
TRPC Channels in the Physiology and Pathophysiology of the Renal Tubular System: What Do We Know?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
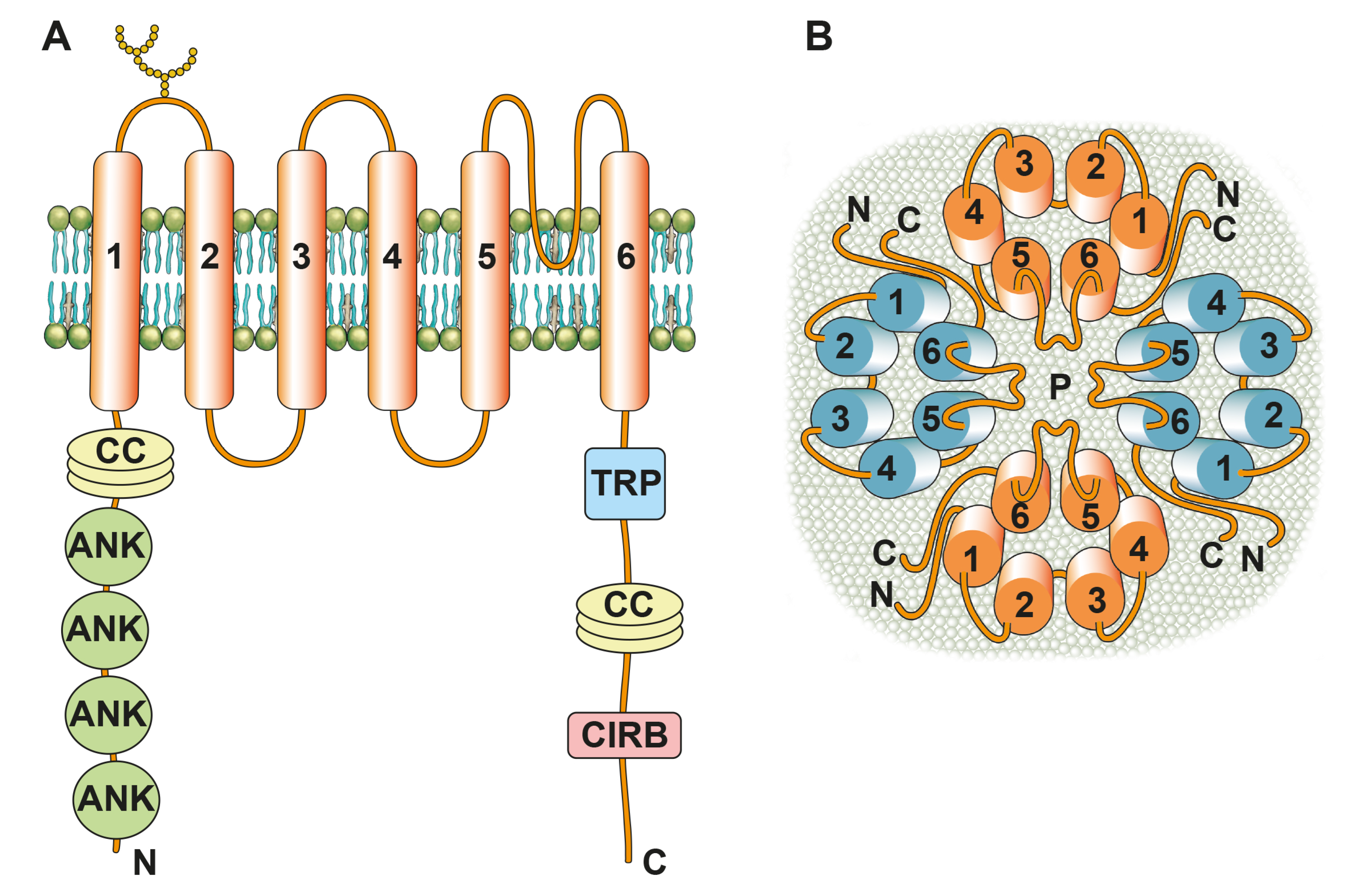
:1. Introduction
2. TRPC6 Is a Controversial Player in Tubular Cells Experiencing Ischemia-Reperfusion Injuries
3. TRPC6 Drives Tumorigenesis and the Progression of Renal Cell Carcinoma
4. TRPC3 Is a Cytoprotective Key Player in Ca2+ Reabsorption of the Proximal Tubule
5. TRPC3 Is Involved in Vasopressin-Dependent AQP-2 Trafficking, Osmosensation, and Ca2+ Reabsorption in the Collecting Duct
6. Mitochondrial TRPC3 Drives Detrimental Calcium Uptake and Mediates Cell Proliferation in Autosomal Dominant Polycystic Kidney Disease-like Conditions
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grayson, T.H.; Murphy, T.V.; Sandow, S.L. Transient receptor potential canonical type 3 channels: Interactions, role and relevance—A vascular focus. Pharmacol. Ther. 2017, 174, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Cheng, X.; Tian, J.; Xiao, Y.; Tian, T.; Xu, F.; Hong, X.; Zhu, M.X. TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. 2020, 209, 107497. [Google Scholar] [CrossRef]
- Tang, Q.; Guo, W.; Zheng, L.; Wu, J.X.; Liu, M.; Zhou, X.; Zhang, X.; Chen, L. Structure of the receptor-activated human TRPC6 and TRPC3 ion channels. Cell Res. 2018, 28, 746–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, S.A.; Guzman, G.A.; Wissenbach, U.; Philipp, S.E.; Zhu, M.X.; Bruns, D.; Cavalie, A. TRPC5 is a Ca2+-activated channel functionally coupled to Ca2+-selective ion channels. J. Biol. Chem. 2009, 284, 34423–34432. [Google Scholar] [CrossRef] [Green Version]
- Gualdani, R.; Gailly, P. How TRPC Channels Modulate Hippocampal Function. Int. J. Mol. Sci. 2020, 21, 3915. [Google Scholar] [CrossRef] [PubMed]
- Canales Coutino, B.; Mayor, R. Mechanosensitive ion channels in cell migration. Cells Dev. 2021, 166, 203683. [Google Scholar] [CrossRef]
- Ma, R.; Chaudhari, S.; Li, W. Canonical Transient Receptor Potential 6 Channel: A New Target of Reactive Oxygen Species in Renal Physiology and Pathology. Antioxid Redox Signal. 2016, 25, 732–748. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, R.; Mori, Y. Transient receptor potential (TRP) channels: Biosensors for redox environmental stimuli and cellular status. Free Radic. Biol. Med. 2020, 146, 36–44. [Google Scholar] [CrossRef]
- Fan, C.; Choi, W.; Sun, W.; Du, J.; Lu, W. Structure of the human lipid-gated cation channel TRPC3. Elife 2018, 7, e36852. [Google Scholar] [CrossRef]
- Hafner, S.; Urban, N.; Schaefer, M. Discovery and characterization of a positive allosteric modulator of transient receptor potential canonical 6 (TRPC6) channels. Cell Calcium 2019, 78, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wie, J.; Jeong, S.; Kwak, M.; Myeong, J.; Chae, M.; Park, J.K.; Lee, S.W.; So, I. The regulation of transient receptor potential canonical 4 (TRPC4) channel by phosphodiesterase 5 inhibitor via the cyclic guanosine 3’5’-monophosphate. Pflugers Arch. 2017, 469, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhuang, Y.; Zhou, Z.; Zhao, J.; Chen, Q.; Zheng, J. NF-kappaB dependent up-regulation of TRPC6 by Abeta in BV-2 microglia cells increases COX-2 expression and contributes to hippocampus neuron damage. Neurosci. Lett. 2017, 651, 1–8. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, C.; Zhang, D.; Xin, Y.; Li, J.; Zhang, Y.; Chu, X. Increased TRPC6 expression is associated with tubular epithelial cell proliferation and inflammation in diabetic nephropathy. Mol. Immunol. 2018, 94, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Spires, D.; Manis, A.D.; Staruschenko, A. Ion channels and transporters in diabetic kidney disease. Curr. Top. Membr. 2019, 83, 353–396. [Google Scholar] [CrossRef] [PubMed]
- Staruschenko, A.; Spires, D.; Palygin, O. Role of TRPC6 in Progression of Diabetic Kidney Disease. Curr. Hypertens. Rep. 2019, 21, 48. [Google Scholar] [CrossRef]
- Zhou, L.F.; Chen, Q.Z.; Yang, C.T.; Fu, Z.D.; Zhao, S.T.; Chen, Y.; Li, S.N.; Liao, L.; Zhou, Y.B.; Huang, J.R.; et al. TRPC6 contributes to LPS-induced inflammation through ERK1/2 and p38 pathways in bronchial epithelial cells. Am. J. Physiol. Cell Physiol. 2018, 314, C278–C288. [Google Scholar] [CrossRef] [Green Version]
- Asghar, M.Y.; Tornquist, K. Transient Receptor Potential Canonical (TRPC) Channels as Modulators of Migration and Invasion. Int. J. Mol. Sci. 2020, 21, 1739. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, G.; Wedel, B.J.; Aziz, O.; Trebak, M.; Putney, J.W., Jr. The mammalian TRPC cation channels. Biochim. Biophys. Acta 2004, 1742, 21–36. [Google Scholar] [CrossRef] [Green Version]
- Eder, P.; Molkentin, J.D. TRPC channels as effectors of cardiac hypertrophy. Circ. Res. 2011, 108, 265–272. [Google Scholar] [CrossRef]
- Shi, J.; Geshi, N.; Takahashi, S.; Kiyonaka, S.; Ichikawa, J.; Hu, Y.; Mori, Y.; Ito, Y.; Inoue, R. Molecular determinants for cardiovascular TRPC6 channel regulation by Ca2+/calmodulin-dependent kinase II. J. Physiol. 2013, 591, 2851–2866. [Google Scholar] [CrossRef]
- Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC Channels in the SOCE Scenario. Cells 2020, 9, 126. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Sooch, G.; Demaree, I.S.; White, F.A.; Obukhov, A.G. Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells 2020, 9, 1983. [Google Scholar] [CrossRef]
- Reilly, R.F.; Ellison, D.H. Mammalian distal tubule: Physiology, pathophysiology, and molecular anatomy. Physiol. Rev. 2000, 80, 277–313. [Google Scholar] [CrossRef] [Green Version]
- Moor, M.B.; Bonny, O. Ways of calcium reabsorption in the kidney. Am. J. Physiol. Renal Physiol. 2016, 310, F1337–F1350. [Google Scholar] [CrossRef] [Green Version]
- Vincent, J.L.; Su, F. Physiology and pathophysiology of the vasopressinergic system. Best Pract. Res. Clin. Anaesthesiol. 2008, 22, 243–252. [Google Scholar] [CrossRef]
- Ames, M.K.; Atkins, C.E.; Pitt, B. The renin-angiotensin-aldosterone system and its suppression. J. Vet. Intern. Med. 2019, 33, 363–382. [Google Scholar] [CrossRef] [Green Version]
- Gumbert, S.D.; Kork, F.; Jackson, M.L.; Vanga, N.; Ghebremichael, S.J.; Wang, C.Y.; Eltzschig, H.K. Perioperative Acute Kidney Injury. Anesthesiology 2020, 132, 180–204. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Keddis, M.T.; Rule, A.D. Nephrolithiasis and loss of kidney function. Curr. Opin. Nephrol. Hypertens. 2013, 22, 390–396. [Google Scholar] [CrossRef]
- Awuah Boadi, E.; Shin, S.; Yeroushalmi, S.; Choi, B.E.; Li, P.; Bandyopadhyay, B.C. Modulation of Tubular pH by Acetazolamide in a Ca2+ Transport Deficient Mice Facilitates Calcium Nephrolithiasis. Int. J. Mol. Sci. 2021, 22, 3050. [Google Scholar] [CrossRef]
- Shin, S.; Ibeh, C.L.; Awuah Boadi, E.; Choi, B.E.; Roy, S.K.; Bandyopadhyay, B.C. Hypercalciuria switches Ca2+ signaling in proximal tubular cells, induces oxidative damage to promote calcium nephrolithiasis. Genes Dis. 2022, 9, 531–548. [Google Scholar] [CrossRef]
- Padala, S.A.; Kallam, A. Clear Cell Renal Carcinoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef] [Green Version]
- Hall, G.; Wang, L.; Spurney, R.F. TRPC Channels in Proteinuric Kidney Diseases. Cells 2019, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Englisch, C.N.; Röhricht, D.; Walz, M.; Junker, K.; Beckmann, A.; Meier, C.; Paulsen, F.; Jung, M.; Tschernig, T. TRPC6 Is Found in Distinct Compartments of the Human Kidney. Int. J. Transl. Med. 2022, 2, 156–163. [Google Scholar] [CrossRef]
- Ramkumar, N.; Gao, Y.; Kohan, D.E. Characterization of flow-regulated cortical collecting duct endothelin-1 production. Physiol. Rep. 2017, 5, e13126. [Google Scholar] [CrossRef]
- Dryer, S.E.; Roshanravan, H.; Kim, E.Y. TRPC channels: Regulation, dysregulation and contributions to chronic kidney disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1041–1066. [Google Scholar] [CrossRef]
- Staruschenko, A.; Ma, R.; Palygin, O.; Dryer, S.E. Ion channels and channelopathies in glomeruli. Physiol. Rev. 2023, 103, 787–854. [Google Scholar] [CrossRef]
- Guan, X.; Qian, Y.; Shen, Y.; Zhang, L.; Du, Y.; Dai, H.; Qian, J.; Yan, Y. Autophagy protects renal tubular cells against ischemia / reperfusion injury in a time-dependent manner. Cell. Physiol. Biochem. 2015, 36, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin. Pract. 2012, 120, c179–c184. [Google Scholar] [CrossRef]
- Zheng, Z.; Tsvetkov, D.; Bartolomaeus, T.U.P.; Erdogan, C.; Krugel, U.; Schleifenbaum, J.; Schaefer, M.; Nurnberg, B.; Chai, X.; Ludwig, F.A.; et al. Role of TRPC6 in kidney damage after acute ischemic kidney injury. Sci. Rep. 2022, 12, 3038. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Livingston, M.J.; Dong, Z. Autophagy in acute kidney injury. Semin. Nephrol. 2014, 34, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Skrypnyk, N.I.; Harris, R.C.; de Caestecker, M.P. Ischemia-reperfusion model of acute kidney injury and post injury fibrosis in mice. J. Vis. Exp. 2013, 9, e50495. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Jurcau, A.; Ardelean, I.A. Molecular pathophysiological mechanisms of ischemia/reperfusion injuries after recanalization therapy for acute ischemic stroke. J. Integr. Neurosci. 2021, 20, 727–744. [Google Scholar] [CrossRef]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef]
- Yan, H.F.; Tuo, Q.Z.; Yin, Q.Z.; Lei, P. The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zool. Res. 2020, 41, 220–230. [Google Scholar] [CrossRef]
- Gores, G.J.; Nieminen, A.L.; Fleishman, K.E.; Dawson, T.L.; Herman, B.; Lemasters, J.J. Extracellular acidosis delays onset of cell death in ATP-depleted hepatocytes. Am. J. Physiol. 1988, 255, C315–C322. [Google Scholar] [CrossRef] [PubMed]
- Raat, N.J.; Shiva, S.; Gladwin, M.T. Effects of nitrite on modulating ROS generation following ischemia and reperfusion. Adv. Drug Deliv. Rev. 2009, 61, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Bassett, A.L.; Gaide, M.S.; Kozlovskis, P.L.; Myerburg, R.J. Regional changes in intracellular potassium and sodium activity after healing of experimental myocardial infarction in cats. Circ. Res. 1986, 58, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P. Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem. Soc. Trans. 2006, 34, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Nayler, W.G. The role of calcium in the ischemic myocardium. Am. J. Pathol. 1981, 102, 262–270. [Google Scholar] [PubMed]
- Weisfeldt, M.L.; Zweier, J.; Ambrosio, G.; Becker, L.C.; Flaherty, J.T. Evidence that free radicals result in reperfusion injury in heart muscle. Basic Life Sci. 1988, 49, 911–919. [Google Scholar] [CrossRef]
- Zweier, J.L.; Flaherty, J.T.; Weisfeldt, M.L. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. USA 1987, 84, 1404–1407. [Google Scholar] [CrossRef] [Green Version]
- Becker, L.C.; Ambrosio, G. Myocardial consequences of reperfusion. Prog. Cardiovasc. Dis. 1987, 30, 23–44. [Google Scholar] [CrossRef]
- Hess, M.L.; Manson, N.H. Molecular oxygen: Friend and foe. The role of the oxygen free radical system in the calcium paradox, the oxygen paradox and ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 1984, 16, 969–985. [Google Scholar] [CrossRef]
- Dare, A.J.; Bolton, E.A.; Pettigrew, G.J.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015, 5, 163–168. [Google Scholar] [CrossRef]
- Tajima, T.; Yoshifuji, A.; Matsui, A.; Itoh, T.; Uchiyama, K.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. beta-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney Int. 2019, 95, 1120–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhou, S.; He, Y.; Zhao, H.; Mei, M.; Wu, X. Revealing the underlying mechanism of ischemia reperfusion injury using bioinformatics approach. Kidney Blood Press. Res. 2013, 38, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Huang, M.; Zeng, X.; Zhang, Y.; Sun, A.; Wu, Q.; Zhu, L.; Zhao, H.; Liao, Y. The Role of TRPC6 in Renal Ischemia/Reperfusion and Cellular Hypoxia/Reoxygenation Injuries. Front. Mol. Biosci 2021, 8, 698975. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Mei, M.; Ai, S.; Liao, X.; Li, N.; Xiang, S.; Wen, C.; Tao, Y.; Dai, H. TRPC6 inhibits renal tubular epithelial cell pyroptosis through regulating zinc influx and alleviates renal ischemia-reperfusion injury. FASEB J. 2022, 36, e22527. [Google Scholar] [CrossRef]
- Shen, B.; Mei, M.; Pu, Y.; Zhang, H.; Liu, H.; Tang, M.; Pan, Q.; He, Y.; Wu, X.; Zhao, H. Necrostatin-1 Attenuates Renal Ischemia and Reperfusion Injury via Meditation of HIF-1alpha/mir-26a/TRPC6/PARP1 Signaling. Mol. Ther. Nucleic Acids 2019, 17, 701–713. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Winters, A.; Ding, M.; Graham, S.; Akopova, I.; Muallem, S.; Wang, Y.; Hong, J.H.; Gryczynski, Z.; Yang, S.H.; et al. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J. Biol. Chem. 2011, 286, 31799–31809. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Xiao, H.; Zhang, Y.; Zeng, X.; Huang, M.; Chen, X.; Birnbaumer, L.; Liao, Y. Transient receptor potential channel 6 knockdown prevents apoptosis of renal tubular epithelial cells upon oxidative stress via autophagy activation. Cell Death Dis. 2018, 9, 1015. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef] [Green Version]
- Havasi, A.; Dong, Z. Autophagy and Tubular Cell Death in the Kidney. Semin. Nephrol. 2016, 36, 174–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F. Autophagy in renal tubular injury and repair. Acta Physiol. 2017, 220, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Zhao, H.; Shen, B.; Zhou, Q.; Xie, P.; Wu, X. TRPC6 ameliorates renal ischemic reperfusion injury by inducing Zn(2+) influx and activating autophagy to resist necrosis. Ann. Transl. Med. 2022, 10, 249. [Google Scholar] [CrossRef]
- Shen, B.; He, Y.; Zhou, S.; Zhao, H.; Mei, M.; Wu, X. TRPC6 May Protect Renal Ischemia-Reperfusion Injury Through Inhibiting Necroptosis of Renal Tubular Epithelial Cells. Med. Sci. Monit. 2016, 22, 633–641. [Google Scholar] [CrossRef]
- Shen, S.; Jin, Y.; Li, W.; Liu, X.; Zhang, T.; Xia, W.; Wang, Y.; Ma, K. Recombinant human erythropoietin pretreatment attenuates acute renal tubular injury against ischemia-reperfusion by restoring transient receptor potential channel-6 expression and function in collecting ducts. Crit. Care Med. 2014, 42, e663–e672. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Gombedza, F.C.; Bandyopadhyay, B.C. l-ornithine activates Ca2+ signaling to exert its protective function on human proximal tubular cells. Cell. Signal. 2020, 67, 109484. [Google Scholar] [CrossRef]
- Livingstone, C. Zinc: Physiology, deficiency, and parenteral nutrition. Nutr. Clin. Pract. 2015, 30, 371–382. [Google Scholar] [CrossRef]
- Chevallet, M.; Jarvis, L.; Harel, A.; Luche, S.; Degot, S.; Chapuis, V.; Boulay, G.; Rabilloud, T.; Bouron, A. Functional consequences of the over-expression of TRPC6 channels in HEK cells: Impact on the homeostasis of zinc. Metallomics 2014, 6, 1269–1276. [Google Scholar] [CrossRef]
- Liu, J.; Kang, Y.; Yin, S.; Chen, A.; Wu, J.; Liang, H.; Shao, L. Key Role of Microtubule and Its Acetylation in a Zinc Oxide Nanoparticle-Mediated Lysosome-Autophagy System. Small 2019, 15, e1901073. [Google Scholar] [CrossRef]
- Gibon, J.; Tu, P.; Bohic, S.; Richaud, P.; Arnaud, J.; Zhu, M.; Boulay, G.; Bouron, A. The over-expression of TRPC6 channels in HEK-293 cells favours the intracellular accumulation of zinc. Biochim. Biophys. Acta 2011, 1808, 2807–2818. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, M.; He, X.; Ouyang, D. A mini-review on ion fluxes that regulate NLRP3 inflammasome activation. Acta Biochim. Biophys. Sin. 2021, 53, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, G.; Yuan, Y.; Wu, G.; Wang, S.; Yuan, L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-kappaB signaling. Cell Death Dis. 2019, 10, 906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, B.; Guo, C.; Kong, A.; Li, K.; Xie, Y.; Shi, H.; Gu, J. Calcium Signaling Mediates Cell Death and Crosstalk with Autophagy in Kidney Disease. Cells 2021, 10, 3204. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium-cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Monteith, G.R.; Davis, F.M.; Roberts-Thomson, S.J. Calcium channels and pumps in cancer: Changes and consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [Green Version]
- Klatte, T.; Rossi, S.H.; Stewart, G.D. Prognostic factors and prognostic models for renal cell carcinoma: A literature review. World J. Urol. 2018, 36, 1943–1952. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Makhov, P.; Joshi, S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol. Cancer Ther. 2018, 17, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Paner, G.P.; Chumbalkar, V.; Montironi, R.; Moch, H.; Amin, M.B. Updates in Grading of Renal Cell Carcinomas Beyond Clear Cell Renal Cell Carcinoma and Papillary Renal Cell Carcinoma. Adv. Anat. Pathol. 2022, 29, 117–130. [Google Scholar] [CrossRef]
- Nandagopal, L.; Sonpavde, G.P.; Agarwal, N. Investigational MET inhibitors to treat Renal cell carcinoma. Expert Opin. Investig. Drugs 2019, 28, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Hwang, K.H.; Eom, M.; Kim, M.; Park, E.Y.; Jeong, Y.; Park, K.S.; Cha, S.K. WNK1 promotes renal tumor progression by activating TRPC6-NFAT pathway. FASEB J. 2019, 33, 8588–8599. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wang, Y.; Li, X.; Shen, Y.; Yin, M.; Guo, Y.; Diao, L.; Liu, Y.; Yue, D. Critical role of TRPC6 channels in the development of human renal cell carcinoma. Mol. Biol. Rep. 2013, 40, 5115–5122. [Google Scholar] [CrossRef] [PubMed]
- Kochevar, J. Blockage of autonomous growth of ACHN cells by anti-renal cell carcinoma monoclonal antibody 5F4. Cancer Res. 1990, 50, 2968–2972. [Google Scholar]
- Zhan, C.; Shi, Y. TRPC Channels and Cell Proliferation. Adv. Exp. Med. Biol. 2017, 976, 149–155. [Google Scholar] [CrossRef]
- Horie, S.; Aruga, S.; Kawamata, H.; Okui, N.; Kakizoe, T.; Kitamura, T. Biological role of HGF/MET pathway in renal cell carcinoma. J. Urol. 1999, 161, 990–997. [Google Scholar] [CrossRef]
- Yamauchi, M.; Kataoka, H.; Itoh, H.; Seguchi, T.; Hasui, Y.; Osada, Y. Hepatocyte growth factor activator inhibitor types 1 and 2 are expressed by tubular epithelium in kidney and down-regulated in renal cell carcinoma. J. Urol. 2004, 171, 890–896. [Google Scholar] [CrossRef]
- Finley, D.S.; Pantuck, A.J.; Belldegrun, A.S. Tumor biology and prognostic factors in renal cell carcinoma. Oncologist 2011, 16 (Suppl. 2), 4–13. [Google Scholar] [CrossRef] [Green Version]
- Bahena-Lopez, J.P.; Gamba, G.; Castaneda-Bueno, M. WNK1 in the kidney. Curr. Opin. Nephrol. Hypertens. 2022, 31, 471–478. [Google Scholar] [CrossRef]
- McCormick, J.A.; Ellison, D.H. The WNKs: Atypical protein kinases with pleiotropic actions. Physiol. Rev. 2011, 91, 177–219. [Google Scholar] [CrossRef] [Green Version]
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017, 25, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Moniz, S.; Jordan, P. Emerging roles for WNK kinases in cancer. Cell Mol. Life Sci. 2010, 67, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragoni, S.; Turin, I.; Laforenza, U.; Potenza, D.M.; Bottino, C.; Glasnov, T.N.; Prestia, M.; Ferulli, F.; Saitta, A.; Mosca, A.; et al. Store-operated Ca2+ entry does not control. proliferation in primary cultures of human metastatic renal cellular carcinoma. Biomed. Res. Int. 2014, 2014, 739494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Shan, G.; Ge, M.; Qian, H.; Xia, Y. Transient Receptor Potential Channel 1 Potentially Serves as a Biomarker Indicating T/TNM Stages and Predicting Long-Term Prognosis in Patients With Renal Cell Carcinoma. Front. Surg. 2022, 9, 853310. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Semwal, D.K.; Mishra, S.P.; Goyal, S.; Marathe, R.; Semwal, R.B. Combination of mTOR and MAPK Inhibitors-A Potential Way to Treat Renal Cell Carcinoma. Med. Sci. 2016, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Goel, M.; Sinkins, W.G.; Zuo, C.D.; Estacion, M.; Schilling, W.P. Identification and localization of TRPC channels in the rat kidney. Am. J. Physiol. Renal Physiol. 2006, 290, F1241–F1252. [Google Scholar] [CrossRef] [Green Version]
- Ibeh, C.L.; Yiu, A.J.; Kanaras, Y.L.; Paal, E.; Birnbaumer, L.; Jose, P.A.; Bandyopadhyay, B.C. Evidence for a regulated Ca2+ entry in proximal tubular cells and its implication in calcium stone formation. J. Cell Sci. 2019, 132, jcs225268. [Google Scholar] [CrossRef] [Green Version]
- Khayyat, N.H.; Tomilin, V.N.; Zaika, O.; Pochynyuk, O. Polymodal roles of TRPC3 channel in the kidney. Channels 2020, 14, 257–267. [Google Scholar] [CrossRef]
- Friedman, P.A. Mechanisms of renal calcium transport. Exp. Nephrol. 2000, 8, 343–350. [Google Scholar] [CrossRef]
- Bandyopadhyay, B.C.; Swaim, W.D.; Liu, X.; Redman, R.S.; Patterson, R.L.; Ambudkar, I.S. Apical localization of a functional TRPC3/TRPC6-Ca2+-signaling complex in polarized epithelial cells. Role in apical Ca2+ influx. J. Biol. Chem. 2005, 280, 12908–12916. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.M.; MacLeod, R.J. Extracellular calcium sensing and extracellular calcium signaling. Physiol. Rev. 2001, 81, 239–297. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Chen, T.H.; Pratt, S.; Shoback, D. Amino acids in the second and third intracellular loops of the parathyroid Ca2+-sensing receptor mediate efficient coupling to phospholipase C. J. Biol. Chem. 2000, 275, 19955–19963. [Google Scholar] [CrossRef] [Green Version]
- Gombedza, F.C.; Shin, S.; Kanaras, Y.L.; Bandyopadhyay, B.C. Abrogation of store-operated Ca2+ entry protects against crystal-induced ER stress in human proximal tubular cells. Cell Death Discov. 2019, 5, 124. [Google Scholar] [CrossRef] [Green Version]
- Yiu, A.J.; Ibeh, C.L.; Roy, S.K.; Bandyopadhyay, B.C. Melamine induces Ca2+-sensing receptor activation and elicits apoptosis in proximal tubular cells. Am. J. Physiol. Cell Physiol. 2017, 313, C27–C41. [Google Scholar] [CrossRef] [Green Version]
- Asplin, J.R.; Mandel, N.S.; Coe, F.L. Evidence of calcium phosphate supersaturation in the loop of Henle. Am. J. Physiol. 1996, 270, F604–F613. [Google Scholar] [CrossRef]
- Tiselius, H.G. A hypothesis of calcium stone formation: An interpretation of stone research during the past decades. Urol. Res. 2011, 39, 231–243. [Google Scholar] [CrossRef]
- Coe, F.L.; Worcester, E.M.; Evan, A.P. Idiopathic hypercalciuria and formation of calcium renal stones. Nat. Rev. Nephrol. 2016, 12, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Bird, V.Y.; Khan, S.R. How do stones form? Is unification of theories on stone formation possible? Arch. Esp. Urol. 2017, 70, 12–27. [Google Scholar]
- Evan, A.P.; Lingeman, J.E.; Coe, F.L.; Parks, J.H.; Bledsoe, S.B.; Shao, Y.; Sommer, A.J.; Paterson, R.F.; Kuo, R.L.; Grynpas, M. Randall’s plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J. Clin. Investig. 2003, 111, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Doroszewicz, J.; Waldegger, P.; Jeck, N.; Seyberth, H.; Waldegger, S. pH dependence of extracellular calcium sensing receptor activity determined by a novel technique. Kidney Int. 2005, 67, 187–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matlaga, B.R.; Shah, O.D.; Assimos, D.G. Drug-induced urinary calculi. Rev. Urol. 2003, 5, 227–231. [Google Scholar] [PubMed]
- Luo, D.D.; Fielding, C.; Phillips, A.; Fraser, D. Interleukin-1 beta regulates proximal tubular cell transforming growth factor beta-1 signalling. Nephrol. Dial Transplant. 2009, 24, 2655–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulay, S.R.; Evan, A.; Anders, H.J. Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol. embolism, crystalline nephropathies and kidney stone disease. Nephrol. Dial Transplant. 2014, 29, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Vesey, D.A.; Cheung, C.W.; Cuttle, L.; Endre, Z.A.; Gobe, G.; Johnson, D.W. Interleukin-1beta induces human proximal tubule cell injury, alpha-smooth muscle actin expression and fibronectin production. Kidney Int. 2002, 62, 31–40. [Google Scholar] [CrossRef]
- Gewin, L.S. Renal fibrosis: Primacy of the proximal tubule. Matrix Biol. 2018, 68–69, 248–262. [Google Scholar] [CrossRef]
- Berg, C.; Tiselius, H.G. The effect of pH on the risk of calcium oxalate crystallization in urine. Eur. Urol. 1986, 12, 59–61. [Google Scholar] [CrossRef]
- Carra, G.; Avalle, L.; Secli, L.; Brancaccio, M.; Morotti, A. Shedding Light on NF-kappaB Functions in Cellular Organelles. Front. Cell Dev. Biol. 2022, 10, 841646. [Google Scholar] [CrossRef]
- Zhu, X.; Huang, L.; Gong, J.; Shi, C.; Wang, Z.; Ye, B.; Xuan, A.; He, X.; Long, D.; Zhu, X.; et al. NF-kappaB pathway link with ER stress-induced autophagy and apoptosis in cervical tumor cells. Cell Death Discov. 2017, 3, 17059. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Hitomi, J.; Katayama, T.; Taniguchi, M.; Honda, A.; Imaizumi, K.; Tohyama, M. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci. Lett. 2004, 357, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Rustom, R.; Wang, B.; McArdle, F.; Shalamanova, L.; Alexander, J.; McArdle, A.; Thomas, C.E.; Bone, J.M.; Shenkin, A.; Jackson, M.J. Oxidative stress in a novel model of chronic acidosis in LLC-PK1 cells. Nephron Exp. Nephrol. 2003, 95, e13–e23. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Scholze, A.; Jankowski, J.; Pedraza-Chaverri, J.; Evenepoel, P. Oxidative Stress in Chronic Kidney Disease. Oxid Med. Cell. Longev. 2016, 2016, 8375186. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.R. Reactive oxygen species, inflammation and calcium oxalate nephrolithiasis. Transl. Androl. Urol. 2014, 3, 256–276. [Google Scholar] [CrossRef]
- Gombedza, F.; Evans, S.; Shin, S.; Awuah Boadi, E.; Zhang, Q.; Nie, Z.; Bandyopadhyay, B.C. Melamine promotes calcium crystal formation in three-dimensional microfluidic device. Sci. Rep. 2019, 9, 875. [Google Scholar] [CrossRef] [Green Version]
- Lashhab, R.; Ullah, A.; Cordat, E. Renal collecting duct physiology and pathophysiology. Biochem. Cell Biol. 2019, 97, 234–242. [Google Scholar] [CrossRef]
- Magaldi, A.J.; van Baak, A.A.; Rocha, A.S. Calcium transport across rat inner medullary collecting duct perfused in vitro. Am. J. Physiol. 1989, 257, F738–F745. [Google Scholar] [CrossRef]
- Goel, M.; Sinkins, W.G.; Zuo, C.D.; Hopfer, U.; Schilling, W.P. Vasopressin-induced membrane trafficking of TRPC3 and AQP2 channels in cells of the rat renal collecting duct. Am. J. Physiol. Renal Physiol. 2007, 293, F1476–F1488. [Google Scholar] [CrossRef] [Green Version]
- Knepper, M.A.; Kwon, T.H.; Nielsen, S. Molecular physiology of water balance. N. Engl. J. Med. 2015, 372, 1349–1358. [Google Scholar] [CrossRef]
- Goel, M.; Zuo, C.D.; Schilling, W.P. Role of cAMP/PKA signaling cascade in vasopressin-induced trafficking of TRPC3 channels in principal cells of the collecting duct. Am. J. Physiol. Renal Physiol. 2010, 298, F988–F996. [Google Scholar] [CrossRef] [Green Version]
- Feinstein, T.N.; Yui, N.; Webber, M.J.; Wehbi, V.L.; Stevenson, H.P.; King, J.D., Jr.; Hallows, K.R.; Brown, D.; Bouley, R.; Vilardaga, J.P. Noncanonical control. of vasopressin receptor type 2 signaling by retromer and arrestin. J. Biol. Chem. 2013, 288, 27849–27860. [Google Scholar] [CrossRef]
- Seaman, M.N. The retromer complex—Endosomal protein recycling and beyond. J. Cell Sci. 2012, 125, 4693–4702. [Google Scholar] [CrossRef] [Green Version]
- Hanouna, G.; Haymann, J.P.; Baud, L.; Letavernier, E. Vasopressin regulates renal calcium excretion in humans. Physiol. Rep. 2015, 3, e12562. [Google Scholar] [CrossRef]
- Goel, M.; Schilling, W.P. Role of TRPC3 channels in ATP-induced Ca2+ signaling in principal cells of the inner medullary collecting duct. Am. J. Physiol. Renal Physiol. 2010, 299, F225–F233. [Google Scholar] [CrossRef] [Green Version]
- Ranieri, M. Renal Ca2+ and Water Handling in Response to Calcium Sensing Receptor Signaling: Physiopathological Aspects and Role of CaSR-Regulated microRNAs. Int. J. Mol. Sci. 2019, 20, 5341. [Google Scholar] [CrossRef] [Green Version]
- Praetorius, H.A.; Leipziger, J. Intrarenal purinergic signaling in the control. of renal tubular transport. Annu. Rev. Physiol. 2010, 72, 377–393. [Google Scholar] [CrossRef]
- Weinbaum, S.; Duan, Y.; Satlin, L.M.; Wang, T.; Weinstein, A.M. Mechanotransduction in the renal tubule. Am. J. Physiol. Renal Physiol. 2010, 299, F1220–F1236. [Google Scholar] [CrossRef] [Green Version]
- McCarty, N.A.; O’Neil, R.G. Calcium signaling in cell volume regulation. Physiol. Rev. 1992, 72, 1037–1061. [Google Scholar] [CrossRef]
- Tomilin, V.N.; Mamenko, M.; Zaika, O.; Ren, G.; Marrelli, S.P.; Birnbaumer, L.; Pochynyuk, O. TRPC3 determines osmosensitive [Ca2+]i signaling in the collecting duct and contributes to urinary concentration. PLoS ONE 2019, 14, e0226381. [Google Scholar] [CrossRef] [Green Version]
- Berrout, J.; Jin, M.; Mamenko, M.; Zaika, O.; Pochynyuk, O.; O’Neil, R.G. Function of transient receptor potential cation channel subfamily V member 4 (TRPV4) as a mechanical transducer in flow-sensitive segments of renal collecting duct system. J. Biol. Chem. 2012, 287, 8782–8791. [Google Scholar] [CrossRef] [Green Version]
- Mamenko, M.V.; Boukelmoune, N.; Tomilin, V.N.; Zaika, O.L.; Jensen, V.B.; O’Neil, R.G.; Pochynyuk, O.M. The renal TRPV4 channel is essential for adaptation to increased dietary potassium. Kidney Int. 2017, 91, 1398–1409. [Google Scholar] [CrossRef]
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef]
- Reiterova, J.; Tesar, V. Autosomal Dominant Polycystic Kidney Disease: From Pathophysiology of Cystogenesis to Advances in the Treatment. Int. J. Mol. Sci. 2022, 23, 3317. [Google Scholar] [CrossRef]
- Chaudhari, S.; Mallet, R.T.; Shotorbani, P.Y.; Tao, Y.; Ma, R. Store-operated calcium entry: Pivotal roles in renal physiology and pathophysiology. Exp. Biol. Med. 2021, 246, 305–316. [Google Scholar] [CrossRef]
- Piazzon, N.; Maisonneuve, C.; Guilleret, I.; Rotman, S.; Constam, D.B. Bicc1 links the regulation of cAMP signaling in polycystic kidneys to microRNA-induced gene silencing. J. Mol. Cell Biol. 2012, 4, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Kuo, I.Y.; DesRochers, T.M.; Kimmerling, E.P.; Nguyen, L.; Ehrlich, B.E.; Kaplan, D.L. Cyst formation following disruption of intracellular calcium signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 14283–14288. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhou, J.; Li, Y.; Yang, F.; Lian, X.; Liu, W. Mitochondrial TRPC3 promotes cell proliferation by regulating the mitochondrial calcium and metabolism in renal polycystin-2 knockdown cells. Int. Urol. Nephrol. 2019, 51, 1059–1070. [Google Scholar] [CrossRef]
- Klawitter, J.; Reed-Gitomer, B.Y.; McFann, K.; Pennington, A.; Klawitter, J.; Abebe, K.Z.; Klepacki, J.; Cadnapaphornchai, M.A.; Brosnahan, G.; Chonchol, M.; et al. Endothelial dysfunction and oxidative stress in polycystic kidney disease. Am. J. Physiol. Renal Physiol. 2014, 307, F1198–F1206. [Google Scholar] [CrossRef] [Green Version]
- Menon, V.; Rudym, D.; Chandra, P.; Miskulin, D.; Perrone, R.; Sarnak, M. Inflammation, oxidative stress, and insulin resistance in polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, Y.; Inagi, R.; Yoshihara, D.; Kugita, M.; Nagao, S.; Shimizu, A.; Takeda, N.; Wake, M.; Honda, K.; Zhou, J.; et al. Mitochondrial Abnormality Facilitates Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. Mol. Cell Biol. 2017, 37, 00337-17. [Google Scholar] [CrossRef] [Green Version]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Feng, S.; Li, H.; Tai, Y.; Huang, J.; Su, Y.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L.; Wang, Y. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 11011–11016. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Xiong, S.; Lin, S.; Xia, W.; Li, Q.; Zhao, Z.; Wei, X.; Lu, Z.; Wei, X.; Gao, P.; et al. Enhanced Mitochondrial Transient Receptor Potential Channel, Canonical Type 3-Mediated Calcium Handling in the Vasculature From Hypertensive Rats. J. Am. Heart Assoc 2017, 6, e005812. [Google Scholar] [CrossRef]
- Kitajima, N.; Numaga-Tomita, T.; Watanabe, M.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; et al. TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci. Rep. 2016, 6, 37001. [Google Scholar] [CrossRef] [Green Version]
- Miyagi, K.; Kiyonaka, S.; Yamada, K.; Miki, T.; Mori, E.; Kato, K.; Numata, T.; Sawaguchi, Y.; Numaga, T.; Kimura, T.; et al. A pathogenic C terminus-truncated polycystin-2 mutant enhances receptor-activated Ca2+ entry via association with TRPC3 and TRPC7. J. Biol. Chem. 2009, 284, 34400–34412. [Google Scholar] [CrossRef] [Green Version]
- Tiapko, O.; Groschner, K. TRPC3 as a Target of Novel Therapeutic Interventions. Cells 2018, 7, 83. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Englisch, C.N.; Paulsen, F.; Tschernig, T. TRPC Channels in the Physiology and Pathophysiology of the Renal Tubular System: What Do We Know? Int. J. Mol. Sci. 2023, 24, 181. https://doi.org/10.3390/ijms24010181
Englisch CN, Paulsen F, Tschernig T. TRPC Channels in the Physiology and Pathophysiology of the Renal Tubular System: What Do We Know? International Journal of Molecular Sciences. 2023; 24(1):181. https://doi.org/10.3390/ijms24010181
Chicago/Turabian StyleEnglisch, Colya N., Friedrich Paulsen, and Thomas Tschernig. 2023. "TRPC Channels in the Physiology and Pathophysiology of the Renal Tubular System: What Do We Know?" International Journal of Molecular Sciences 24, no. 1: 181. https://doi.org/10.3390/ijms24010181