
The Landscapes of Gluten Regulatory Network in Elite Wheat Cultivars Contrasting in Gluten Strength
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Nitrogen Treatment Increased SDS Sedimentation and Crude Protein Content
2.2. Construction of High-Quality Isoforms
2.3. Functional Annotations for Identified Isoforms
2.4. Isoforms Differentially Expressed under Nitrogen Treatment
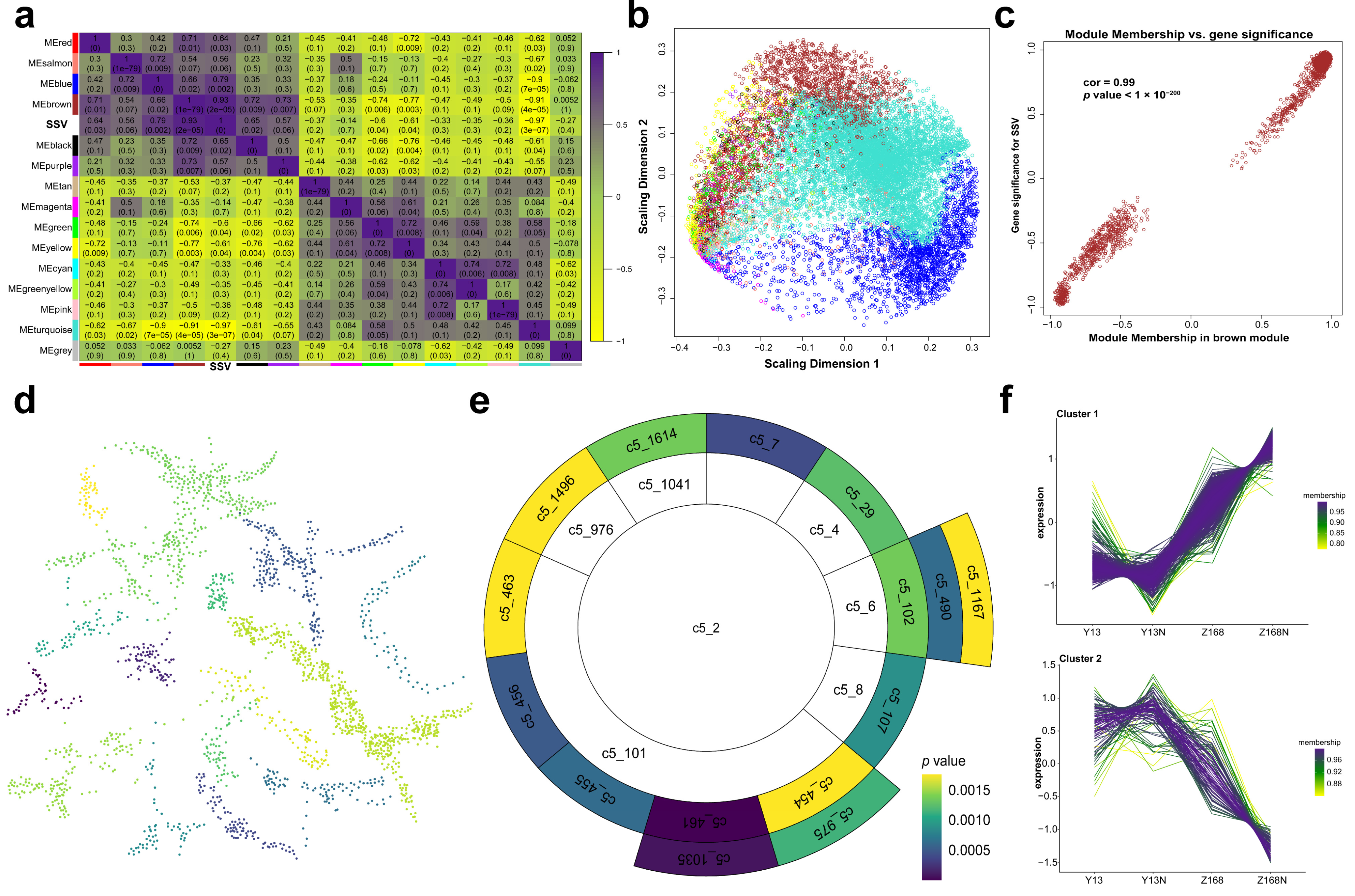
2.5. Co-Expression Network for SSV
2.6. Alternative Splicing Events in Significant Modules
2.7. Identification of TFs, lncRNAs, and APA
3. Discussion
3.1. High-Quality Data Obtained from PacBio Transcriptome Sequencing
3.2. The Key Modules Show Strong Associations with SSV and Are Well-Constructed and Annotated
3.3. Hub Genes Involved in Regulatory of Strong Gluten Aggregates
4. Materials and Methods
4.1. Plant Materials and Nitrogen Treatment
4.2. Qualitative Traits Measurement
4.3. PacBio Full-Length Data Processing and Illumina RNA Sequencing
4.4. Structure Analysis
4.5. Functional Annotation and Enrichment Analysis
4.6. WGCNA and MEGENA
4.7. RT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- OECD/FAO. OECD-FAO Agricultural Outlook 2021–2030; OECD: Montpelhièr, France, 2021. [Google Scholar]
- Brandner, S.; Becker, T.; Jekle, M. Classification of starch-gluten networks into a viscoelastic liquid or solid, based on rheological aspects—A review. Int. J. Biol. Macromol. 2019, 136, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.J.; MacMillan, J.; Shewry, P.R.; Tatham, A. Elastomeric proteins: Structures, biomechanical properties and biological roles: Preface. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2002, 357, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Payne, P.I. Genetics of Wheat Storage Proteins and the Effect of Allelic Variation on Bread-Making Quality. Annu. Rev. Plant Physiol. 1987, 38, 141–153. [Google Scholar] [CrossRef]
- Shewry, P.R. Wheat. J. Exp. Bot. 2009, 60, 1537–1553. [Google Scholar] [CrossRef]
- Payne, P.I.; Nightingale, M.A.; Krattiger, A.F.; Holt, L.M. The relationship between HMW glutenin subunit composition and the bread-making quality of British-grown wheat varieties. J. Sci. Food Agric. 1987, 40, 51–65. [Google Scholar] [CrossRef]
- Payne, P.I.; Holt, L.M.; Jackson, E.A.; Law, C.N.; Damania, A.B.; Fowden, L.; Miflin, B.J. Wheat storage proteins: Their genetics and their potential for manipulation by plant breeding. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1984, 304, 359–371. [Google Scholar]
- Chen, Q.; Zhang, W.; Gao, Y.; Yang, C.; Gao, X.; Peng, H.; Hu, Z.; Xin, M.; Ni, Z.; Zhang, P.; et al. High Molecular Weight Glutenin Subunits 1Bx7 and 1By9 Encoded by Glu-B1 Locus Affect Wheat Dough Properties and Sponge Cake Quality. J. Agric. Food Chem. 2019, 67, 11796–11804. [Google Scholar] [CrossRef]
- Mao, X.; Li, Y.; Zhao, S.; Zhang, J.; Lei, Q.; Meng, D.; Ma, F.; Hu, W.; Chen, M.; Chang, J.; et al. The interactive effects of transgenically overexpressed 1Ax1 with various HMW-GS combinations on dough quality by introgression of exogenous subunits into an elite Chinese Wheat variety. PLoS ONE 2013, 8, e78451. [Google Scholar] [CrossRef]
- Altpeter, F.; Vasil, V.; Srivastava, V.; Vasil, I.K. Integration and expression of the high-molecular-weight glutenin subunit 1Ax1 gene into wheat. Nat. Biotechnol. 1996, 14, 1155–1159. [Google Scholar] [CrossRef]
- Li, Y.; Fu, J.; Shen, Q.; Yang, D. High-Molecular-Weight Glutenin Subunits: Genetics, Structures, and Relation to End Use Qualities. Int. J. Mol. Sci. 2020, 22, 184. [Google Scholar] [CrossRef]
- Li, S.; Liu, Y.; Tong, J.; Yu, L.; Ding, M.; Zhang, Z.; Rehman, A.U.; Majzoobi, M.; Wang, Z.; Gao, X. The overexpression of high-molecular-weight glutenin subunit Bx7 improves the dough rheological properties by altering secondary and micro-structures of wheat gluten. Food Res. Int. 2020, 130, 108914. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, L.; Song, L.; Liu, Z.; Li, X.; Li, X. HMW-GS at Glu-B1 Locus Affects Gluten Quality Possibly Regulated by the Expression of Nitrogen Metabolism Enzymes and Glutenin-Related Genes in Wheat. J. Agric. Food Chem. 2020, 68, 5426–5436. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhou, Q.; Wang, Q.; Wang, X.; Cai, J.; Huang, M.; Jiang, D. Effects of Nitrogen Fertilizer on Quality Characteristics of Wheat with the Absence of Different Individual High-Molecular-Weight Glutenin Subunits (HMW-GSs). Int. J. Mol. Sci. 2022, 23, 2178. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Beom, H.-R.; Altenbach, S.B.; Lim, S.-H.; Kim, Y.-T.; Kang, C.-S.; Yoon, U.-H.; Gupta, R.; Kim, S.-T.; Ahn, S.-N.; et al. Comprehensive identification of LMW-GS genes and their protein products in a common wheat variety. Funct. Integr. Genomics 2016, 16, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, D.; Jiang, W.; Guo, X.; Yang, W.; Sun, J.; Ling, H.; Zhang, A. PCR-based isolation and identification of full-length low-molecular-weight glutenin subunit genes in bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2011, 123, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Franaszek, S.; Salmanowicz, B. Composition of low-molecular-weight glutenin subunits in common wheat (Triticum aestivum L.) and their effects on the rheological properties of dough. Open Life Sci. 2021, 16, 641–652. [Google Scholar] [CrossRef]
- Huang, J.; Dai, S.; Song, Z.; Liu, L.; Liang, L.; Li, J.; Yan, Z. Characterization of novel LMW-i genes with nine cysteine residues from Chinese wheat landraces (Triticum aestivum L.) and analysis of their functional properties on dough mixing. 3 Biotech 2021, 11, 499. [Google Scholar] [CrossRef]
- Du, X.; Wei, J.; Luo, X.; Liu, Z.; Qian, Y.; Zhu, B.; Weng, Q.; Tang, H. Low-molecular-weight glutenin subunit LMW-N13 improves dough quality of transgenic wheat. Food Chem. 2020, 327, 127048. [Google Scholar] [CrossRef]
- D’Ovidio, R.; Masci, S. The low-molecular-weight glutenin subunits of wheat gluten. J. Cereal Sci. 2004, 39, 321–339. [Google Scholar] [CrossRef]
- Pogna, N.E.; Autran, J.C.; Mellini, F.; Lafiandra, D.; Feillet, P. Chromosome 1B-encoded gliadins and glutenin subunits in durum wheat: Genetics and relationship to gluten strength. J. Cereal Sci. 1990, 11, 15–34. [Google Scholar] [CrossRef]
- Huo, N.; Zhu, T.; Altenbach, S.; Dong, L.; Wang, Y.; Mohr, T.; Liu, Z.; Dvorak, J.; Luo, M.-C.; Gu, Y.Q. Dynamic Evolution of α-Gliadin Prolamin Gene Family in Homeologous Genomes of Hexaploid Wheat. Sci. Rep. 2018, 8, 5181. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, V.; Khatkar, B.S. Effects of Gliadin/Glutenin and HMW-GS/LMW-GS Ratio on Dough Rheological Properties and Bread-Making Potential of Wheat Varieties. J. Food Qual. 2015, 38, 71–82. [Google Scholar] [CrossRef]
- Li, X.; Liu, T.; Song, L.; Zhang, H.; Li, L.; Gao, X. Influence of high-molecular-weight glutenin subunit composition at Glu-A1 and Glu-D1 loci on secondary and micro structures of gluten in wheat (Triticum aestivum L.). Food Chem. 2016, 213, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Singh, N.; Ahlawat, A.K.; Kaur, S.; Singh, A.M.; Chauhan, H.; Singh, G.P. Diversity in grain, flour, dough and gluten properties amongst Indian wheat cultivars varying in high molecular weight subunits (HMW-GS). Food Res. Int. 2013, 53, 63–72. [Google Scholar] [CrossRef]
- Peña, R.J.; Trethowan, R.; Pfeiffer, W.H.; Ginkel, M.V. Quality (End-Use) Improvement in Wheat. J. Crop Produc. 2002, 5, 1–37. [Google Scholar] [CrossRef]
- Xue, C.; Auf’m Erley, G.S.; Rossmann, A.; Schuster, R.; Koehler, P.; Muhling, K.H. Split Nitrogen Application Improves Wheat Baking Quality by Influencing Protein Composition Rather Than Concentration. Front. Plant Sci. 2016, 7, 738. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, W.; Huang, X.; Liu, M.; Hebelstrup, K.H.; Yang, D.; Cai, J.; Wang, X.; Zhou, Q.; Cao, W.; et al. Nitrogen topdressing timing modifies the gluten quality and grain hardness related protein levels as revealed by iTRAQ. Food Chem. 2019, 277, 135–144. [Google Scholar] [CrossRef]
- Yu, Z.; Islam, S.; She, M.; Diepeveen, D.; Zhang, Y.; Tang, G.; Zhang, J.; Juhasz, A.; Yang, R.; Ma, W. Wheat grain protein accumulation and polymerization mechanisms driven by nitrogen fertilization. Plant J. 2018, 96, 1160–1177. [Google Scholar] [CrossRef]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef]
- Li, Z.; Bai, D.; Zhong, Y.; Lin, M.; Sun, L.; Qi, X.; Hu, C.; Fang, J. Full-Length Transcriptome and RNA-Seq Analyses Reveal the Mechanisms Underlying Waterlogging Tolerance in Kiwifruit (Actinidia valvata). Int. J. Mol. Sci. 2022, 23, 3237. [Google Scholar] [CrossRef]
- Zheng, X.; Shi, M.; Wang, J.; Yang, N.; Wang, K.; Xi, J.; Wu, C.; Xi, T.; Zheng, J.; Zhang, J. Isoform Sequencing Provides Insight Into Freezing Response of Common Wheat (Triticum aestivum L.). Front. Genet. 2020, 11, 462. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Chen, M.; Zhong, Z.; Tang, W.; Lin, L.; Zhang, X.; Jiang, H.; Zhang, D.; Miao, C.; Tang, H.; et al. PacBio Sequencing Reveals Transposable Elements as a Key Contributor to Genomic Plasticity and Virulence Variation in Magnaporthe oryzae. Mol. Plant 2017, 10, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, Z.; Feng, H.; Yang, J.; Li, C. Characterization of the Gene Expression Profile Response to Drought Stress in Populus ussuriensis Using PacBio SMRT and Illumina Sequencing. Int. J. Mol. Sci. 2022, 23, 3840. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinf. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Liu, Z.; Qin, J.; Tian, X.; Xu, S.; Wang, Y.; Li, H.; Wang, X.; Peng, H.; Yao, Y.; Hu, Z.; et al. Global profiling of alternative splicing landscape responsive to drought, heat and their combination in wheat (Triticum aestivum L.). Plant Biotechnol. J. 2018, 16, 714–726. [Google Scholar] [CrossRef]
- Chang, C.Y.; Lin, W.D.; Tu, S.L. Genome-Wide Analysis of Heat-Sensitive Alternative Splicing in Physcomitrella patens. Plant Physiol. 2014, 165, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Zhou, Y.; Liu, Z.; Zhang, L.; Song, G.; Guo, Z.; Wang, W.; Qu, X.; Zhu, Y.; Yang, D. An alternatively spliced heat shock transcription factor, OsHSFA2dI, functions in the heat stress-induced unfolded protein response in rice. Plant Biol. 2015, 17, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Cui, P.; Wang, Z.; Zhang, S.; Ali, S.; Xiong, L. Genome-wide analysis of alternative splicing of pre-mRNA under salt stress in Arabidopsis. BMC Genom. 2014, 15, 431. [Google Scholar] [CrossRef]
- Reddy, A.S.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef]
- Gupta, P.K. Single-molecule DNA sequencing technologies for future genomics research. Trends Biotechnol. 2008, 26, 602–611. [Google Scholar] [CrossRef]
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Wang, Z.; Rasko, D.A.; McCombie, W.R.; Jarvis, E.D.; et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012, 30, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D. Homology between DUF784, DUF1278 domains and the plant prolamin superfamily typifies evolutionary changes of disulfide bonding patterns. Cell Cycle 2009, 8, 3428–3430. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E. Hsp90: Structure and function. Top. Curr. Chem. 2013, 328, 155–240. [Google Scholar] [PubMed]
- Wang, R.Y.; Noddings, C.M.; Kirschke, E.; Myasnikov, A.G.; Johnson, J.L.; Agard, D.A. Structure of Hsp90-Hsp70-Hop-GR reveals the Hsp90 client-loading mechanism. Nature 2022, 601, 460–464. [Google Scholar] [CrossRef]
- Kumar, R.R.; Goswami, S.; Shamim, M.; Dubey, K.; Singh, K.; Singh, S.; Kala, Y.K.; Niraj, R.R.K.; Sakhrey, A.; Singh, G.P.; et al. Exploring the heat-responsive chaperones and microsatellite markers associated with terminal heat stress tolerance in developing wheat. Funct. Integr. Genom. 2017, 17, 621–640. [Google Scholar] [CrossRef]
- Tomas, D.; Viegas, W.; Silva, M. Grain Transcriptome Dynamics Induced by Heat in Commercial and Traditional Bread Wheat Genotypes. Front. Plant Sci. 2022, 13, 842599. [Google Scholar] [CrossRef]
- Jacob, P.; Hirt, H.; Bendahmane, A. The heat-shock protein/chaperone network and multiple stress resistance. Plant Biotechnol. J. 2017, 15, 405–414. [Google Scholar] [CrossRef]
- Tian, S.; Zhang, M.; Li, J.; Wen, S.; Bi, C.; Zhao, H.; Wei, C.; Chen, Z.; Yu, J.; Shi, X.; et al. Identification and Validation of Stable Quantitative Trait Loci for SDS-Sedimentation Volume in Common Wheat (Triticum aestivum L.). Front. Plant Sci. 2021, 12, 747775. [Google Scholar] [CrossRef]
- Yang, Y.; Chai, Y.; Zhang, X.; Lu, S.; Zhao, Z.; Wei, D.; Chen, L.; Hu, Y.G. Multi-Locus GWAS of Quality Traits in Bread Wheat: Mining More Candidate Genes and Possible Regulatory Network. Front. Plant Sci. 2020, 11, 1091. [Google Scholar] [CrossRef]
- Cubadda, R.; Carcea, M.; Pasqui, L.A. Suitability of the gluten index method for assessing gluten strength in durum wheat and semolina. Cereal Food World 1992, 37, 866–869. [Google Scholar]
- Hao, S.; Lou, H.; Wang, H.; Shi, J.; Liu, D.; Baogerile; Tao, J.; Miao, S.; Pei, Q.; Yu, L.; et al. Genome-Wide Association Study Reveals the Genetic Basis of Five Quality Traits in Chinese Wheat. Front. Plant Sci. 2022, 13, 835306. [Google Scholar] [CrossRef] [PubMed]
- Wegele, H.; Muller, L.; Buchner, J. Hsp70 and Hsp90--a relay team for protein folding. Rev. Physiol. Biochem. Pharmacol. 2004, 151, 1–44. [Google Scholar] [PubMed]
- Bracher, A.; Verghese, J. GrpE, Hsp110/Grp170, HspBP1/Sil1 and BAG Domain Proteins: Nucleotide Exchange Factors for Hsp70 Molecular Chaperones. In The Networking of Chaperones by Co-chaperones: Control of Cellular Protein Homeostasis; Blatch, G.L., Edkins, A.L., Eds.; Springer: Cham, Switzerland, 2015; pp. 1–33. [Google Scholar]
- Dong, G.; Ni, Z.; Yao, Y.; Nie, X.; Sun, Q. Wheat Dof transcription factor WPBF interacts with TaQM and activates transcription of an alpha-gliadin gene during wheat seed development. Plant Mol. Biol. 2007, 63, 73–84. [Google Scholar] [CrossRef]
- Albani, D.; Hammond-Kosack, M.C.; Smith, C.; Conlan, S.; Colot, V.; Holdsworth, M.; Bevan, M.W. The wheat transcriptional activator SPA: A seed-specific bZIP protein that recognizes the GCN4-like motif in the bifactorial endosperm box of prolamin genes. Plant Cell 1997, 9, 171–184. [Google Scholar]
- Gao, Y.; An, K.; Guo, W.; Chen, Y.; Zhang, R.; Zhang, X.; Chang, S.; Rossi, V.; Jin, F.; Cao, X.; et al. The endosperm-specific transcription factor TaNAC019 regulates glutenin and starch accumulation and its elite allele improves wheat grain quality. Plant Cell 2021, 33, 603–622. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, J.; Zhao, Y.; Liu, Q.; Islam, S.; Yang, W.; Ma, W. Wheat glutamine synthetase TaGSr-4B is a candidate gene for a QTL of thousand grain weight on chromosome 4B. Theor. Appl. Genet. 2022, 135, 2369–2384. [Google Scholar] [CrossRef]
- GB/T 15685-2011; Inspection of Grain and Oils—Determination of Sedimentation Index of Wheat—SDS Test. National Standard of China: Beijing, China, 2011.
- Levin, J.Z.; Yassour, M.; Adiconis, X.; Nusbaum, C.; Thompson, D.A.; Friedman, N.; Gnirke, A.; Regev, A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 2010, 7, 709–715. [Google Scholar] [CrossRef]
- Alaux, M.; Rogers, J.; Letellier, T.; Flores, R.; Alfama, F.; Pommier, C.; Mohellibi, N.; Durand, S.; Kimmel, E.; Michotey, C.; et al. Linking the International Wheat Genome Sequencing Consortium bread wheat reference genome sequence to wheat genetic and phenomic data. Genome Biol. 2018, 19, 111. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simao, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Matlin, A.J.; Clark, F.; Smith, C.W. Understanding alternative splicing: Towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005, 6, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Arefeen, A.; Liu, J.; Xiao, X.; Jiang, T. TAPAS: Tool for alternative polyadenylation site analysis. Bioinformatics 2018, 34, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Song, W.M.; Zhang, B. Multiscale Embedded Gene Co-expression Network Analysis. PLoS Comput. Biol. 2015, 11, e1004574. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Li, D.; Zhu, P.; Qiu, S.; Yao, K.; Zhuang, Y.; Chen, C.; Liu, G.; Wen, M.; Guo, R.; et al. The Landscapes of Gluten Regulatory Network in Elite Wheat Cultivars Contrasting in Gluten Strength. Int. J. Mol. Sci. 2023, 24, 9447. https://doi.org/10.3390/ijms24119447
Liu J, Li D, Zhu P, Qiu S, Yao K, Zhuang Y, Chen C, Liu G, Wen M, Guo R, et al. The Landscapes of Gluten Regulatory Network in Elite Wheat Cultivars Contrasting in Gluten Strength. International Journal of Molecular Sciences. 2023; 24(11):9447. https://doi.org/10.3390/ijms24119447
Chicago/Turabian StyleLiu, Jiajun, Dongsheng Li, Peng Zhu, Shi Qiu, Kebing Yao, Yiqing Zhuang, Chen Chen, Guanqing Liu, Mingxing Wen, Rui Guo, and et al. 2023. "The Landscapes of Gluten Regulatory Network in Elite Wheat Cultivars Contrasting in Gluten Strength" International Journal of Molecular Sciences 24, no. 11: 9447. https://doi.org/10.3390/ijms24119447