
Single-Cell RNA-Seq Analysis Reveals Ferroptosis in the Tumor Microenvironment of Clear Cell Renal Cell Carcinoma
Abstract
:1. Introduction
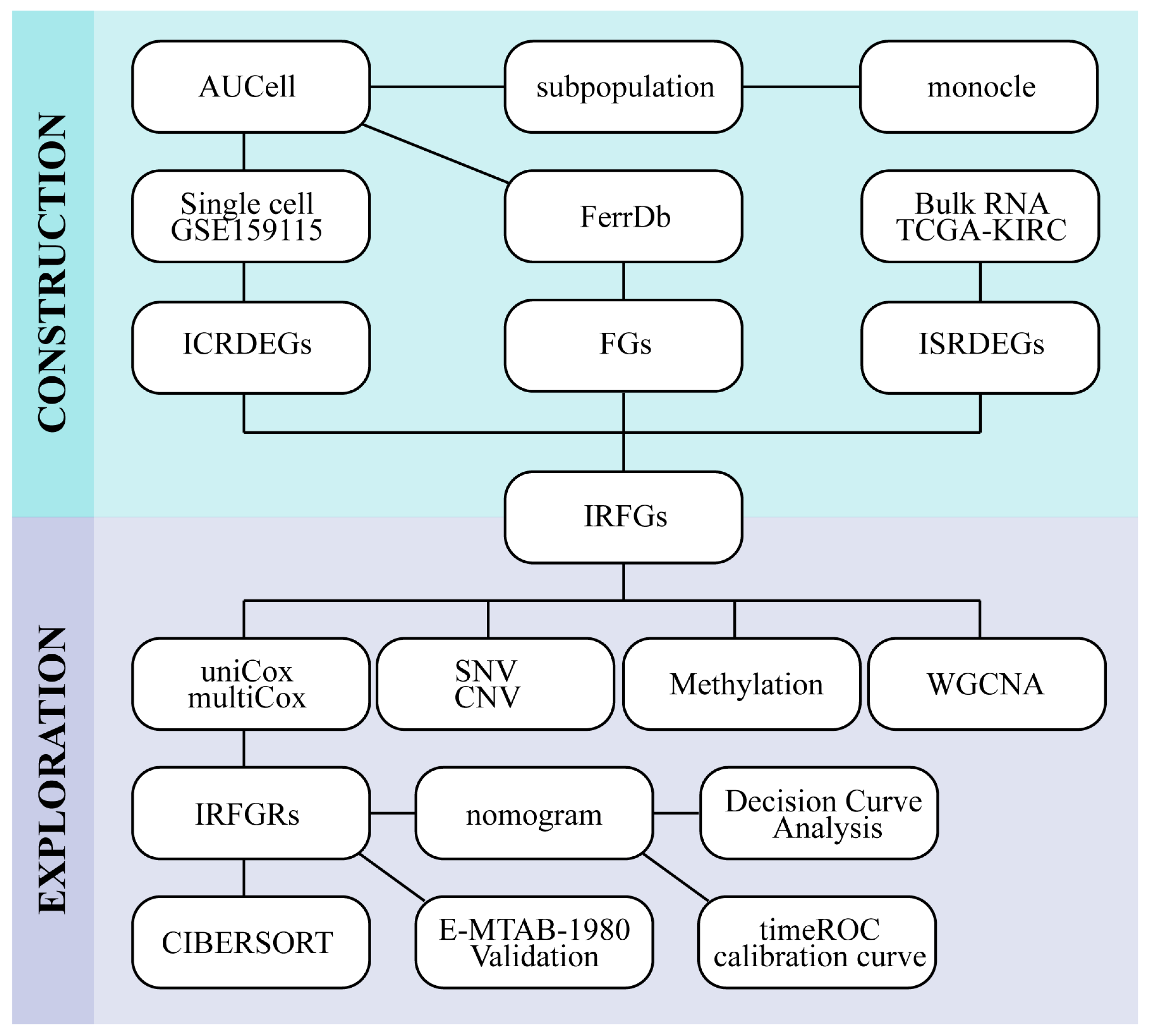
2. Results
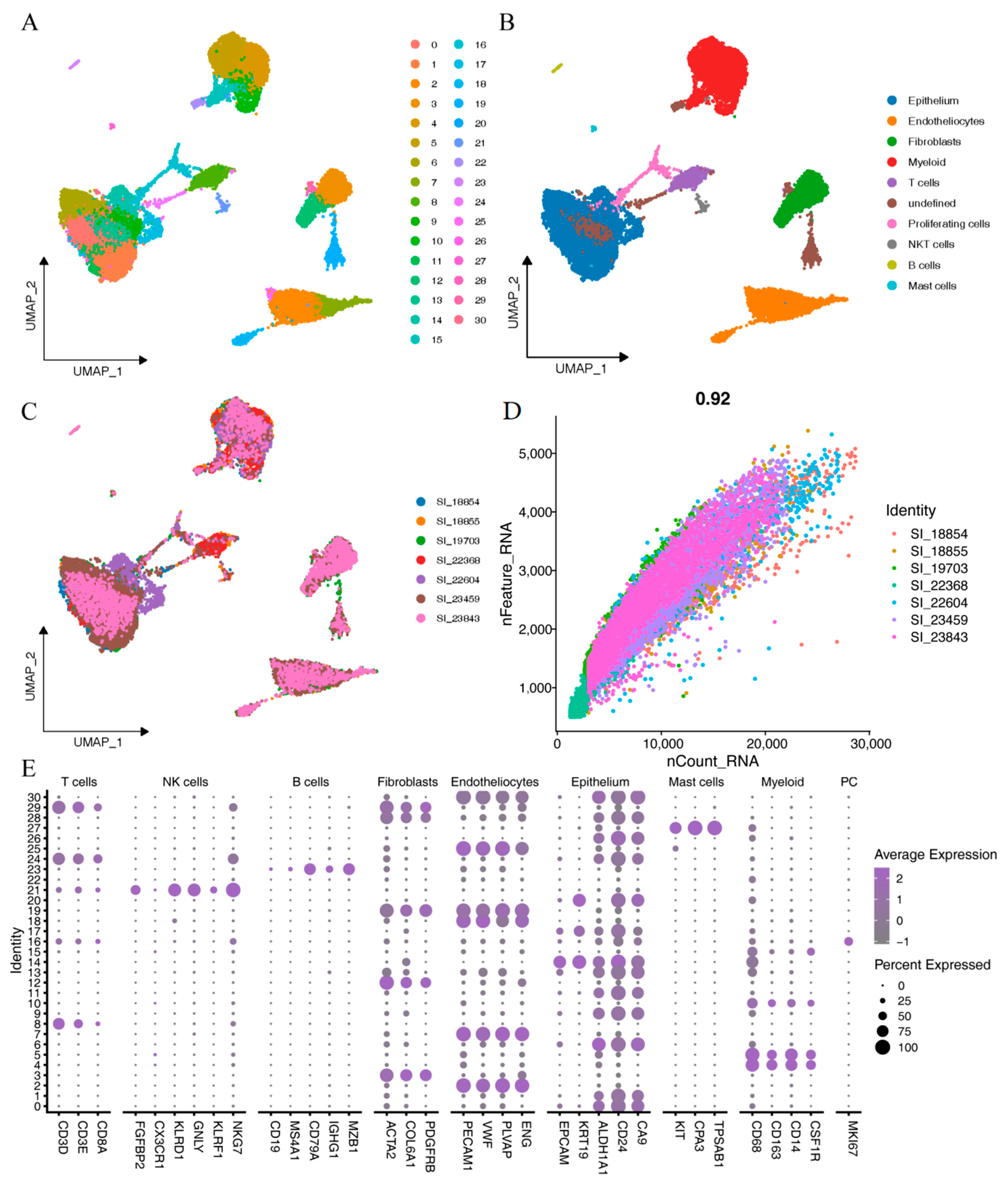
2.1. Identification of ccRCC Single-Cell Subpopulations
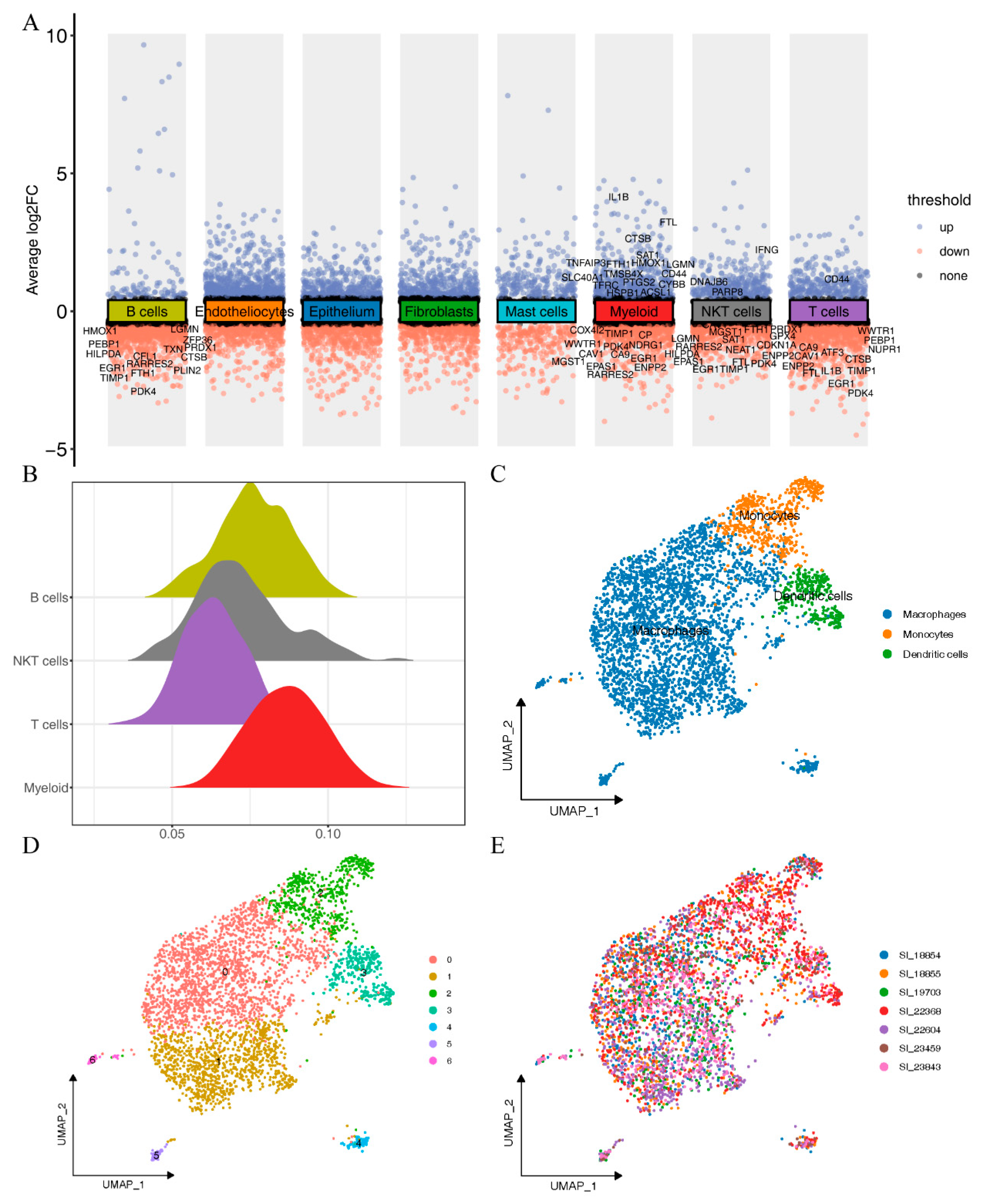
2.2. Ferroptosis Activity in Different Immune Cell Types
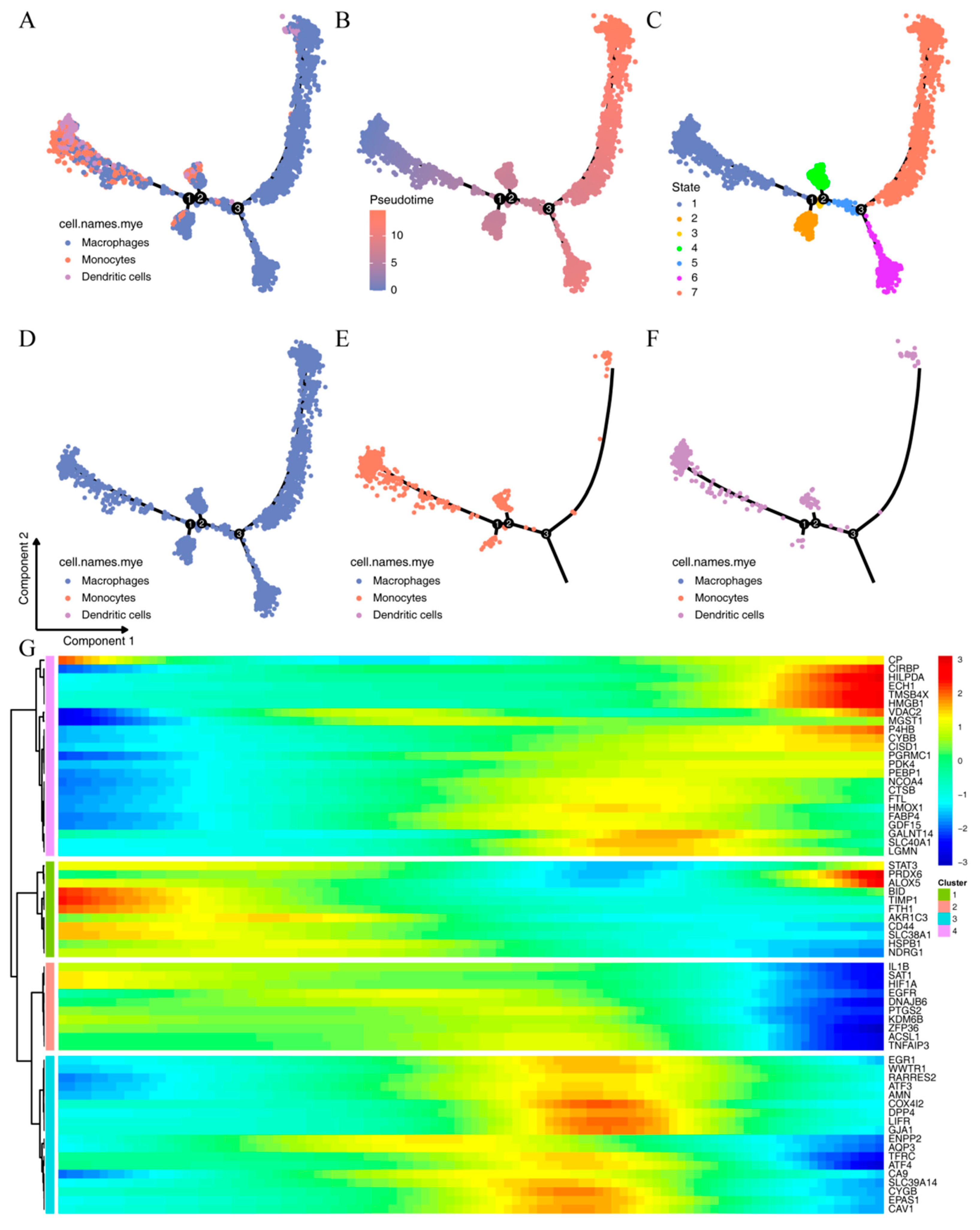
2.3. Subtyping of Myeloid
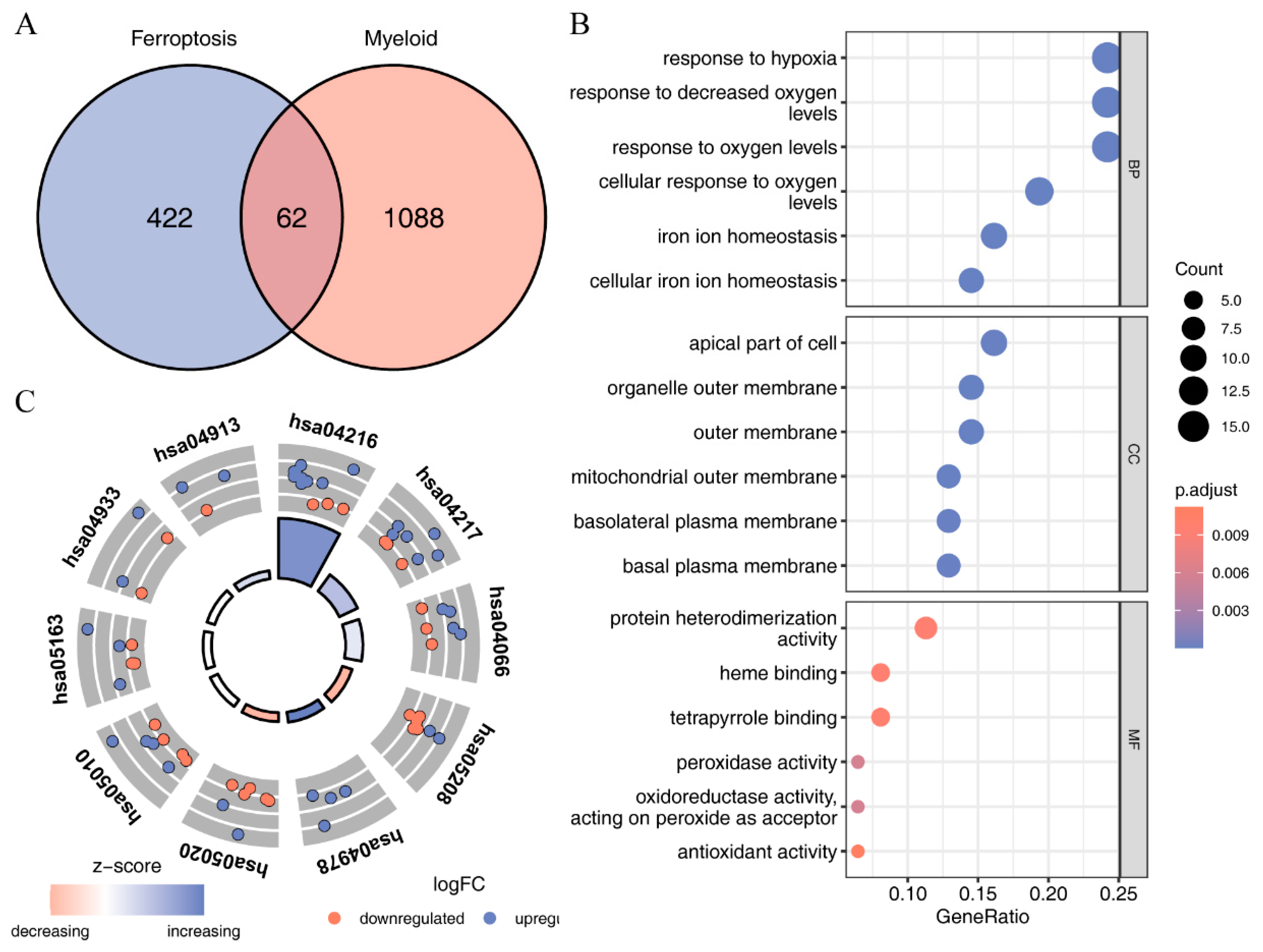
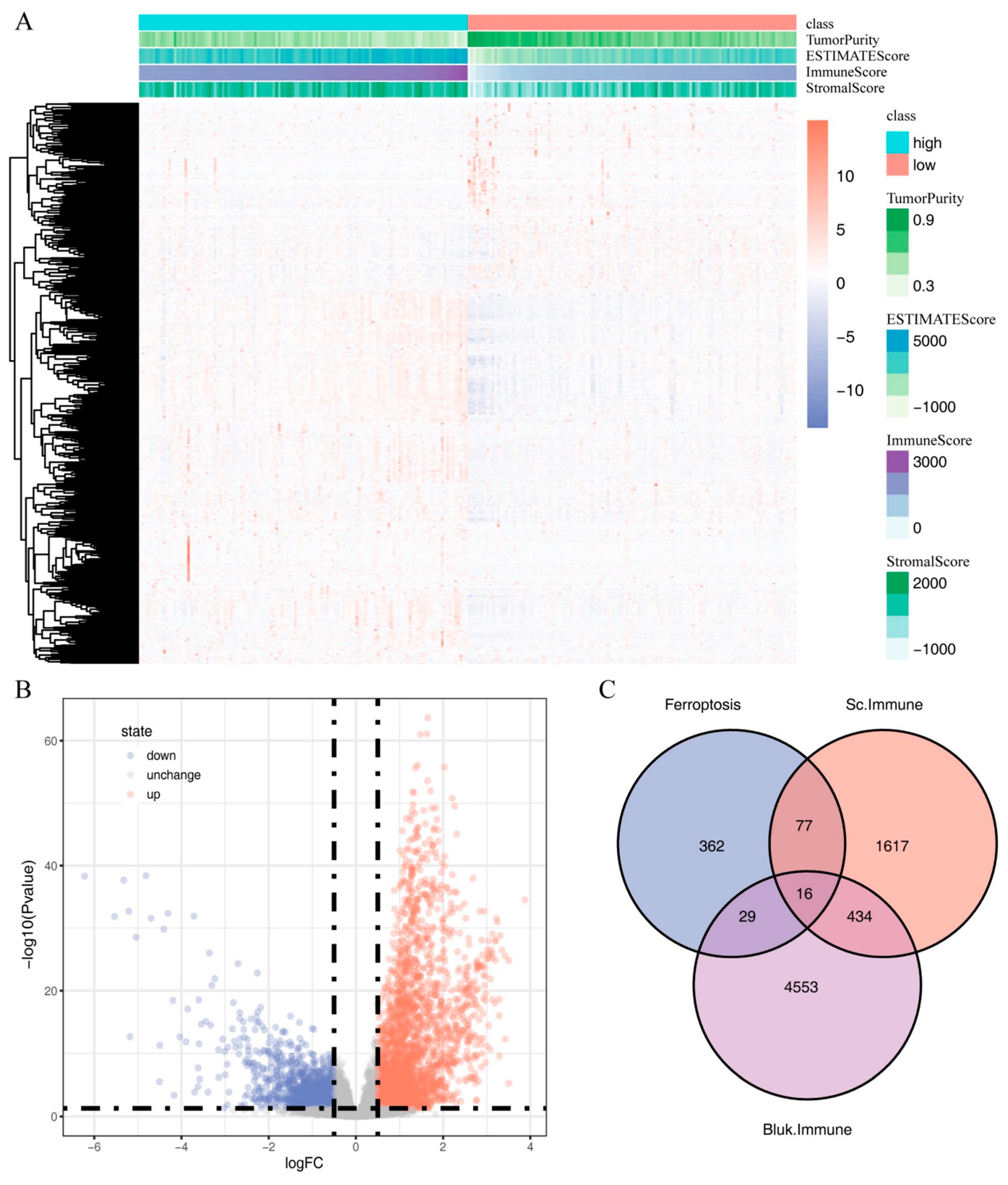
2.4. Identification of Immune-Related Ferroptosis Genes (IRFGs)
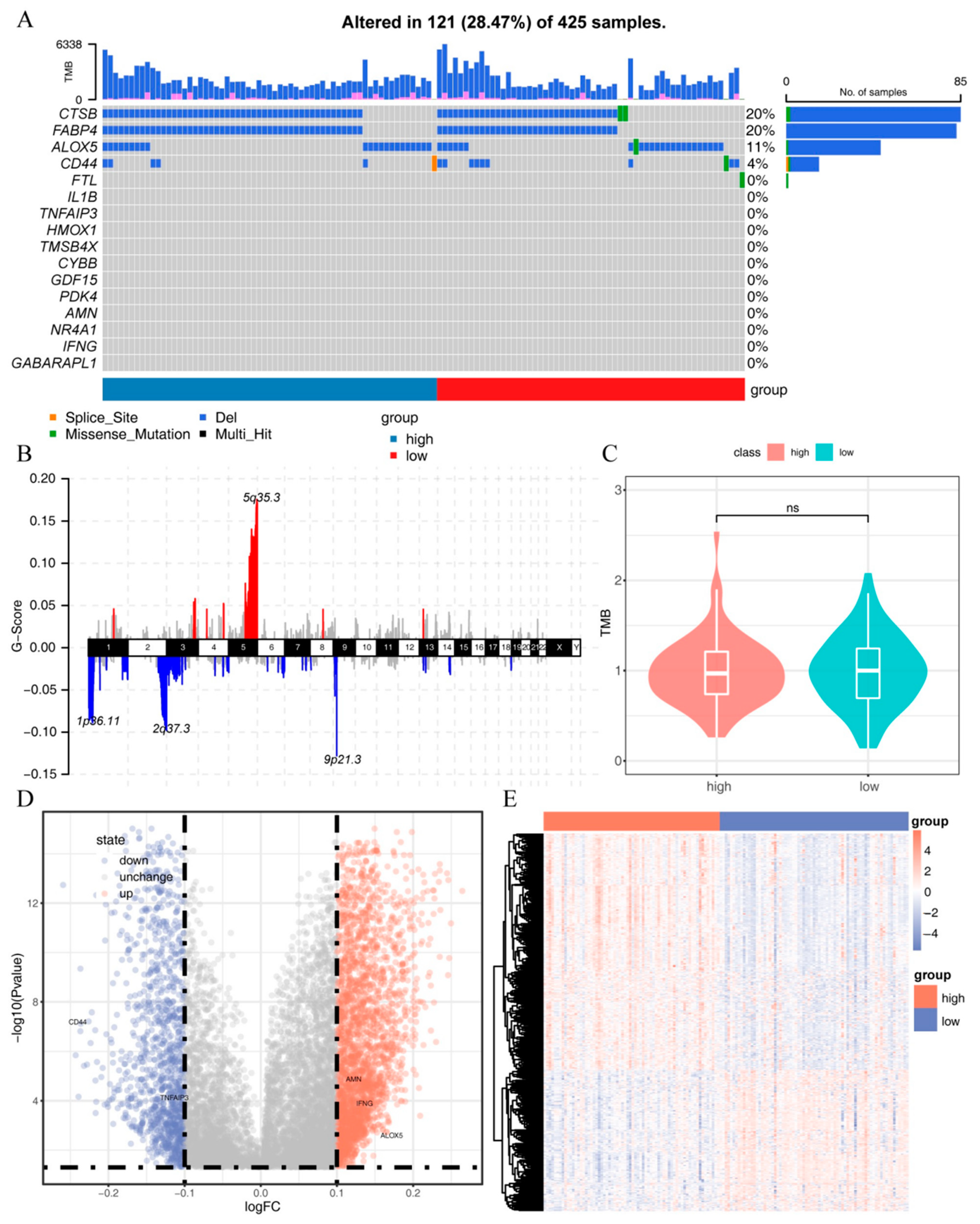
2.5. Mutation and Methylation Signatures of IRFGs
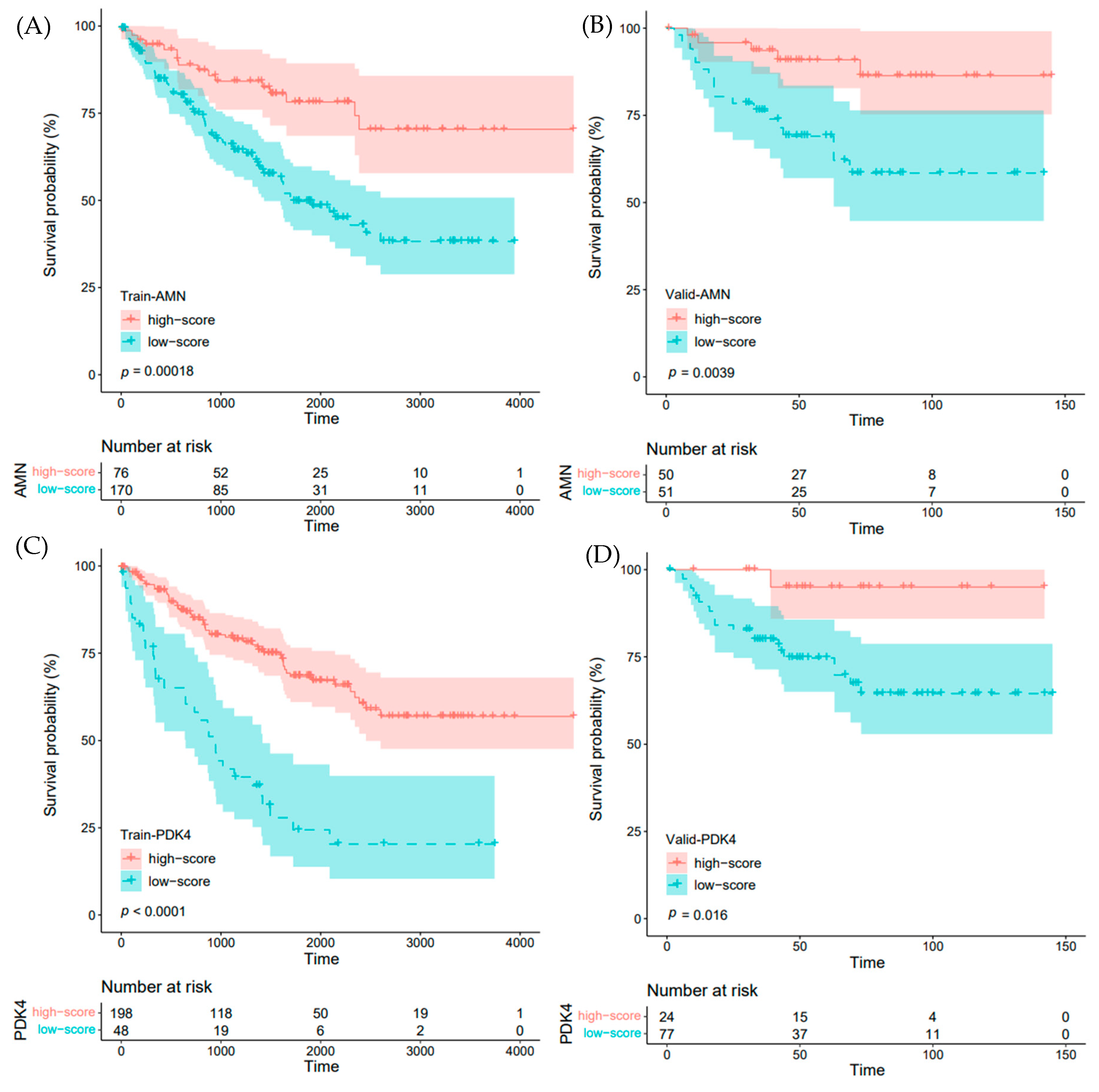
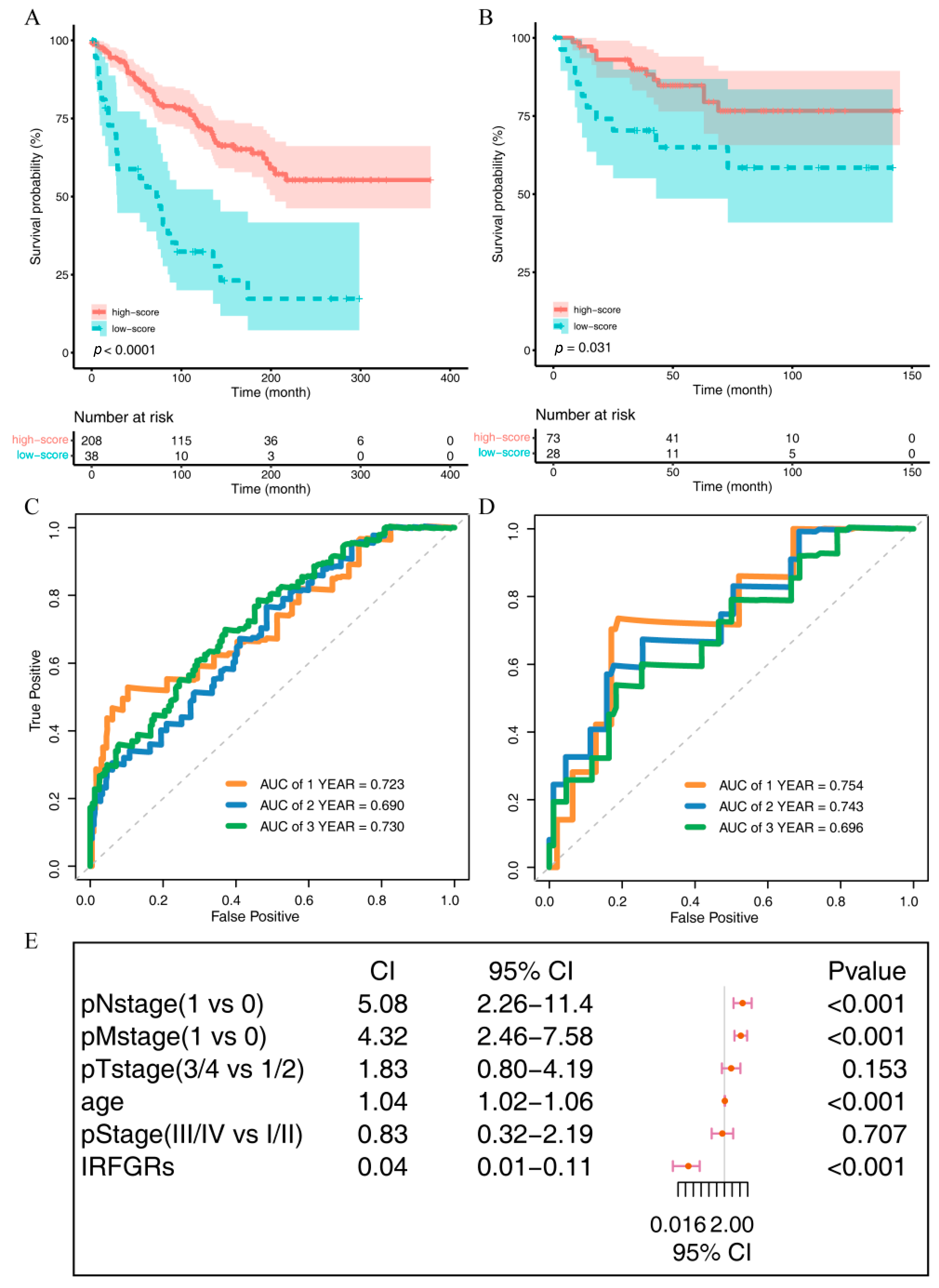
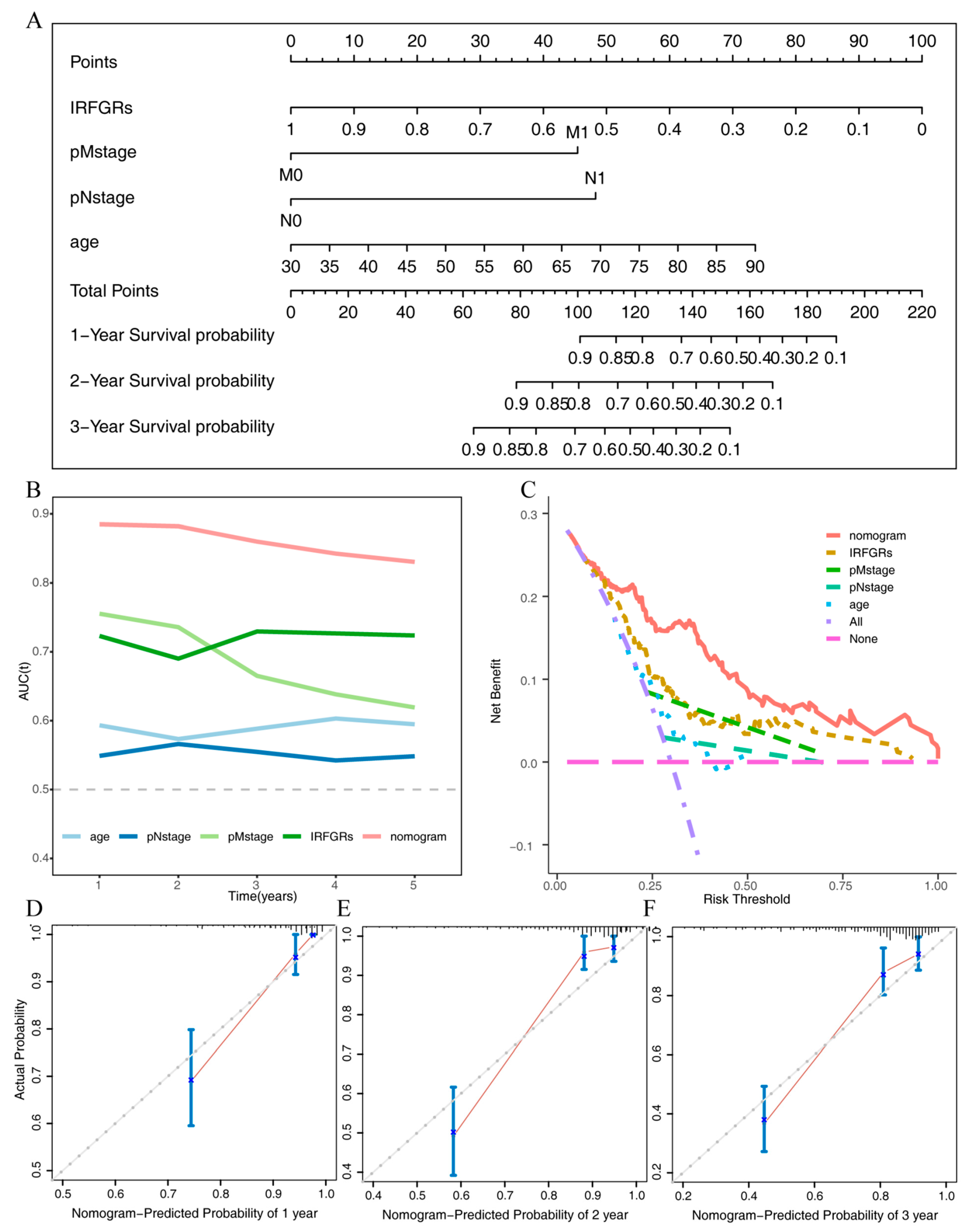
2.6. Impact of IRFGs on ccRCC Prognosis
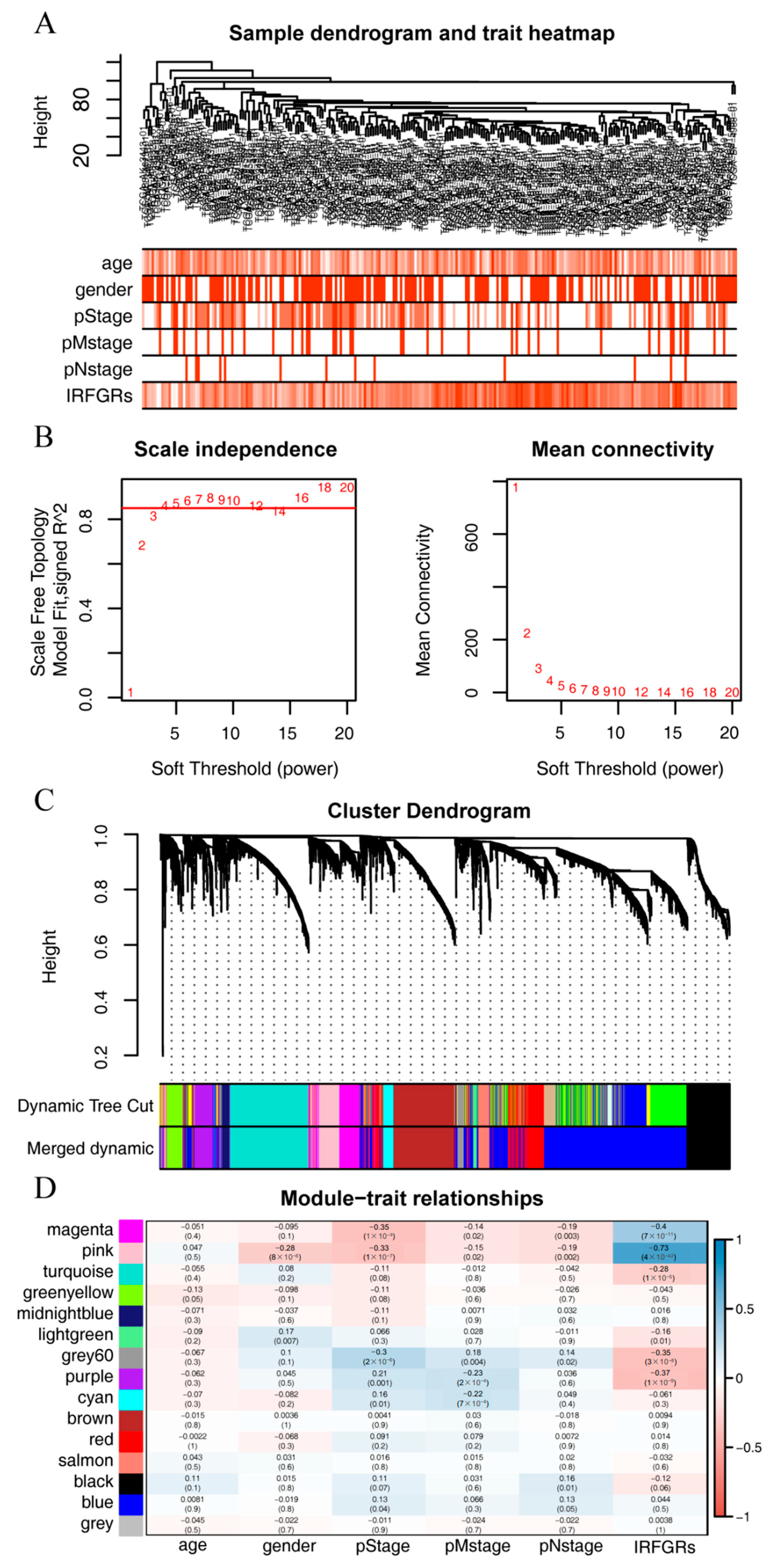
2.7. Exploring the Gene Modules and Their Functions Associated with the IRFGRs
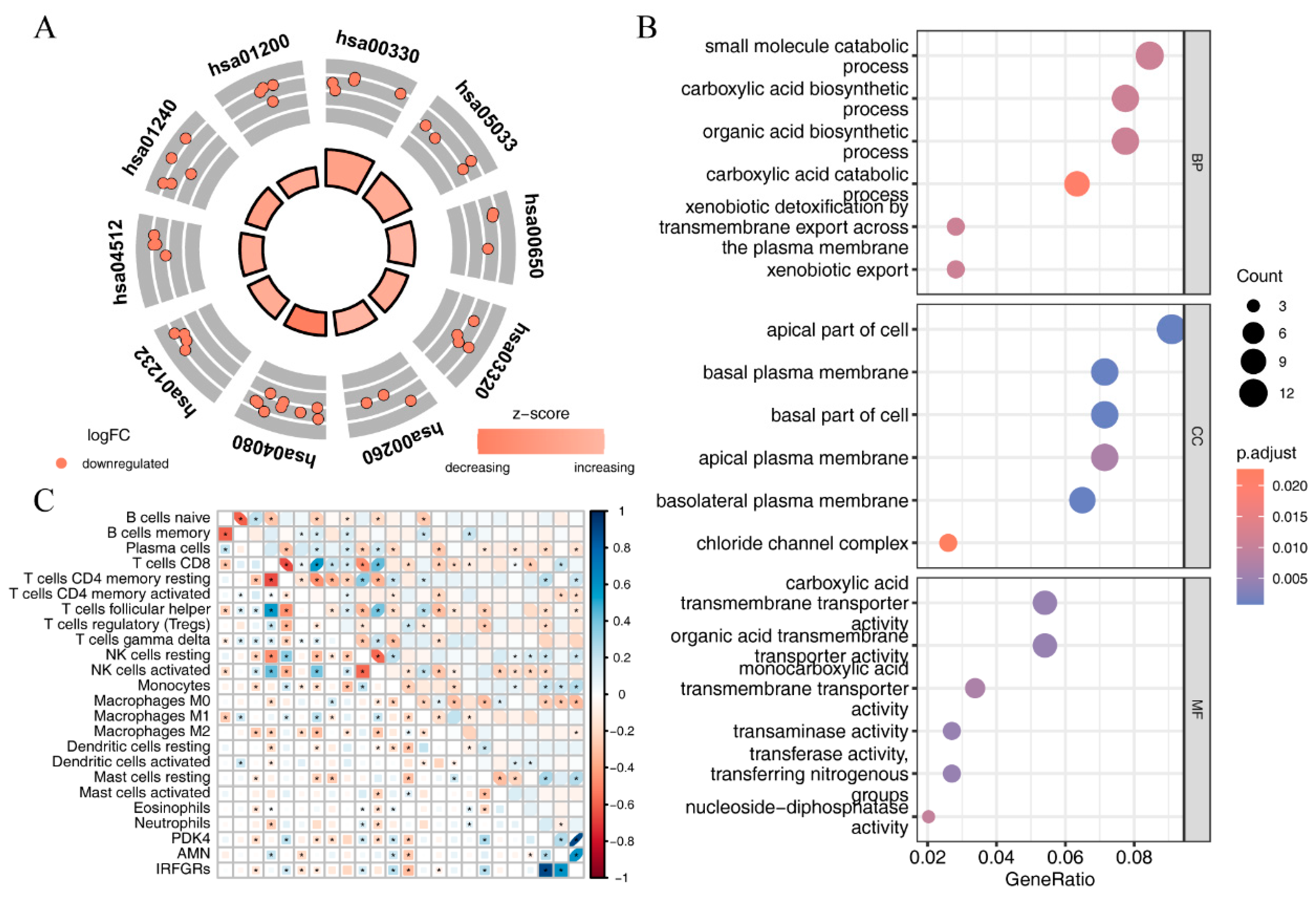
2.8. Investigating the Relationship between IRFGRs and Immune-Cell Infiltration
3. Discussion
4. Materials and Methods
4.1. Single-Cell Data Acquisition
4.2. Acquisition of Ferroptosis-Related Genes
4.3. Bulk RNA-Seq Data Acquisition
4.4. Mutation Data Acquisition and Processing
4.5. Methylation Data Acquisition and Processing
4.6. Single-Cell Data Processing
4.7. Pseudotime Analysis
4.8. Identification of Immune-Related Ferroptosis Genes
4.9. Mutations and Methylation of IRFGs
4.10. Constructing of the IRFGRs and Exploring Their Role in Prognosis
4.11. Weighted Gene Correlation Network Analysis
4.12. Relationship between Immune Cell Enrichment Scores and IRFGs
4.13. Enrichment Analysis
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.T.; McGovern, F.J. Renal-cell carcinoma. N. Engl. J. Med. 2005, 353, 2477–2490. [Google Scholar] [CrossRef]
- Escudier, B.; Porta, C.; Schmidinger, M.; Rioux-Leclercq, N.; Bex, A.; Khoo, V.; Grunwald, V.; Gillessen, S.; Horwich, A.; Esmo Guidelines Committee. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-updagger. Ann. Oncol. 2019, 30, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, R.R.; Motzer, R.J.; Voss, M.H. Towards individualized therapy for metastatic renal cell carcinoma. Nat. Rev. Clin. Oncol. 2019, 16, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, B.; Albiges, L.; Abu-Ghanem, Y.; Bedke, J.; Capitanio, U.; Dabestani, S.; Fernandez-Pello, S.; Giles, R.H.; Hofmann, F.; Hora, M.; et al. European association of urology guidelines on renal cell carcinoma: The 2022 update. Eur. Urol. 2022, 82, 399–410. [Google Scholar] [CrossRef]
- Singh, A.K.; Winslow, T.B.; Kermany, M.H.; Goritz, V.; Heit, L.; Miller, A.; Hoffend, N.C.; Stein, L.C.; Kumaraswamy, L.K.; Warren, G.W.; et al. A Pilot Study of Stereotactic Body Radiation Therapy Combined with Cytoreductive Nephrectomy for Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5055–5065. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Yangyun, W.; Guowei, S.; Shufen, S.; Jie, Y.; Rui, Y.; Yu, R. Everolimus accelerates Erastin and RSL3-induced ferroptosis in renal cell carcinoma. Gene 2022, 809, 145992. [Google Scholar] [CrossRef]
- Wang, J.; Yin, X.; He, W.; Xue, W.; Zhang, J.; Huang, Y. SUV39H1 deficiency suppresses clear cell renal cell carcinoma growth by inducing ferroptosis. Acta Pharm. Sin. B 2021, 11, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Qin, H.; Jiang, B.; Lu, W.; Hao, J.; Cao, W.; Du, L.; Chen, W.; Zhao, X.; Guo, H. KLF2 inhibits cancer cell migration and invasion by regulating ferroptosis through GPX4 in clear cell renal cell carcinoma. Cancer Lett. 2021, 522, 1–13. [Google Scholar] [CrossRef]
- Li, Y.Z.; Zhu, H.C.; Du, Y.; Zhao, H.C.; Wang, L. Silencing lncRNA SLC16A1-AS1 induced ferroptosis in renal cell carcinoma through miR-143-3p/SLC7A11 signaling. Technol. Cancer Res. Treat. 2022, 21, 15330338221077803. [Google Scholar] [CrossRef]
- Yang, W.H.; Ding, C.C.; Sun, T.; Rupprecht, G.; Lin, C.C.; Hsu, D.; Chi, J.T. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019, 28, 2501–2508.e4. [Google Scholar] [CrossRef] [PubMed]
- Vuong, L.; Kotecha, R.R.; Voss, M.H.; Hakimi, A.A. Tumor microenvironment dynamics in clear-cell renal cell carcinoma. Cancer Discov. 2019, 9, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lonnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef]
- Young, M.D.; Mitchell, T.J.; Braga, F.A.V.; Tran, M.G.B.; Stewart, B.J.; Ferdinand, J.R.; Collord, G.; Botting, R.A.; Popescu, D.M.; Loudon, K.W.; et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 2018, 361, 594–599. [Google Scholar] [CrossRef]
- Hu, J.; Chen, Z.; Bao, L.; Zhou, L.; Hou, Y.; Liu, L.; Xiong, M.; Zhang, Y.; Wang, B.; Tao, Z.; et al. Single-cell transcriptome analysis reveals intratumoral heterogeneity in ccRCC, which results in different clinical outcomes. Mol. Ther. 2020, 28, 1658–1672. [Google Scholar] [CrossRef]
- Braun, D.A.; Street, K.; Burke, K.P.; Cookmeyer, D.L.; Denize, T.; Pedersen, C.B.; Gohil, S.H.; Schindler, N.; Pomerance, L.; Hirsch, L.; et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell 2021, 39, 632–648.e8. [Google Scholar] [CrossRef]
- Krishna, C.; DiNatale, R.G.; Kuo, F.; Srivastava, R.M.; Vuong, L.; Chowell, D.; Gupta, S.; Vanderbilt, C.; Purohit, T.A.; Liu, M.; et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 2021, 39, 662–677.e6. [Google Scholar] [CrossRef] [PubMed]
- Bi, K.; He, M.X.; Bakouny, Z.; Kanodia, A.; Napolitano, S.; Wu, J.; Grimaldi, G.; Braun, D.A.; Cuoco, M.S.; Mayorga, A.; et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 2021, 39, 649–661.e5. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Ascierto, M.L.; McMiller, T.L.; Berger, A.E.; Danilova, L.; Anders, R.A.; Netto, G.J.; Xu, H.; Pritchard, T.S.; Fan, J.; Cheadle, C.; et al. The intratumoral balance between metabolic and immunologic gene expression is associated with anti-PD-1 response in patients with renal cell carcinoma. Cancer Immunol. Res. 2016, 4, 726–733. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, X.; Xie, F.; Zhang, L.; Yan, H.; Huang, J.; Zhang, C.; Zhou, F.; Chen, J.; Zhang, L. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. 2022, 42, 88–116. [Google Scholar] [CrossRef]
- Dang, Q.; Sun, Z.; Wang, Y.; Wang, L.; Liu, Z.; Han, X. Ferroptosis: A double-edged sword mediating immune tolerance of cancer. Cell Death Dis. 2022, 13, 925. [Google Scholar] [CrossRef]
- Xu, H.; Ye, D.; Ren, M.; Zhang, H.; Bi, F. Ferroptosis in the tumor microenvironment: Perspectives for immunotherapy. Trends Mol. Med. 2021, 27, 856–867. [Google Scholar] [CrossRef]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef]
- Tan, S.K.; Mahmud, I.; Fontanesi, F.; Puchowicz, M.; Neumann, C.K.A.; Griswold, A.J.; Patel, R.; Dispagna, M.; Ahmed, H.H.; Gonzalgo, M.L.; et al. Obesity-dependent adipokine chemerin suppresses fatty acid oxidation to confer ferroptosis resistance. Cancer Discov. 2021, 11, 2072–2093. [Google Scholar] [CrossRef] [PubMed]
- Green, Y.S.; Ferreira Dos Santos, M.C.; Fuja, D.G.; Reichert, E.C.; Campos, A.R.; Cowman, S.J.; Acuña Pilarte, K.; Kohan, J.; Tripp, S.R.; Leibold, E.A.; et al. ISCA2 inhibition decreases HIF and induces ferroptosis in clear cell renal carcinoma. Oncogene 2022, 41, 4709–4723. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.Y.; Hsieh, P.C.; Chiu, V.; Lan, C.C.; Lu, K.C. The von Hippel-Lindau Tumor Suppressor Gene Mutations Modulate Lipocalin-2 Expression in Ferroptotic-Inflammatory Pathways. Oxid. Med. Cell Longev. 2023, 2023, 7736638. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhu, C.; Chen, X.; Guan, G.; Zou, C.; Shen, S.; Wu, J.; Wang, Y.; Lin, Z.; Chen, L.; et al. Ferroptosis, as the most enriched programmed cell death process in glioma, induces immunosuppression and immunotherapy resistance. Neuro-Oncology 2022, 24, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Z.; Yang, Y.; Di, T.; Wu, Y.; Bian, T. NCOA4-Mediated Ferroptosis in Bronchial Epithelial Cells Promotes Macrophage M2 Polarization in COPD Emphysema. Int. J. Chron. Obstruct. Pulmon. Dis. 2022, 17, 667–681. [Google Scholar] [CrossRef]
- Li, R.; Ferdinand, J.R.; Loudon, K.W.; Bowyer, G.S.; Laidlaw, S.; Muyas, F.; Mamanova, L.; Neves, J.B.; Bolt, L.; Fasouli, E.S.; et al. Mapping single-cell transcriptomes in the intra-tumoral and associated territories of kidney cancer. Cancer Cell 2022, 40, 1583–1599.e10. [Google Scholar] [CrossRef]
- Chen, C.H.; Bhasin, S.; Khanna, P.; Joshi, M.; Joslin, P.M.; Saxena, R.; Amin, S.; Liu, S.; Sindhu, S.; Walker, S.R.; et al. Study of Cathepsin B inhibition in VEGFR TKI treated human renal cell carcinoma xenografts. Oncogenesis 2019, 8, 15. [Google Scholar] [CrossRef]
- Li, L.; Zhao, S.; Liu, Z.; Zhang, N.; Pang, S.; Liu, J.; Liu, C.; Fan, Y. Sunitinib treatment promotes metastasis of drug-resistant renal cell carcinoma via TFE3 signaling pathway. Cell Death Dis. 2021, 12, 220. [Google Scholar] [CrossRef]
- Hu, Z.W.; Chen, L.; Ma, R.Q.; Wei, F.Q.; Wen, Y.H.; Zeng, X.L.; Sun, W.; Wen, W.P. Comprehensive analysis of ferritin subunits expression and positive correlations with tumor-associated macrophages and T regulatory cells infiltration in most solid tumors. Aging 2021, 13, 11491–11506. [Google Scholar] [CrossRef]
- Mou, Y.; Wu, J.; Zhang, Y.; Abdihamid, O.; Duan, C.; Li, B. Low expression of ferritinophagy-related NCOA4 gene in relation to unfavorable outcome and defective immune cells infiltration in clear cell renal carcinoma. BMC Cancer 2021, 21, 18. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Wang, X. Prognosis-related genes participate in immunotherapy of renal clear cell carcinoma possibly by targeting dendritic cells. Front. Cell Dev. Biol. 2022, 10, 892616. [Google Scholar] [CrossRef] [PubMed]
- Martini, J.F.; Plimack, E.R.; Choueiri, T.K.; McDermott, D.F.; Puzanov, I.; Fishman, M.N.; Cho, D.C.; Vaishampayan, U.; Rosbrook, B.; Fernandez, K.C.; et al. Angiogenic and Immune-Related Biomarkers and Outcomes Following Axitinib/Pembrolizumab Treatment in Patients with Advanced Renal Cell Carcinoma. Clin. Cancer Res. 2020, 26, 5598–5608. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Niu, Z.; Si, S.; Zhao, G.; Wang, J.; Yao, Z.; Cheng, F.; He, W. Comprehensive analysis of new prognostic signature based on ferroptosis-related genes in clear cell renal cell carcinoma. Aging 2021, 13, 19789–19804. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Lin, M.; Ou, D.; Huang, Z.; Shen, P. A novel ferroptosis-related 12-gene signature predicts clinical prognosis and reveals immune relevancy in clear cell renal cell carcinoma. BMC Cancer 2021, 21, 831. [Google Scholar] [CrossRef]
- Zhao, G.J.; Wu, Z.; Ge, L.; Yang, F.; Hong, K.; Zhang, S.; Ma, L. Ferroptosis-related gene-based prognostic model and immune infiltration in clear cell renal cell carcinoma. Front. Genet. 2021, 12, 650416. [Google Scholar] [CrossRef]
- Xing, Q.; Zeng, T.; Liu, S.; Cheng, H.; Ma, L.; Wang, Y. A novel 10 glycolysis-related genes signature could predict overall survival for clear cell renal cell carcinoma. BMC Cancer 2021, 21, 381. [Google Scholar] [CrossRef]
- Xing, X.L.; Liu, Y.; Liu, J.H.; Zhou, H.F.; Zhang, H.R.; Zuo, Q.; Bu, P.; Duan, T.; Zhou, Y.; Xiao, Z.Q. A nomogram integrating ferroptosis-and immune-related biomarker for prediction of prognosis and diagnosis in kidney renal clear cell carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 6176–6186. [Google Scholar]
- Chang, K.; Yuan, C.; Liu, X. Ferroptosis-related gene signature accurately predicts survival outcomes in patients with clear-cell renal cell carcinoma. Front. Oncol. 2021, 11, 649347. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Zhu, Z.; Chen, J.; Wang, C.; Chen, X.; Zhu, S.; Zhang, A. A novel anoikis-related prognostic signature associated with prognosis and immune infiltration landscape in clear cell renal cell carcinoma. Front. Genet. 2022, 13, 1039465. [Google Scholar] [CrossRef]
- Rini, B.I.; Battle, D.; Figlin, R.A.; George, D.J.; Hammers, H.; Hutson, T.; Jonasch, E.; Joseph, R.W.; McDermott, D.F.; Motzer, R.J.; et al. The society for immunotherapy of cancer consensus statement on immunotherapy for the treatment of advanced renal cell carcinoma (RCC). J. Immunother. Cancer 2019, 7, 354. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, J.S.; Lipson, E.J.; Danilova, L.; Topalian, S.L.; Jedrych, J.; Baraban, E.; Ged, Y.; Singla, N.; Choueiri, T.K.; Gupta, S.; et al. Combinatorial biomarker for predicting outcomes to anti-PD-1 therapy in patients with metastatic clear cell renal cell carcinoma. Cell Rep. Med. 2023, 4, 100947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Narayanan, S.P.; Mannan, R.; Raskind, G.; Wang, X.; Vats, P.; Su, F.; Hosseini, N.; Cao, X.; Kumar-Sinha, C.; et al. Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proc. Natl. Acad. Sci. USA 2021, 118, e2103240118. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Bao, J. FerrDb: A manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database 2020, 2020, baaa021. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- Athar, A.; Fullgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019, 8, 329–337.e4. [Google Scholar] [CrossRef] [PubMed]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Heagerty, P.J.; Lumley, T.; Pepe, M.S. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 2000, 56, 337–344. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Gene Ontology Consortium, Gene ontology consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age | Gender | pTstage | pNstage | pMstage | pStage | Treatment |
|---|---|---|---|---|---|---|---|
| 1 | 71 | Male | T2 | N0 | M0 | Stage II | Radical nephrectomy |
| 2 | 70 | Male | T1 | N0 | M0 | Stage I | Partial nephrectomy |
| 3 | 69 | Male | T1 | N0 | M0 | Stage I | Radical nephrectomy |
| 4 | 76 | Male | T2 | N0 | M0 | Stage II | Partial nephrectomy |
| 5 | 74 | Male | T2 | N0 | M0 | Stage II | Radical nephrectomy |
| 6 | 66 | Male | T1 | N0 | M0 | Stage I | Partial nephrectomy |
| 7 | 71 | Male | T2 | N0 | M0 | Stage II | Radical nephrectomy |
| Factors | Level | E-MTAB-1980 | TCGA |
|---|---|---|---|
| 101 | 246 | ||
| age (mean (SD)) | 63.48 (11.50) | 61.54 (11.91) | |
| gender (%) | female | 24 (23.8) | 100 (40.7) |
| male | 77 (76.2) | 146 (59.3) | |
| race (%) | asian | - | 4 (1.6) |
| black or african american | - | 17 (6.9) | |
| unkown | - | 4 (1.6) | |
| white | - | 221 (89.8) | |
| tumor.history (%) | no | - | 212 (86.2) |
| yes | - | 34 (13.8) | |
| pStage (%) | Stage I | 66 (65.3) | 102 (41.5) |
| Stage II | 10 (9.9) | 34 (13.8) | |
| Stage III | 13 (12.9) | 71 (28.9) | |
| Stage IV | 12 (11.9) | 39 (15.9) | |
| pTstage (%) | T1 | 68 (67.3) | 106 (43.1) |
| T2 | 11 (10.9) | 42 (17.1) | |
| T3 | 21 (20.8) | 93 (37.8) | |
| T4 | 1 (1.0) | 5 (2.0) | |
| pNstage (%) | N0 | 94 (93.1) | 233 (94.7) |
| N1 | 3 (3.0) | 13 (5.3) | |
| N2 | 4 (4.0) | 0 (0.0) | |
| pMstage (%) | M0 | 89 (88.1) | 207 (84.1) |
| M1 | 12 (11.9) | 39 (15.9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Deng, Y.; Zhang, H.; Zhang, Z.; Jin, X.; Xuan, Y.; Zhang, Z.; Ma, X. Single-Cell RNA-Seq Analysis Reveals Ferroptosis in the Tumor Microenvironment of Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 9092. https://doi.org/10.3390/ijms24109092
Zhang J, Deng Y, Zhang H, Zhang Z, Jin X, Xuan Y, Zhang Z, Ma X. Single-Cell RNA-Seq Analysis Reveals Ferroptosis in the Tumor Microenvironment of Clear Cell Renal Cell Carcinoma. International Journal of Molecular Sciences. 2023; 24(10):9092. https://doi.org/10.3390/ijms24109092
Chicago/Turabian StyleZhang, Jing, Yun Deng, Hui Zhang, Zhiyuan Zhang, Xin Jin, Yan Xuan, Zhen Zhang, and Xuejun Ma. 2023. "Single-Cell RNA-Seq Analysis Reveals Ferroptosis in the Tumor Microenvironment of Clear Cell Renal Cell Carcinoma" International Journal of Molecular Sciences 24, no. 10: 9092. https://doi.org/10.3390/ijms24109092