
Increasing Charge Carrier Mobility through Modifications of Terminal Groups of Y6: A Theoretical Study

Abstract
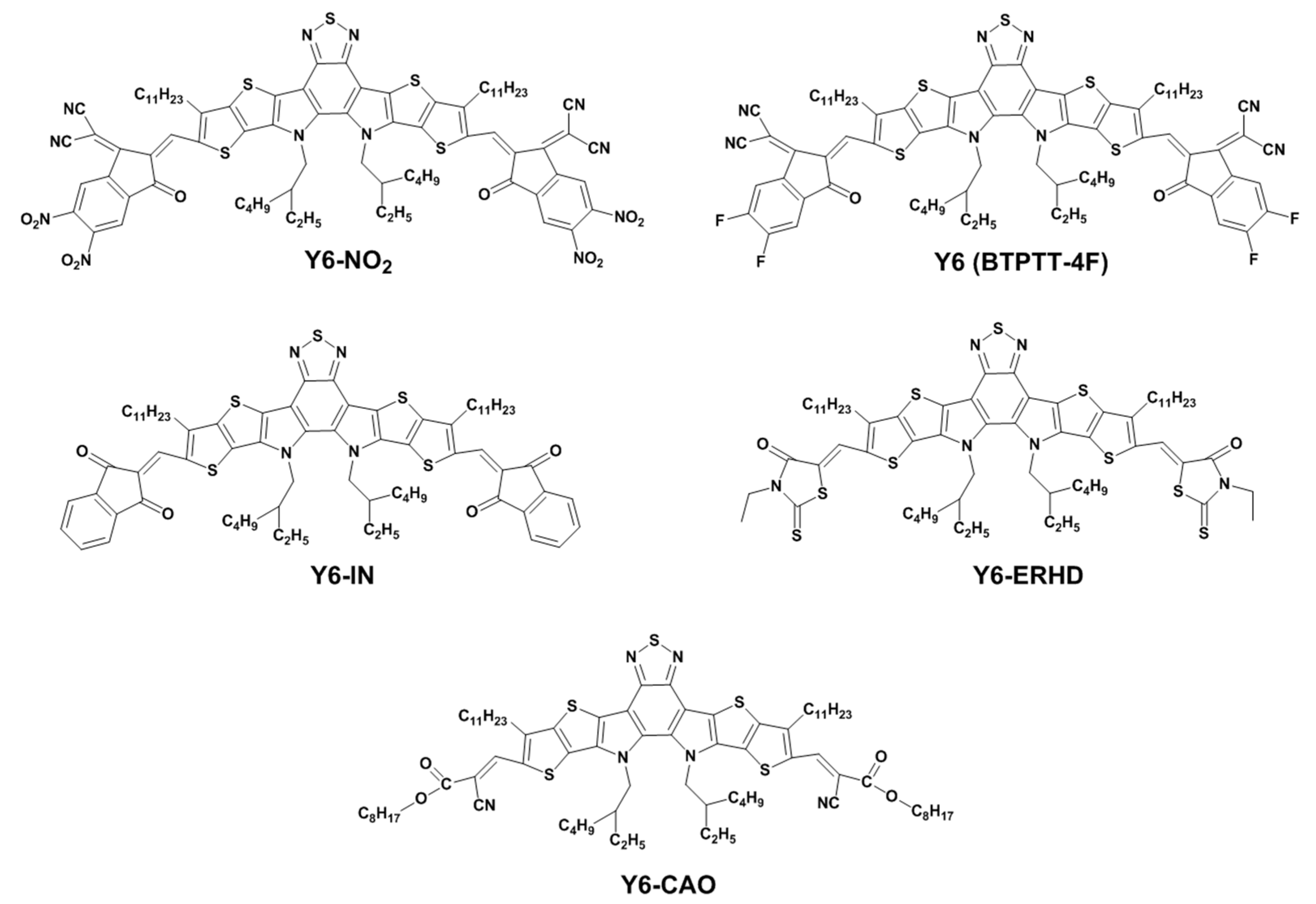
:1. Introduction
- (1)
- To investigate the effects of different terminal acceptor units on the photovoltaic properties of Y6.
- (2)
- To find new promising acceptors (if possible).
2. Results and Discussions
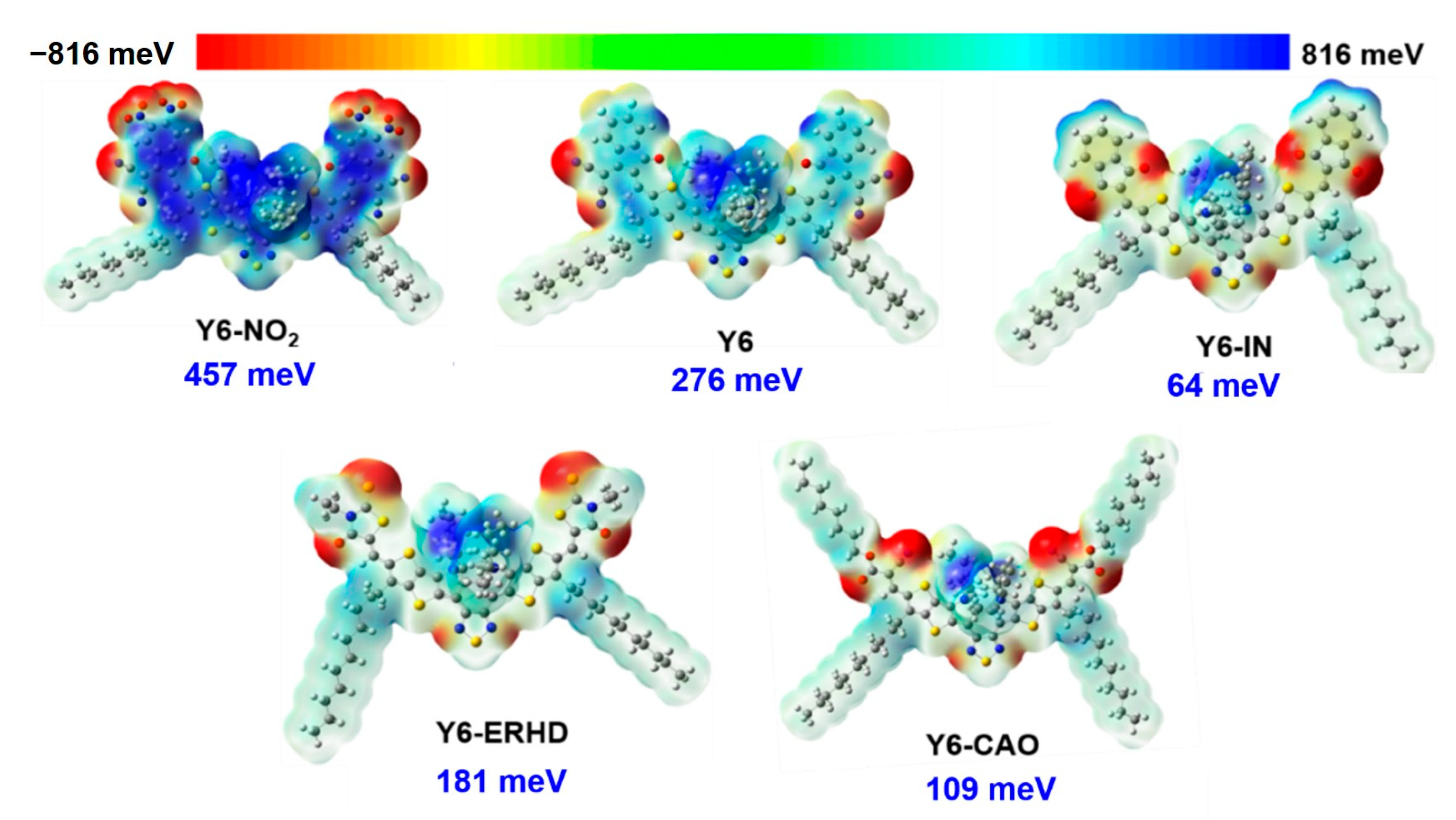
2.1. Dipole Moments and Electrostatic Potential of the Modified Molecules
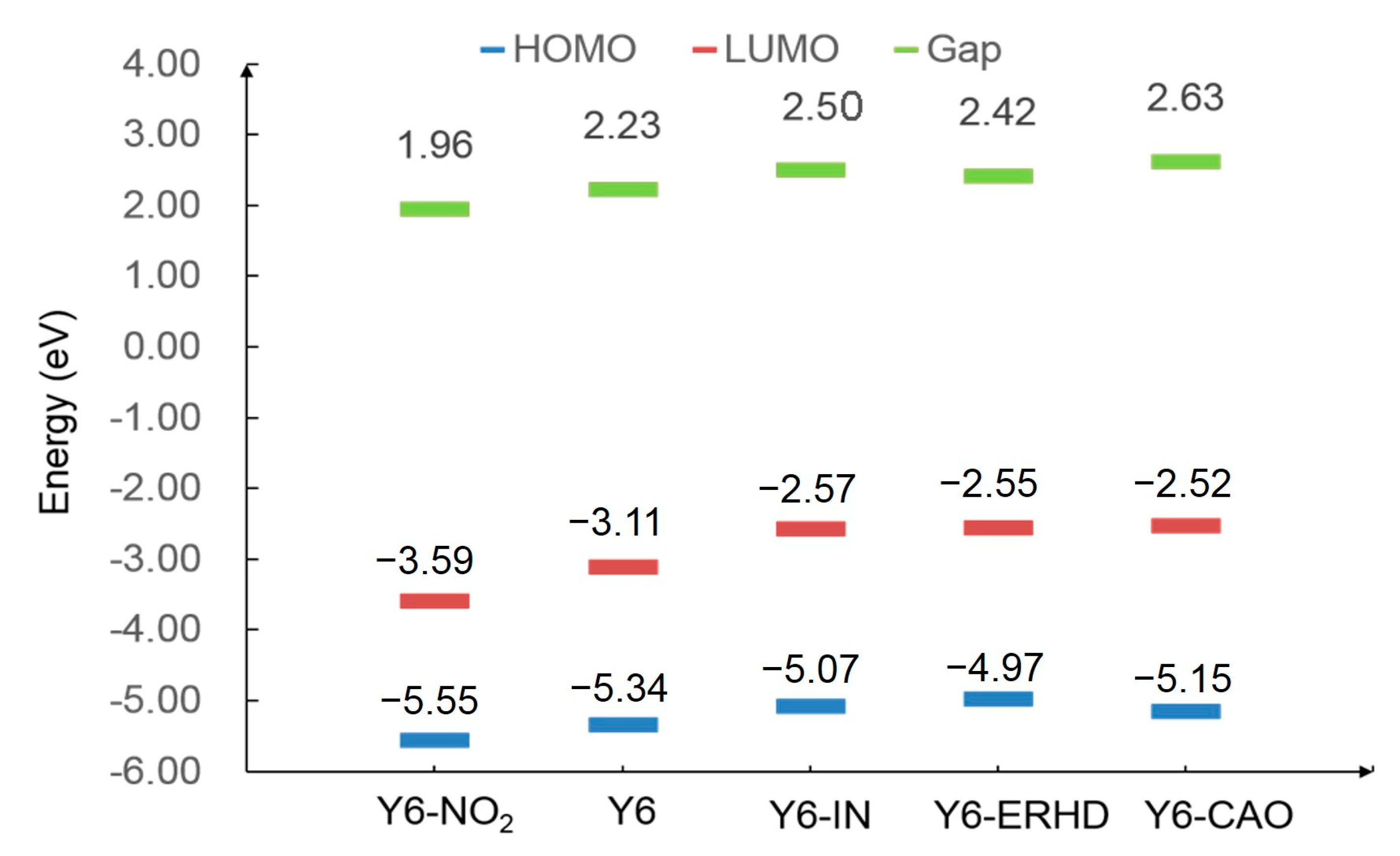
2.2. FMO and Gap Energy
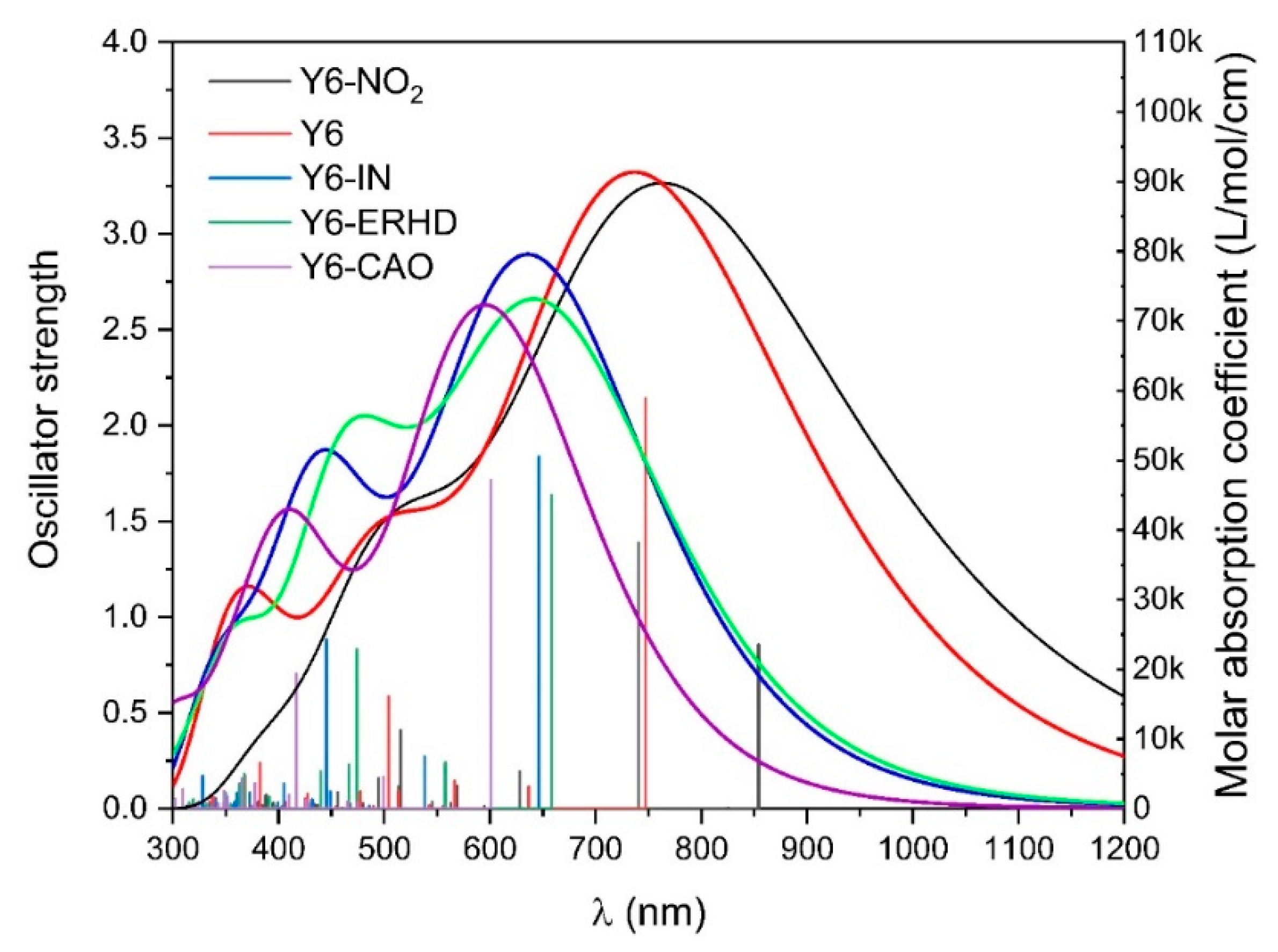
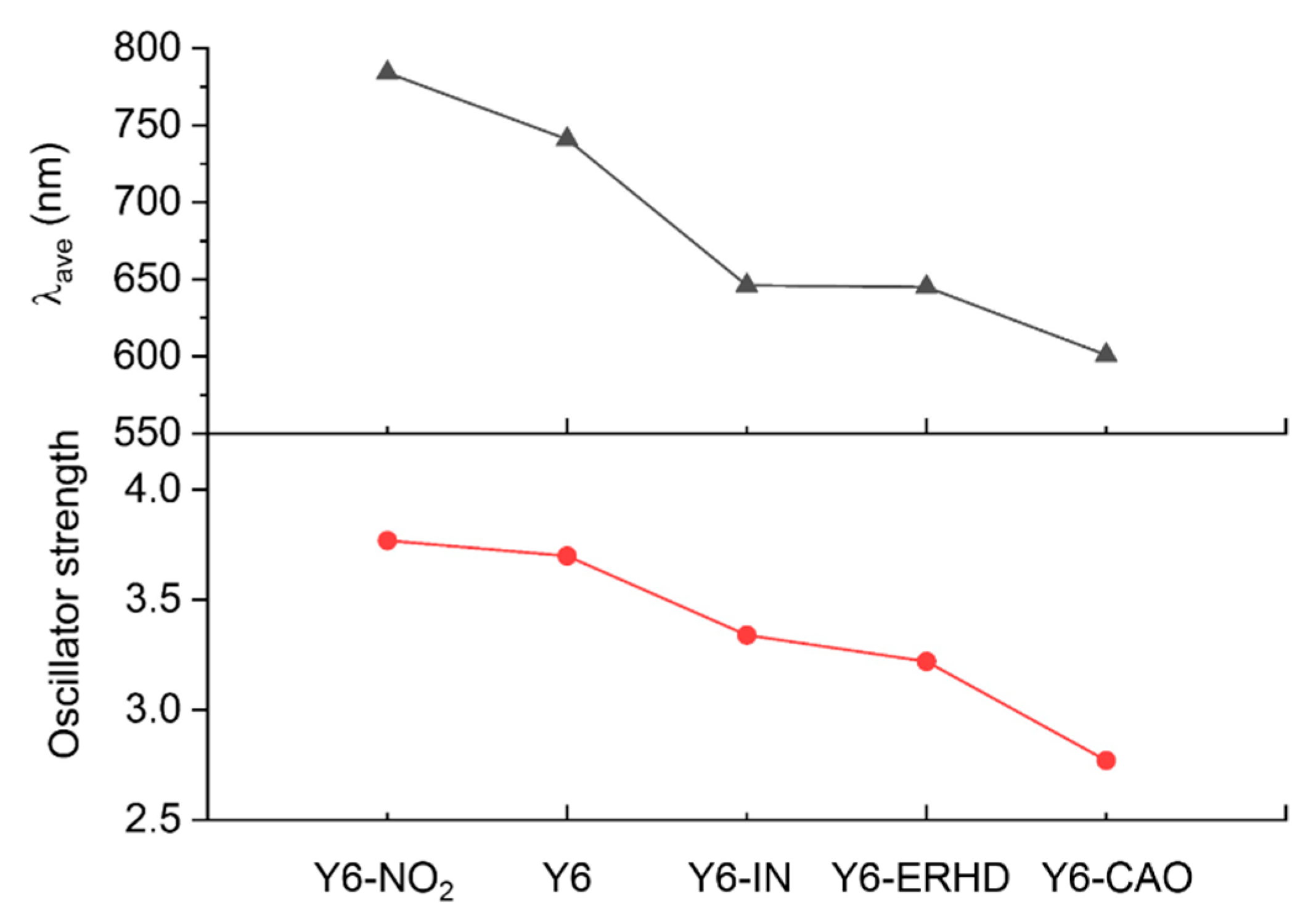
2.3. UV-Vis Spectra
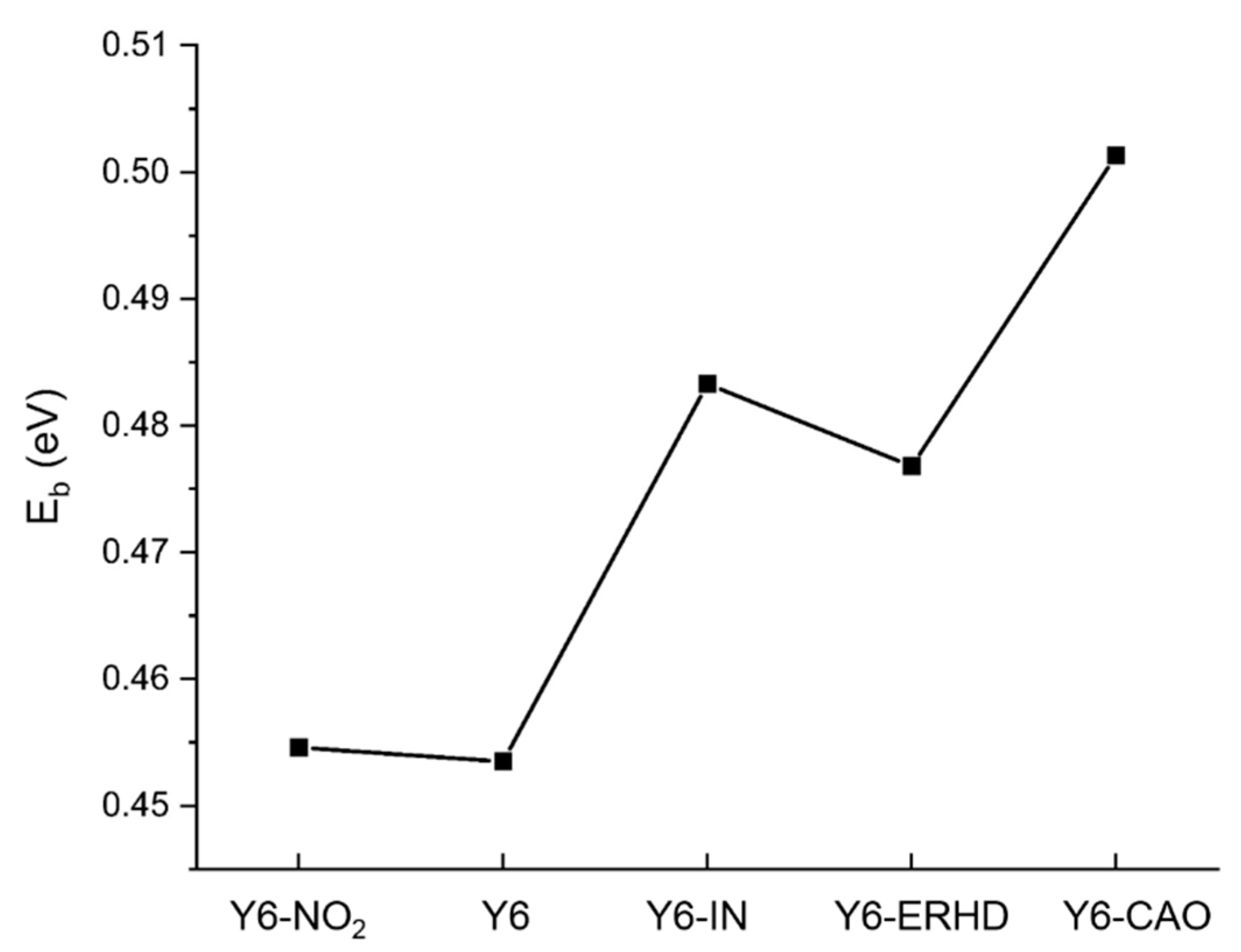
2.4. Exciton Binding Energy
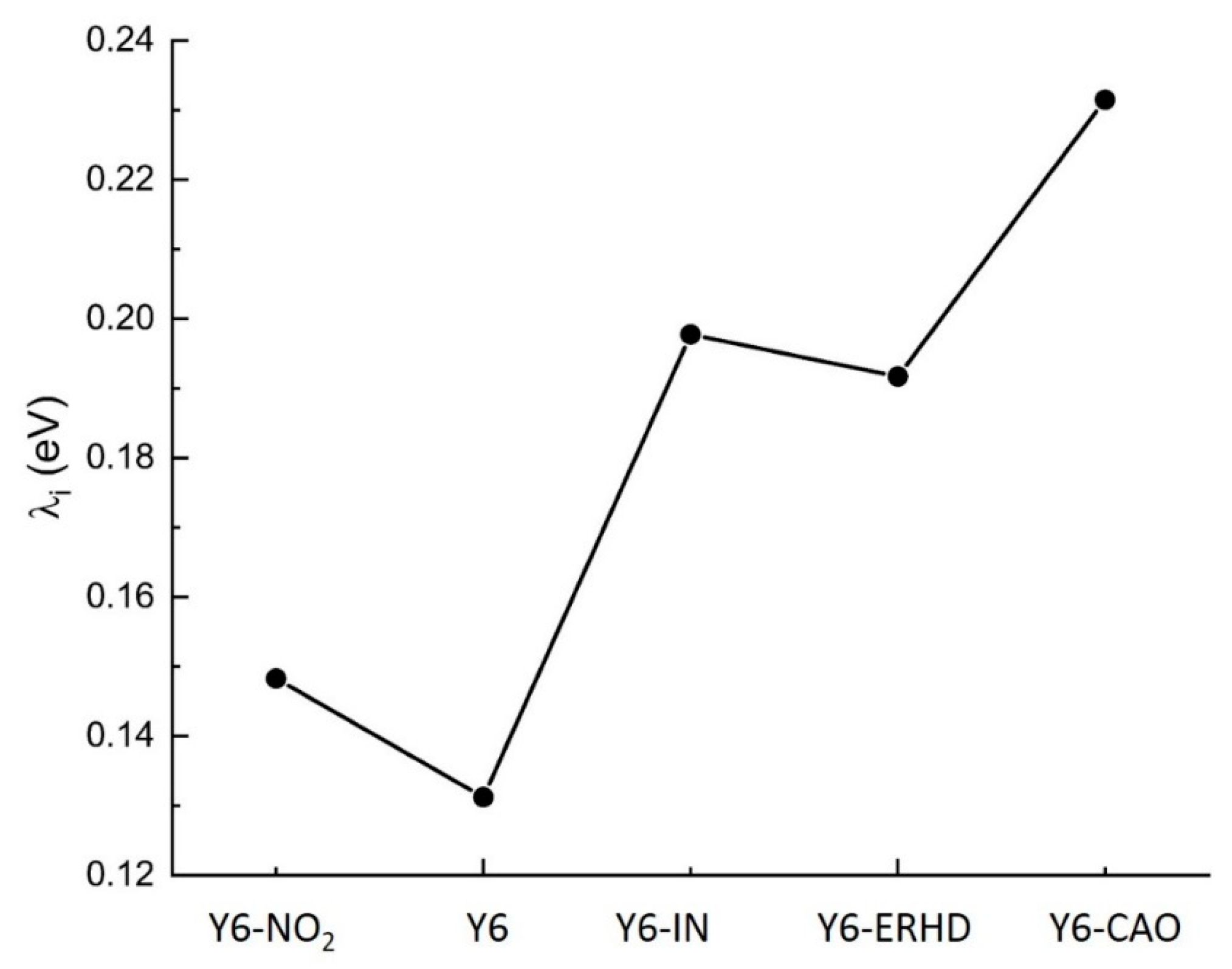
2.5. Reorganization Energy of Electron Transfer
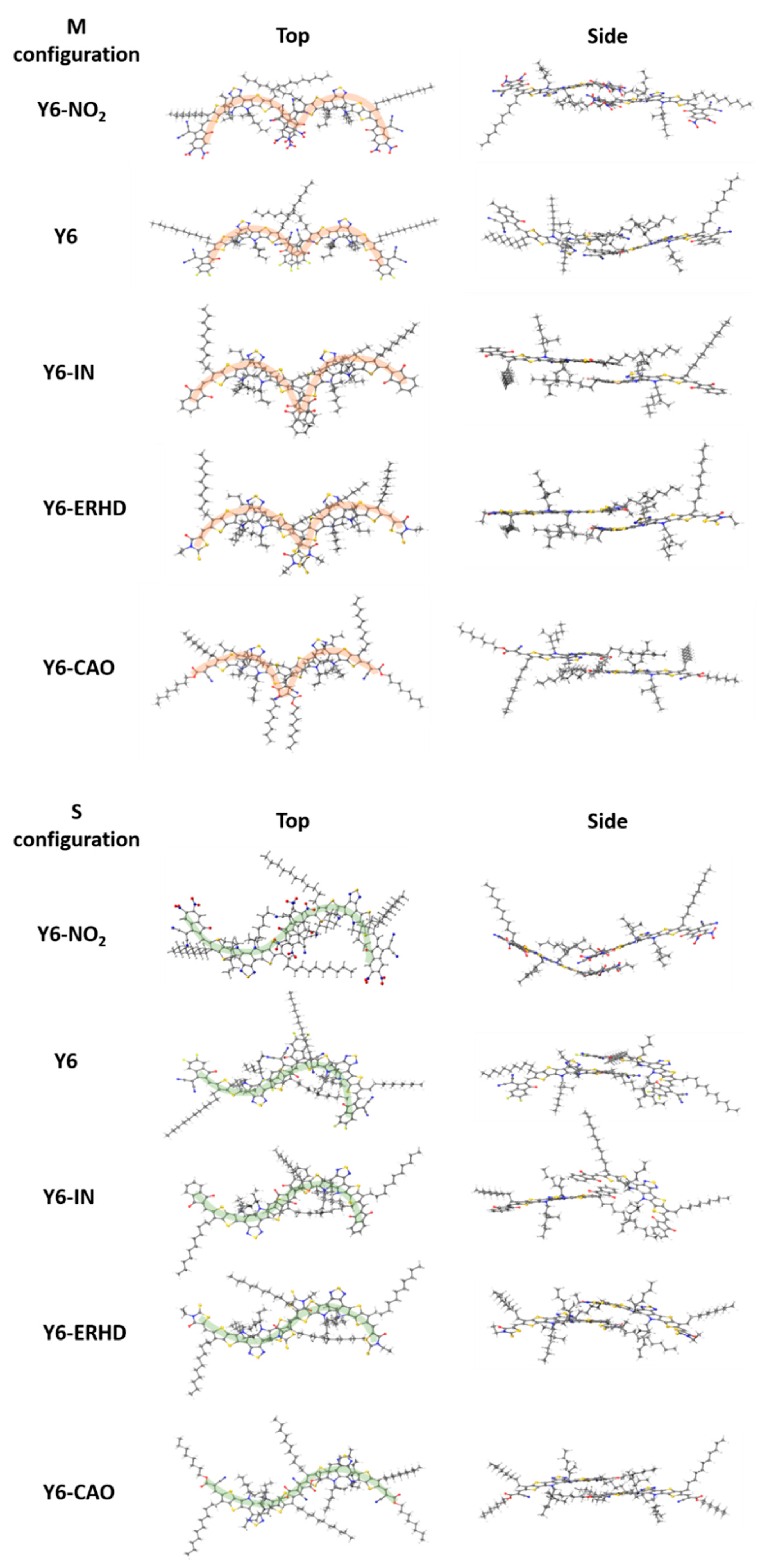
2.6. Electron/Hole Distribution of Monomers
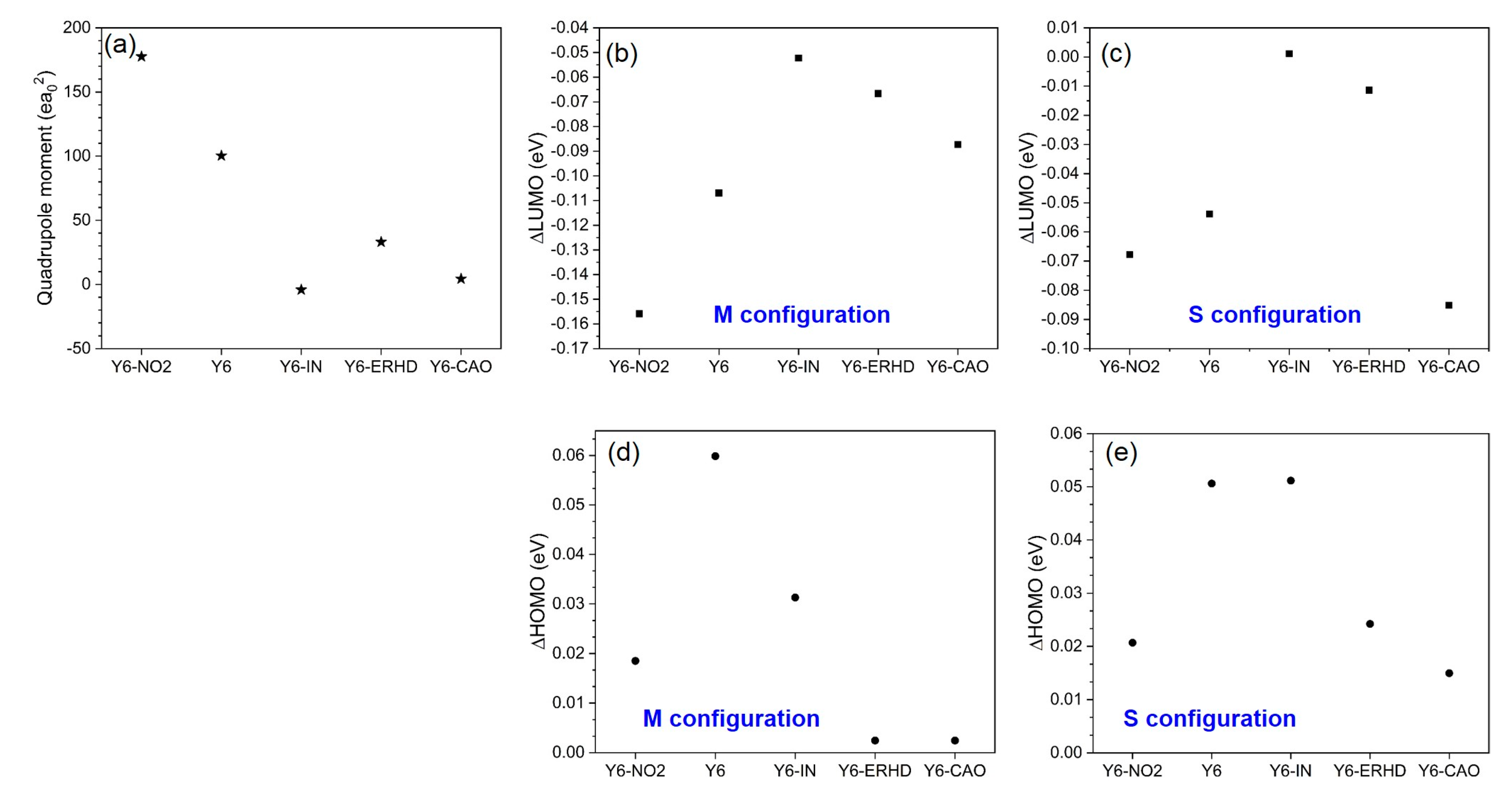
2.7. Quadrupole Moment along π-π Stacking Direction, ΔLUMO and ΔHOMO
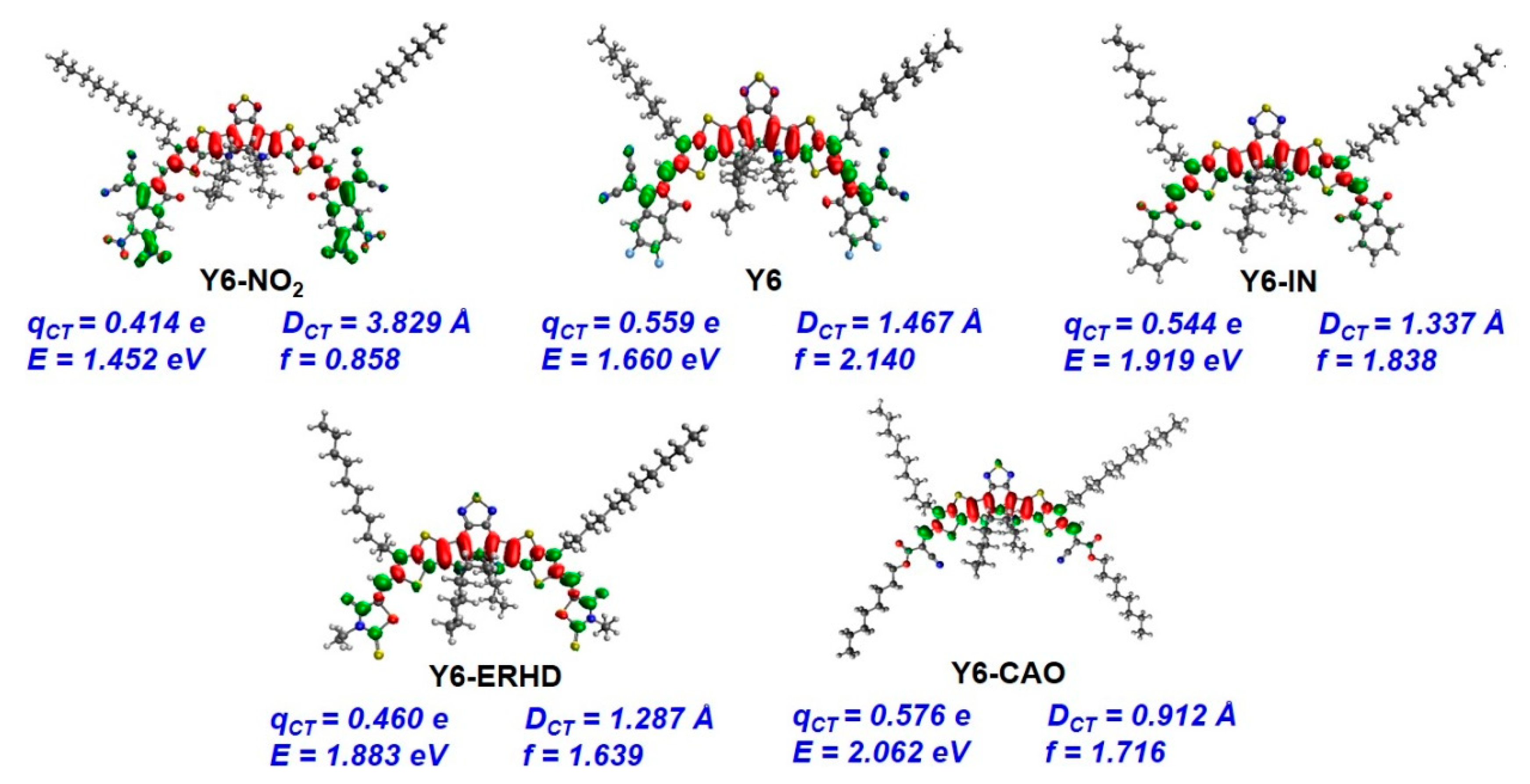
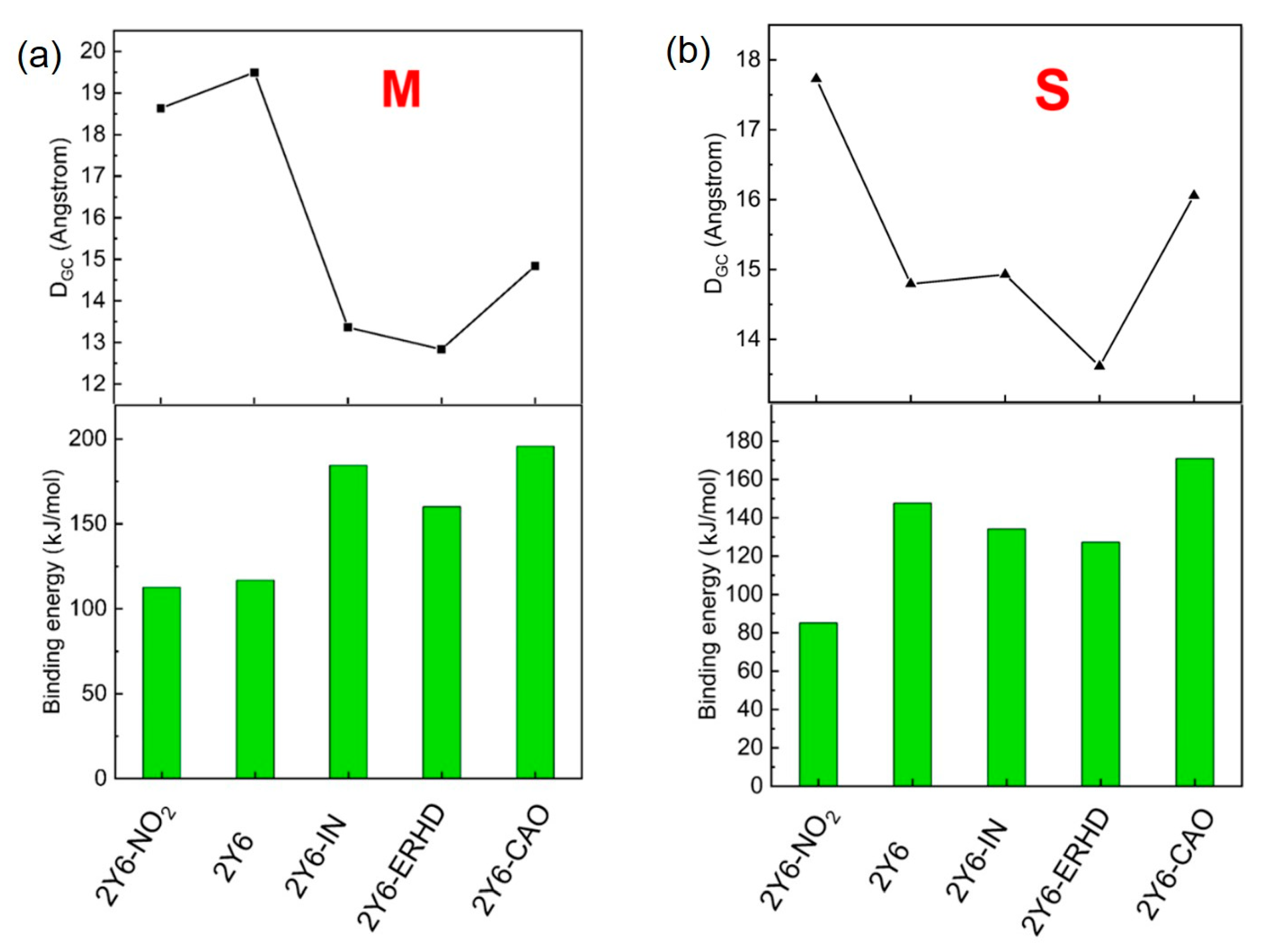
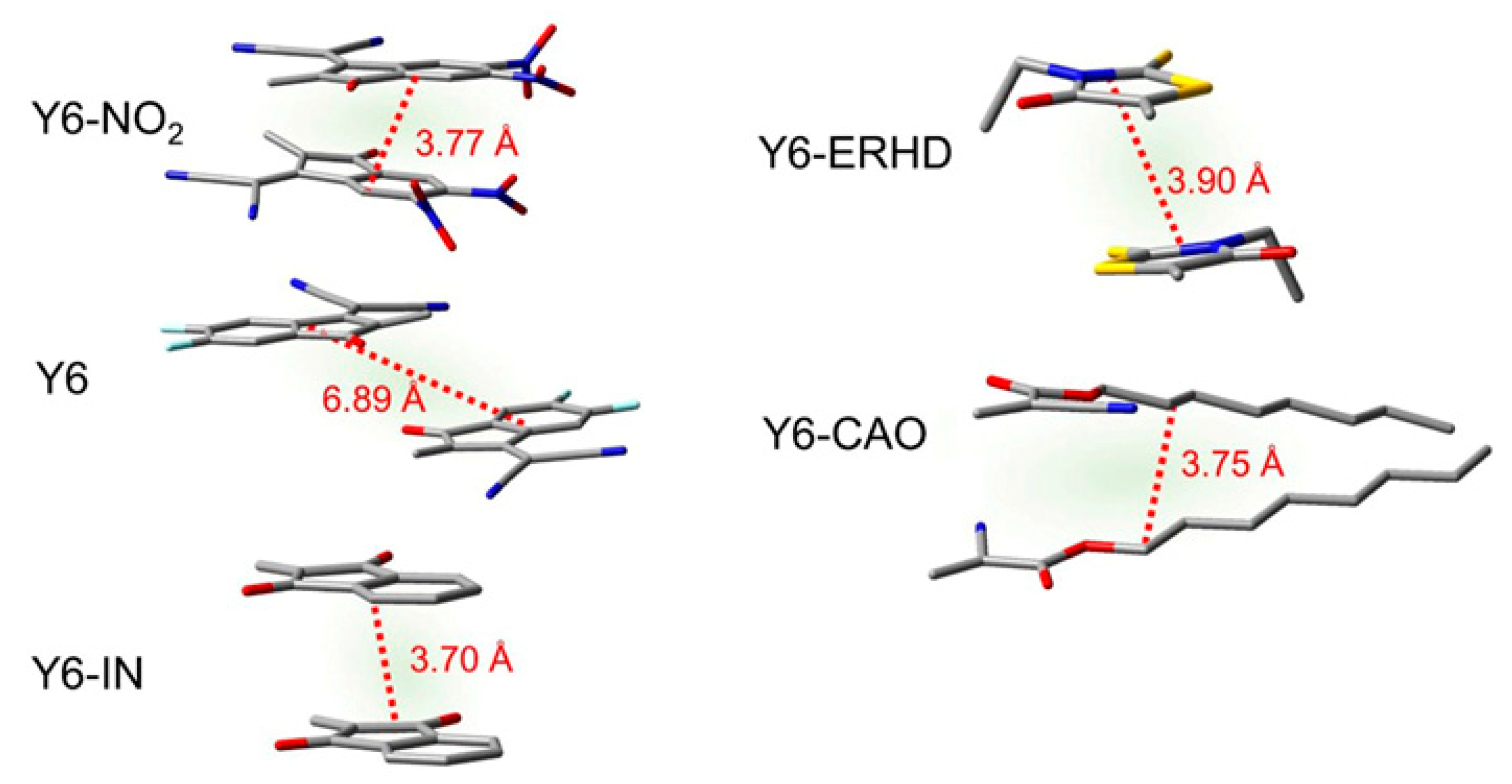
2.8. Binding Energy of Dimers and Intermolecular Distance of Charge Transfer
2.9. Charge Carrier Mobility
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, Y.; Xu, Y.; Yao, H.; Bi, P.; Hong, L.; Zhang, J.; Zu, Y.; Zhang, T.; Qin, J.; Ren, J.; et al. Single-Junction Organic Photovoltaic Cell with 19% Efficiency. Adv. Mater. 2021, 33, e2102420. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, Y.; Zhou, L.; Zhang, G.; Yip, H.-L.; Lau, T.-K.; Lu, X.; Zhu, C.; Peng, H.; Johnson, P.A.; et al. Single-Junction Organic Solar Cell with over 15% Efficiency Using Fused-Ring Acceptor with Electron-Deficient Core. Joule 2019, 3, 1140–1151. [Google Scholar] [CrossRef]
- Hou, J.; Inganas, O.; Friend, R.H.; Gao, F. Organic solar cells based on non-fullerene acceptors. Nat. Mater. 2018, 17, 119–128. [Google Scholar] [CrossRef]
- Fei, Z.; Eisner, F.D.; Jiao, X.; Azzouzi, M.; Rohr, J.A.; Han, Y.; Shahid, M.; Chesman, A.S.R.; Easton, C.D.; McNeill, C.R.; et al. An Alkylated Indacenodithieno[3,2-b]thiophene-Based Nonfullerene Acceptor with High Crystallinity Exhibiting Single Junction Solar Cell Efficiencies Greater than 13% with Low Voltage Losses. Adv. Mater. 2018, 30, 1705209. [Google Scholar] [CrossRef]
- Hong, L.; Yao, H.; Wu, Z.; Cui, Y.; Zhang, T.; Xu, Y.; Yu, R.; Liao, Q.; Gao, B.; Xian, K.; et al. Eco-Compatible Solvent-Processed Organic Photovoltaic Cells with Over 16% Efficiency. Adv. Mater. 2019, 31, 1903441. [Google Scholar] [CrossRef]
- Li, C.; Zhou, J.; Song, J.; Xu, J.; Zhang, H.; Zhang, X.; Guo, J.; Zhu, L.; Wei, D.; Han, G.; et al. Non-fullerene acceptors with branched side chains and improved molecular packing to exceed 18% efficiency in organic solar cells. Nat. Energy 2021, 6, 605–613. [Google Scholar] [CrossRef]
- Gao, W.; Fu, H.; Li, Y.; Lin, F.; Sun, R.; Wu, Z.; Wu, X.; Zhong, C.; Min, J.; Luo, J.; et al. Asymmetric Acceptors Enabling Organic Solar Cells to Achieve an over 17% Efficiency: Conformation Effects on Regulating Molecular Properties and Suppressing Nonradiative Energy Loss. Adv. Energy Mater. 2021, 11, 2003177. [Google Scholar] [CrossRef]
- Wang, R.; Yuan, J.; Wang, R.; Han, G.; Huang, T.; Huang, W.; Xue, J.; Wang, H.C.; Zhang, C.; Zhu, C.; et al. Rational Tuning of Molecular Interaction and Energy Level Alignment Enables High-Performance Organic Photovoltaics. Adv. Mater. 2019, 31, 1904215. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Y.; Cai, G.; Zhang, Y.; Lu, X.; Lin, Y. Selenium Heterocyclic Electron Acceptor with Small Urbach Energy for As-Cast High-Performance Organic Solar Cells. J. Am. Chem. Soc. 2020, 142, 18741–18745. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yao, H.; Zhang, J.; Zhang, T.; Wang, Y.; Hong, L.; Xian, K.; Xu, B.; Zhang, S.; Peng, J.; et al. Over 16% efficiency organic photovoltaic cells enabled by a chlorinated acceptor with increased open-circuit voltages. Nat. Commun. 2019, 10, 2515. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Yang, Q.; Zheng, Y.; Tang, H.; Chung, S.; Singh, R.; Lv, J.; Fu, J.; Kan, Z.; Qin, B.; et al. 15.3% Efficiency All-Small-Molecule Organic Solar Cells Achieved by a Locally Asymmetric F, Cl Disubstitution Strategy. Adv. Sci. 2021, 8, 2004262. [Google Scholar] [CrossRef]
- Lu, H.; Jin, H.; Huang, H.; Liu, W.; Tang, Z.; Zhang, J.; Bo, Z. High-Efficiency Organic Solar Cells Based on Asymmetric Acceptors Bearing One 3D Shape-Persistent Terminal Group. Adv. Funct. Mater. 2021, 31, 2103445. [Google Scholar] [CrossRef]
- Li, S.; Zhan, L.; Yao, N.; Xia, X.; Chen, Z.; Yang, W.; He, C.; Zuo, L.; Shi, M.; Zhu, H.; et al. Unveiling structure-performance relationships from multi-scales in non-fullerene organic photovoltaics. Nat. Commun. 2021, 12, 4627. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Lai, H.; Chen, H.; Zhu, Y.; Pu, M.; Zheng, N.; He, F. Over 17.5% efficiency ternary organic solar cells with enhanced photon utilization via a medium band gap non-fullerene acceptor. J. Mater. Chem. A 2021, 9, 16418–16426. [Google Scholar] [CrossRef]
- Jones, R.O.; Gunnarsson, O. The density functional formalism, its applications and prospects. Rev. Mod. Phys. 1989, 61, 689–746. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density functional theory for time-dependent systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084–106. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Yi, X.; Huang, T.Y.; Zheng, Z.; Zhang, J.; Salehi, A.; Coropceanu, V.; Ho, C.H.Y.; Marder, S.R.; Toney, M.F.; et al. Donor Conjugated Polymers with Polar Side Chain Groups: The Role of Dielectric Constant and Energetic Disorder on Photovoltaic Performance. Adv. Funct. Mater. 2018, 28, 1803418. [Google Scholar] [CrossRef]
- Takacs, C.J.; Sun, Y.; Welch, G.C.; Perez, L.A.; Liu, X.; Wen, W.; Bazan, G.C.; Heeger, A.J. Solar cell efficiency, self-assembly, and dipole-dipole interactions of isomorphic narrow-band-gap molecules. J. Am. Chem. Soc. 2012, 134, 16597–16606. [Google Scholar] [CrossRef]
- Kraner, S.; Prampolini, G.; Cuniberti, G. Exciton Binding Energy in Molecular Triads. J. Phys. Chem. C 2017, 121, 17088–17095. [Google Scholar] [CrossRef]
- Fu, Y.; Lee, T.H.; Chin, Y.C.; Pacalaj, R.A.; Labanti, C.; Park, S.Y.; Dong, Y.; Cho, H.W.; Kim, J.Y.; Minami, D.; et al. Molecular orientation-dependent energetic shifts in solution-processed non-fullerene acceptors and their impact on organic photovoltaic performance. Nat. Commun. 2023, 14, 1870. [Google Scholar] [CrossRef]
- Zhu, W.; Spencer, A.P.; Mukherjee, S.; Alzola, J.M.; Sangwan, V.K.; Amsterdam, S.H.; Swick, S.M.; Jones, L.O.; Heiber, M.C.; Herzing, A.A.; et al. Crystallography, Morphology, Electronic Structure, and Transport in Non-Fullerene/Non-Indacenodithienothiophene Polymer:Y6 Solar Cells. J. Am. Chem. Soc. 2020, 142, 14532–14547. [Google Scholar] [CrossRef]
- Zhong, S.; Yap, B.K.; Zhong, Z.; Ying, L. Review on Y6-Based Semiconductor Materials and Their Future Development via Machine Learning. Crystals 2022, 12, 168. [Google Scholar] [CrossRef]
- Cornil, J.; Lemaur, V.; Calbert, J.P.; Brédas, J.L. Charge Transport in Discotic Liquid Crystals: A Molecular Scale Description. Adv. Mater. 2002, 14, 726–729. [Google Scholar] [CrossRef]
- Dennington, R.K.T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 2009 Revision D.01; Gaussian: Wallingford, CT, USA, 2009. [Google Scholar]
- Jacobson, L.D.; Herbert, J.M. Polarization-Bound Quasi-Continuum States Are Responsible for the “Blue Tail” in the Optical Absorption Spectrum of the Aqueous Electron. J. Am. Chem. Soc. 2010, 132, 10000–10002. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Geva, E.; Dunietz, B.D. Solvated Charge Transfer States of Functionalized Anthracene and Tetracyanoethylene Dimers: A Computational Study Based on a Range Separated Hybrid Functional and Charge Constrained Self-Consistent Field with Switching Gaussian Polarized Continuum Models. J. Chem. Theory Comput. 2013, 9, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab-Initio Calculation of Vibrational Absorption and Circular-Dichroism Spectra Using Density-Functional Force-Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular-Orbital Methods.XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular-Orbital Studies of Organic-Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Al-shamiri, H.A.S.; Melhi, S.; Alosaimi, E.H.; El-Gammal, B.; Elhouichet, H.; Sakr, M.A.S.; Abou Kana, M.T.H.; Kandel, H.M. Experimental and theoretical study of optical properties of pyrromethene (PM-597) laser dye in binary eco-friendly solvent. J. Phys. Org. Chem. 2023, 36, e4445. [Google Scholar] [CrossRef]
- McCarthy, M.; Lee, K.L.K. Molecule Identification with Rotational Spectroscopy and Probabilistic Deep Learning. J. Phys. Chem. A 2020, 124, 3002–3017. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.T.; Zhong, C.; Sun, Z.R. Recent Advances in the Optimally “Tuned” Range-Separated Density Functional Theory. Acta Phys.-Chim. Sin. 2016, 32, 2197–2208. [Google Scholar] [CrossRef]
- Qiu, W.; Zheng, S. Designing and Screening High-Performance Non-Fullerene Acceptors: A Theoretical Exploration of Modified Y6. Solar Rrl 2021, 5, 2100023. [Google Scholar] [CrossRef]
- Shukla, S.; Srivastava, A.; Srivastava, K.; Tandon, P.; Jamalis, J.; Singh, R.B. Non-covalent interactions and spectroscopic study of chalcone derivative 1-(4-chlorophenyl)-3-(5-methylfuran-2-yl) prop-2-en-1-one. J. Mol. Struct. 2020, 1201, 127145. [Google Scholar] [CrossRef]
- Lee, J.C.; Chai, J.D.; Lin, S.T. Assessment of density functional methods for exciton binding energies and related optoelectronic properties. RSC Adv. 2015, 5, 101370–101376. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Blackstock, S.C.; Kim, Y. Estimation of Inner Shell Marcus Terms for Amino Nitrogen Compounds by Molecular Orbital Calculations. J. Am. Chem. Soc. 1987, 109, 677–682. [Google Scholar] [CrossRef]
- Sammut, D.; Bugeja, N.; Szacilowski, K.; Magri, D.C. Molecular engineering of fluorescent bichromophore 1,3,5-triaryl-Delta(2)-pyrazoline and 4-amino-1,8-naphthalimide molecular logic gates. New J. Chem. 2022, 46, 15042–15051. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Marcus, R.A.; Sutin, N. Electron transfer in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Komornicki, A.; Dixon, D.A. Accurate proton affinities:Abinitioproton binding energies for N2, CO, CO2, and CH4. J. Chem. Phys. 1992, 97, 1087–1094. [Google Scholar] [CrossRef]
- Valeev, E.F.; Coropceanu, V.; da Silva Filho, D.A.; Salman, S.; Brédas, J.L. Effect of Electronic Polarization on Charge-Transport Parameters in Molecular Organic Semiconductors. J. Am. Chem. Soc. 2006, 128, 9882–9886. [Google Scholar] [CrossRef]
- Mkoma, S.L.; Msambwa, Y.; Jacob, F.R.; Kiruri, L.W.; Kinunda, G.A.; Mlowe, S.; Deogratias, G. Optical and electronic properties of para-functionalized triphenylamine-based dyes: A theoretical study. Struct. Chem. 2022, 33, 409–419. [Google Scholar] [CrossRef]
- Coropceanu, V.; Cornil, J.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Bredas, J.L. Charge Transport in Organic Semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef] [PubMed]
- An, B.; Wen, K.; Feng, S.; Pan, X.; Wu, W.; Guo, X.; Zhang, J. Theoretical insights into the 1D-charge transport properties in a series of hexaazatrinaphthylene-based discotic molecules. J. Comput. Chem. 2018, 39, 773–779. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Y6-NO2 | Y6 | Y6-IN | Y6-ERHD | Y6-CAO |
|---|---|---|---|---|
| 15.56 | 0.82 | 4.16 | 6.31 | 2.96 |
| System | λi | λo | λ |
|---|---|---|---|
| Y6-NO2 | 0.15 | 0.19 | 0.34 |
| Y6 | 0.13 | 0.16 | 0.29 |
| Y6-IN | 0.20 | 0.15 | 0.35 |
| Y6-ERHD | 0.19 | 0.14 | 0.33 |
| Y6-CAO | 0.23 | 0.15 | 0.38 |
| r | |Ve| | λ | ke | μe | |
|---|---|---|---|---|---|
| Y6-NO2-M-dimer | 18.63 | 27.89 | 0.33 | 8.69 × 1011 | 5.87 × 10−1 |
| Y6-S-dimer | 14.79 | 11.43 | 0.29 | 2.42 × 1011 | 1.03 × 10−1 |
| Y6-IN-M-dimer | 13.37 | 33.75 | 0.35 | 1.11 × 1012 | 3.84 × 10−1 |
| Y6-ERHD-M-dimer | 12.84 | 1.57 | 0.33 | 2.83 × 109 | 1.00 × 10−3 |
| Y6-CAO-M-dimer | 14.84 | 37.00 | 0.38 | 8.77 × 1011 | 3.76 × 10−1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, Y.; Xu, C.; Zheng, S. Increasing Charge Carrier Mobility through Modifications of Terminal Groups of Y6: A Theoretical Study. Int. J. Mol. Sci. 2023, 24, 8610. https://doi.org/10.3390/ijms24108610
Xiang Y, Xu C, Zheng S. Increasing Charge Carrier Mobility through Modifications of Terminal Groups of Y6: A Theoretical Study. International Journal of Molecular Sciences. 2023; 24(10):8610. https://doi.org/10.3390/ijms24108610
Chicago/Turabian StyleXiang, Yunjie, Chunlin Xu, and Shaohui Zheng. 2023. "Increasing Charge Carrier Mobility through Modifications of Terminal Groups of Y6: A Theoretical Study" International Journal of Molecular Sciences 24, no. 10: 8610. https://doi.org/10.3390/ijms24108610