordenave, N. Emerging science on benefits of whole grain oat and barley and their soluble dietary fibers for heart health, glycemic response, and gut microbiota. Nutr. Rev. 2020, 78, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Suriano, S.; Iannucci, A.; Codianni, P.; Fares, C.; Menga, V.; Russo MMarciello, U.; Troccoli, A. Carotenoids and tocols content in genotypes of colored barley. J. Cereal Sci. 2020, 96, 103110. [Google Scholar] [CrossRef]
- Abdel-Aal, E.M.; Choo, T.M.; Dhillon, S.; Rabalski, I. Free and bound phenolic acids and total phenolics in black, blue, and yellow barley and their contribution to free radical scavenging capacity. Cereal Chem. 2012, 89, 198–204. [Google Scholar] [CrossRef]
- Abdel-Aal, E.M.; Young, J.C.; Rabalski, I. Anthocyanin composition in black, blue, pink, purple, and red cereal grains. J. Agric. Food Chem. 2006, 54, 4696–4704. [Google Scholar] [CrossRef]
- Harlan, H. Some Distinctions in Our Cultivated Barleys with Reference to Their Use in Plant Breeding; U.S. Department of Agriculture: Washington, DC, USA, 1914.
- Lundqvist, U.; Franckowiak, J.; Konishi, T. New and revised descriptions of barley genes. Barley Genet. Newsl. 1997, 26, 22–516. [Google Scholar]
- Choo, T.M.; Vigier, B.; Ho, K.M.; Ceccarelli, S.; Grando, S.; Franckowiak, J.D. Comparison of black, purple, and yellow barleys. Genet. Resour. Crop Evol. 2005, 52, 121–126. [Google Scholar] [CrossRef]
- Solano, F. Melanins: Skin pigments and much more-types, structural models, biological functions, and formation routes. New J. Sci. 2014, 2014, 498276. [Google Scholar] [CrossRef]
- Glagoleva, A.Y.; Shoeva, O.Y.; Khlestkina, E.K. Melanin Pigment in Plants: Current Knowledge and Future Perspectives. Front. Plant Sci. 2020, 11, 770. [Google Scholar] [CrossRef]
- Bell, A.A.; Wheeler, M.H. Biosynthesis and functions of fungal melanins. Annu. Rev. Phytopathol. 1986, 24, 411–451. [Google Scholar] [CrossRef]
- Elobeid, A.S.; Kamal-Eldin, A.; Abdelhalim MA, K.; Haseeb, A.M. Pharmacological properties of melanin and its function in health. Basic Clin. Pharmacol. Toxicol. 2017, 120, 515–522. [Google Scholar] [CrossRef]
- Vahidzadeh, E.; Kalra, A.P.; Shankar, K. Melanin-based electronics: From proton conductors to photovoltaics and beyond. Biosens. Bioelectron. 2018, 122, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Gracheva, N.V.; Zheltobryukhov, V.F. Sorption properties of sunflower husk melanins. Pharm. Chem. J. 2019, 53, 337–341. [Google Scholar] [CrossRef]
- Kablov, V.F.; Novopoltseva, O.M.; Gracheva, N.V.; Zheltobryukho, V.F.; Dao, P.K. Prospects of application of melanins as antiaging agents in elastomer compositions. Vietnam J. Chem. 2019, 57, 255–260. [Google Scholar] [CrossRef]
- Mullick, R.M.; Faris, D.G.; Brink, V.C.; Acheson, R.M. Anthocyanins and anthocyanidins of the barley pericarp and aleurone tissues. Can. J. Plant Sci. 1958, 38, 445–456. [Google Scholar] [CrossRef]
- Bungartz, A.; Klaus, M.; Mathew, B.; Léon, J.; Naz, A.A. Development of new SNP derived cleaved amplified polymorphic sequence marker set and its successful utilization in the genetic analysis of seed color variation in barley. Genomics 2016, 107, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.M.; Corey, A.; Hayes, P.M.; Jobet, C.; Kleinhofs, A.; Kopisch-Obusch, A.; Kramer, S.F.; Kudrna, D.; Li, M.; Riera-Lizarazu, O.; et al. Molecular mapping of the OregonWolfe Barleys: A phenotypically polymorphic doubled-haploid population. Theor. Appl. Genet. 2001, 103, 415–424. [Google Scholar] [CrossRef]
- Shoeva, O.Y.; Mock, H.P.; Kukoeva, T.V.; Borner, A.; Khlestkina, E.K. Regulation of the flavonoid biosynthesis pathway genes in purple and black grains of Hordeum vulgare. PLoS ONE 2016, 11, e0163782. [Google Scholar] [CrossRef] [PubMed]
- Shoeva, O.Y.; Mursalimov, S.R.; Gracheva, N.V.; Glagoleva, A.Y.; Börner, A.; Khlestkina, E.K. Melanin formation in barley grain occurs within plastids of pericarp and husk cells. Sci. Rep. 2020, 10, 179. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Wang, J.; Zhu, J.; Hua, W.; Shang, Y.; Yang, J.; Liang, Z. Toward identification of black lemma and pericarp gene Blp1 in barley combining bulked segregant analysis and specific-locus amplified fragment sequencing. Front. Plant Sci. 2017, 8, 1414. [Google Scholar] [CrossRef]
- Long, Z.; Jia, Y.; Tan, C.; Zhang, X.-Q.; Angessa, T.; Broughton, S.; Westcott, S.; Dai, F.; Zhang, G.; Sun, D.; et al. Genetic mapping and evolutionary analyses of the black grain trait in barley. Front. Plant Sci. 2019, 9, 1921. [Google Scholar] [CrossRef]
- Glagoleva, A.Y.; Shmakov, N.A.; Shoeva, O.Y.; Vasiliev, G.V.; Shatskaya, N.V.; Börner, A.; Afonnikov, D.A.; Khlestkina, E.K. Metabolic pathways and genes identified by RNA-seq analysis of barley near-isogenic lines differing by allelic state of the Black lemma and pericarp (Blp) gene. BMC Plant Biol. 2017, 17, 182. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, Y. Bulk segregation analysis in the NGS era: A review of its teenage years. Plant J. 2022, 109, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Majeed, A.; Johar, P.; Raina, A.; Salgotra, R.K.; Feng, X.; Bhat, J.A. Harnessing the potential of bulk segregant analysis sequencing and its related approaches in crop breeding. Front. Genet. 2022, 13, 944501. [Google Scholar] [CrossRef]
- Edae, E.A.; Rouse, M.N. Bulked segregant analysis RNA-seq (BSRSeq) validated a stem resistance locus in Aegilops umbellulata, a wild relative of wheat. PLoS ONE 2019, 14, e0215492. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Chen, J.D.; Wang, S.L.; Chen, L.; Ma, C.L.; Yao, M.Z. Repressed gene expression of photosynthetic antenna proteins associated with yellow leaf variation as revealed by bulked segregant RNA-seq in tea plant Camellia sinensis. J. Agric. Food Chem. 2020, 68, 8068–8079. [Google Scholar] [CrossRef]
- Li, R.; Hou, Z.; Gao, L.; Xiao, D.; Hou, X.; Zhang, C.; Yan, J.; Song, L. Conjunctive Analyses of BSA-Seq and BSR-Seq to Reveal the Molecular Pathway of Leafy Head Formation in Chinese Cabbage. Plants 2019, 8, 603. [Google Scholar] [CrossRef]
- Gao, Y.; Du, L.; Ma, Q.; Yuan, Y.; Liu, J.; Song, H.; Feng, B. Conjunctive Analyses of Bulk Segregant Analysis Sequencing and Bulk Segregant RNA Sequencing to Identify Candidate Genes Controlling Spikelet Sterility of Foxtail Millet. Front. Plant Sci. 2022, 13, 842336. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Y.; Xu, X.; Yan, D.; Yu, W.; Miao, Y.; Xu, B. Conjunctive Analyses of BSA-Seq and BSR-Seq Unveil the Msβ-GAL and MsJMT as Key Candidate Genes for Cytoplasmic Male Sterility in Alfalfa (Medicago sativa L.). Int. J. Mol. Sci. 2022, 23, 7172. [Google Scholar] [CrossRef]
- Glagoleva, A.Y.; Vikhorev, A.V.; Shmakov, N.A.; Morozov, S.V.; Chernyak, E.I.; Vasiliev, G.V.; Shatskaya, N.V.; Khlestkina, E.K.; Shoeva, O.Y. Features of Activity of the Phenylpropanoid Biosynthesis Pathway in Melanin-Accumulating Barley Grains. Front. Plant Sci. 2022, 13, 923717. [Google Scholar] [CrossRef]
- Isla, I.M.; Moreno, A.M.; Álvarez, A.M.; Zampini, C.I. Chapter 9—Some plants of the Monte region from Argentina: Phytochemistry and its use in health care. In Studies in Natural Products Chemistry; Rahman, A.U., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 349–369. [Google Scholar]
- Widhalm, J.R.; Dudareva, N. A familiar ring to it: Biosynthesis of plant benzoic acids. Mol. Plant 2015, 8, 83–97. [Google Scholar] [CrossRef]
- Wu, J.; Zhu, W.; Zhao, Q. Salicylic acid biosynthesis is not from phenylalanine in Arabidopsis. J. Integr. Plant Biol. 2022, 65, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, K.; Tsao, C.C.; Li, L.G.; Popko, J.L.; Umezawa, T.; Carraway, D.T.; Smeltzer, R.H.; Joshi, C.P.; Chiang, V.L. Coniferyl aldehyde 5-hydroxylation and methylation direct syringyl lignin biosynthesis in angiosperms. Proc. Natl. Acad. Sci. USA 1999, 96, 8955–8960. [Google Scholar] [CrossRef]
- Zhou, J.M.; Fukushi, Y.; Wollenweber, E.; Ibrahim, R.K. Characterization of Two O.-Methyltransferase-like Genes in Barley and Maize. Pharm. Biol. 2008, 46, 26–34. [Google Scholar] [CrossRef]
- Vanholme, R.; Cesarino, I.; Rataj, K.; Xiao, Y.; Sundin, L.; Goeminne, G.; Kim, H.; Cross, J.; Morreel, K.; Araujo, P.; et al. Caffeoyl shikimate esterase (CSE) is an enzyme in the lignin biosynthetic pathway in Arabidopsis. Science 2013, 341, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Welti, R.; Li, W.; Li, M.; Sang, Y.; Biesiada, H.; Zhou, H.E.; Rajashekar, C.B.; Williams, T.D.; Wang, X. Profiling membrane lipids in plant stress responses: Role of phospholipase D in freezing-induced lipid changes in Arabidopsis. J. Biol. Chem. 2002, 277, 31994–32002. [Google Scholar] [CrossRef] [PubMed]
- Ryu, B.S. Phospholipid-derived signaling mediated by phospholipase A in plants. Trends Plant Sci. 2004, 9, 229–235. [Google Scholar] [CrossRef]
- Winter, D.; Vinegar, B.; Nahal, H.; Ammar, R.; Wilson, G.V.; Provart, N.J. An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2007, 2, e718. [Google Scholar] [CrossRef]
- Prota, G. Melanins and Melanogenesis; Academic Press: London, UK, 1992. [Google Scholar]
- Birková, A.; Hubková, B.; Bolerázska, B.; Mareková, M.; Čižmárová, B. Caffeic acid: A brief overview of its presence, metabolism, and bioactivity. Bioact. Compd. Health Dis. 2020, 3, 74–81. [Google Scholar] [CrossRef]
- Ceccarelli, S.; Grando, S.; Van Leur, J.A.G. Genetic diversity in barley landraces from Syria and Jordan. Euphytica 1987, 36, 389–405. [Google Scholar] [CrossRef]
- Weltzien, E. Evaluation of barley (Hordeum vulgare L.) landrace populations originating from different growing regions in the near east. Plant Breed. 1988, 101, 95–106. [Google Scholar] [CrossRef]
- Zhou, X.; Chao, M.; Liang, X. Screening and Testing of Barley Varieties for Scab Resistance. Acta Phytophylacica Sin. 1991, 18, 261–265. [Google Scholar]
- Ogawa, D.; Nakajima, N.; Seo, S.; Mitsuhara, I.; Kamada, H.; Ohashi, Y.J.P.B. The phenylalanine pathway is the main route of salicylic acid Biosynthesis in Tobacco mosaic virus-infected tobacco leaves. Plant Biotechnol. 2006, 23, 395–398. [Google Scholar] [CrossRef]
- Kim, D.S.; Hwang, B.K. An important role of the pepper phenylalanine ammonia-lyase gene (PAL1) in salicylic acid-dependent signalling of the defence response to microbial pathogens. J. Exp. Bot. 2014, 65, 2295–2306. [Google Scholar] [CrossRef]
- Hao, Q.; Wang, W.; Han, X.; Wu, J.; Lyu, B.; Chen, F.; Caplan, A.; Li, C.; Wu, J.; Wang, W.; et al. Isochorismate-based salicylic acid biosynthesis confers basal resistance to Fusarium graminearum in barley. Mol. Plant Pathol. 2018, 19, 1995–2010. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X. Salicylic acid: Biosynthesis, perception, and contributions to plant immunity. Curr. Opin. Plant Biol. 2019, 50, 29–36. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L.; Doyle, J.A.; Doyle, F.J. A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | ZB | EH | F2Y (ng/g) | F2B (ng/g) | Category |
|---|---|---|---|---|---|
| (ng/g) | (ng/g) | ||||
| Caffeic acid | 205.67 | 545.79 | 362.01 | 1956.38 | Phenylpropanoids |
| Ferulic acid | 1056.49 | 2046.86 | 2877.01 | 4901.19 | Phenylpropanoids |
| 2,4-dihydroxybenzoic acid | 4.75 | 23.20 | 42.79 | 84.32 | Benzoic acid derivatives |
| Gallic acid | 3.08 | 5.12 | 2.30 | 11.06 | Benzoic acid derivatives |
| Gentisic acid | 5.24 | 21.02 | 45.00 | 82.99 | Benzoic acid derivatives |
| Protocatechuic acid | 39.37 | 264.94 | 75.93 | 422.88 | Benzoic acid derivatives |
| Salicylic acid | 11.54 | 121.50 | 27.16 | 79.77 | Benzoic acid derivatives |
| Syringic acid | 147.69 | 285.26 | 299.53 | 517.16 | Benzoic acid derivatives |
| Vanillic acid | 1101.53 | 1538.71 | 1750.13 | 3492.70 | Benzoic acid derivatives |
| Cryptochlorogenic acid | 26.89 | 147.85 | 64.98 | 1161.05 | Alcohols and polyols |
| Protocatechuic aldehyde | 13.78 | 173.09 | 12.41 | 207.28 | Catechols |
| Syringaldehyde | 14.15 | 49.05 | 17.05 | 65.25 | Benzaldehydes |
| Narcissin | 69.76 | 196.26 | 227.58 | 333.83 | Flavonols |
| 3,4-dihydrocoumarin | 0.00 | 1.11 | 0.00 | 1.04 | Coumarins |
| Aesculetin | 1.93 | 12.06 | 9.41 | 84.72 | Coumarins |
| Aesculin | 4.77 | 10.57 | 30.62 | 58.08 | Coumarins |
| Daphnetin | 1.43 | 14.93 | 8.66 | 91.28 | Coumarins |
| Sample | Total Reads | Mapping Rate (%) | Q30 (%) | Coverage (%) | Average Depth (×) |
|---|---|---|---|---|---|
| ZN | 169,059,857 | 56.86275128 | 94.85 | 87 | 6.54 |
| EH | 188,406,475 | 48.39110174 | 93.94 | 87.94 | 6.3 |
| F2Y | 705,192,609 | 88.6376719 | 95.56 | 95.14 | 40.53 |
| F2B | 476,694,215 | 78.9104508 | 93.09 | 95.1 | 24.89 |
| Method | Chromosome | Start (Mb) | End (Mb) | Length (Mb) | Gene Numbers |
|---|---|---|---|---|---|
| BSA SNP-index | Chromosome 1H | 494.45 | 504.58 | 10.12 | 283 |
| BSR SNP-index | Chromosome 1H | 490.29 | 506.55 | 16.26 | 455 |
| Sample | Total Reads | Mapping Rate (%) | Q30 (%) | Coverage (%) | Average Depth (×) |
|---|---|---|---|---|---|
| F2Y | 35,950,689 | 88.19726932 | 92.56 | 2.59 | 83.99 |
| F2B | 44,476,127 | 87.70702314 | 93.71 | 2.72 | 98.27 |
| Gene_ID | Start | End | Functional Annotations | Num. of SNP |
|---|---|---|---|---|
| HORVU.MOREX.r3.1HG0085470 | 495,853,418 | 495,855,011 | 3-ketoacyl-CoA synthase 11 | 1 |
| HORVU.MOREX.r3.1HG0087150 | 499,006,879 | 499,010,239 | Purple acid phosphatase | 5 |
| HORVU.MOREX.r3.1HG0088450 | 500,843,053 | 500,843,565 | Coiled-coil domain-containing protein 167 | 1 |
| HORVU.MOREX.r3.1HG0089480 | 503,466,785 | 503,471,314 | Subtilisin-like protease | 2 |
| HORVU.MOREX.r3.1HG0089900 | 504,155,100 | 504,157,234 | Caffeic acid-O-methyltransferase | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Chen, P.; Li, W.; Liu, X.; Yu, G.; Zhao, H.; Zeng, S.; Li, M.; Sun, G.; Feng, Z. Conjunctive Analyses of BSA-Seq and BSR-Seq to Identify Candidate Genes Controlling the Black Lemma and Pericarp Trait in Barley. Int. J. Mol. Sci. 2023, 24, 9473. https://doi.org/10.3390/ijms24119473
Liu Y, Chen P, Li W, Liu X, Yu G, Zhao H, Zeng S, Li M, Sun G, Feng Z. Conjunctive Analyses of BSA-Seq and BSR-Seq to Identify Candidate Genes Controlling the Black Lemma and Pericarp Trait in Barley. International Journal of Molecular Sciences. 2023; 24(11):9473. https://doi.org/10.3390/ijms24119473
Chicago/Turabian StyleLiu, Yajie, Pengzheng Chen, Wenshuo Li, Xinchun Liu, Guowu Yu, Hui Zhao, Shuhua Zeng, Mao Li, Genlou Sun, and Zongyun Feng. 2023. "Conjunctive Analyses of BSA-Seq and BSR-Seq to Identify Candidate Genes Controlling the Black Lemma and Pericarp Trait in Barley" International Journal of Molecular Sciences 24, no. 11: 9473. https://doi.org/10.3390/ijms24119473