
New Frontiers on ER Stress Modulation: Are TRP Channels the Leading Actors?

 , , , , ,
, , , , ,  ,
,  and
and 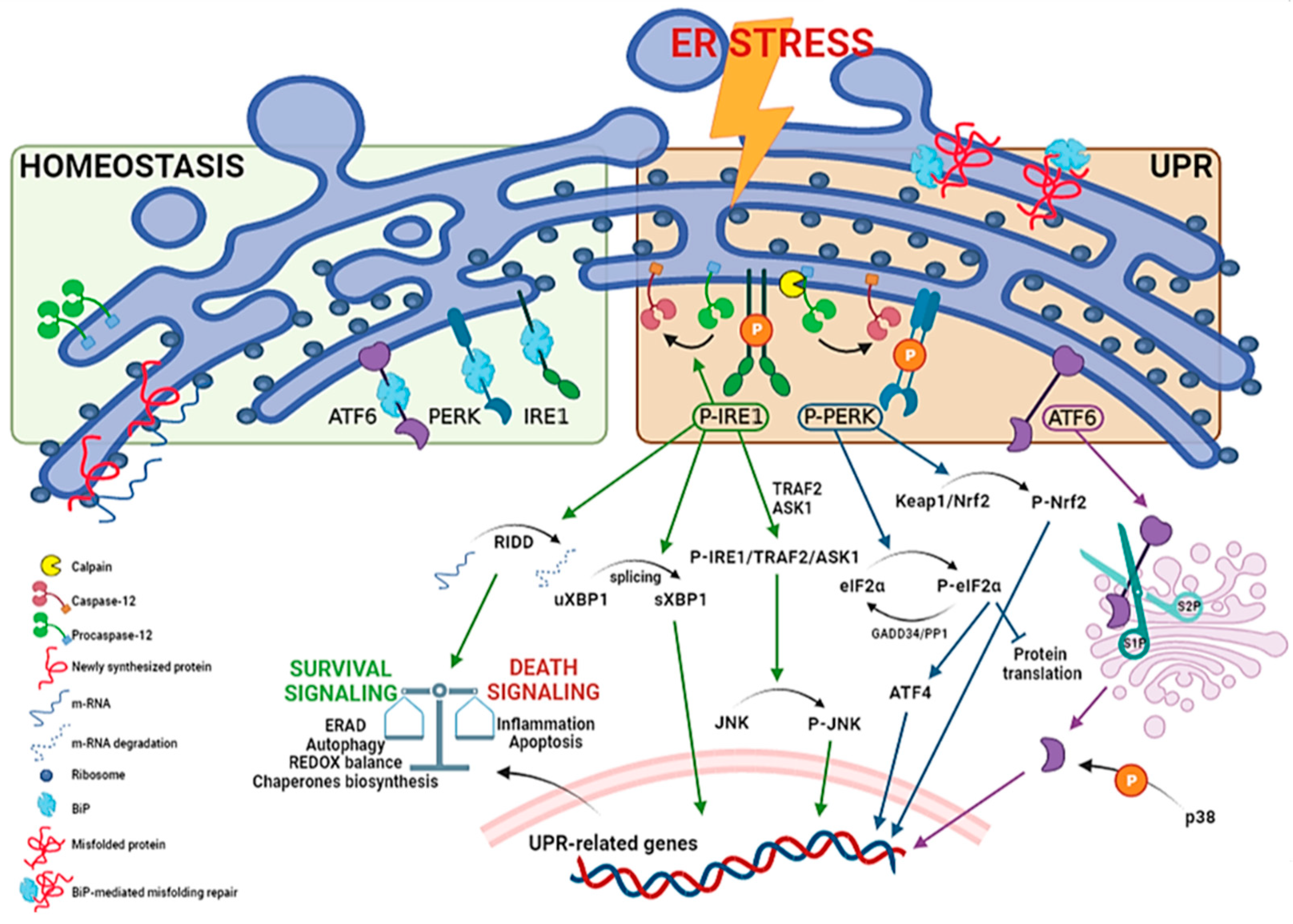{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
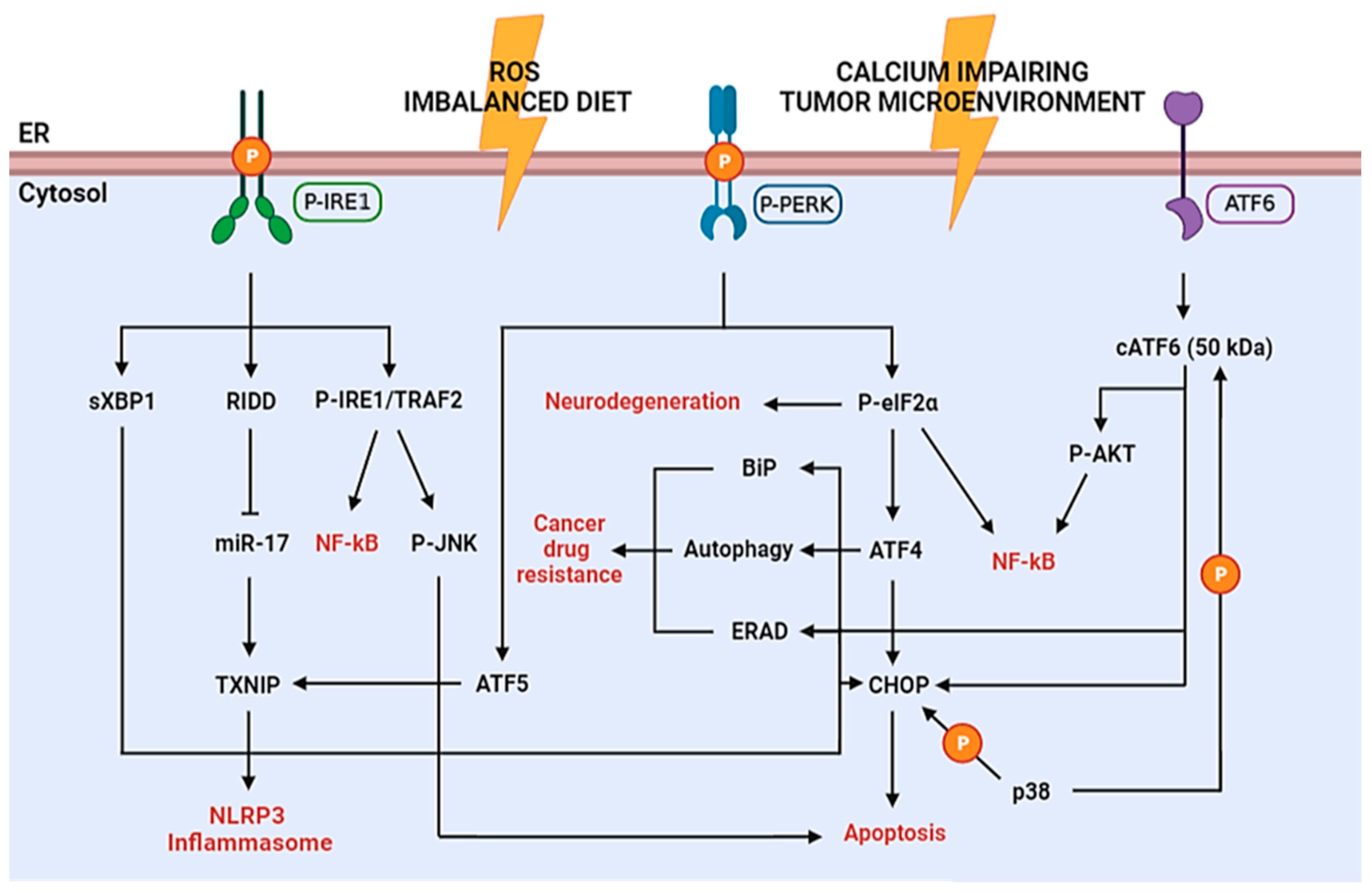
1.1. ER Stress and UPR Overview

1.2. ER Stress in Diseases Pathogenesis

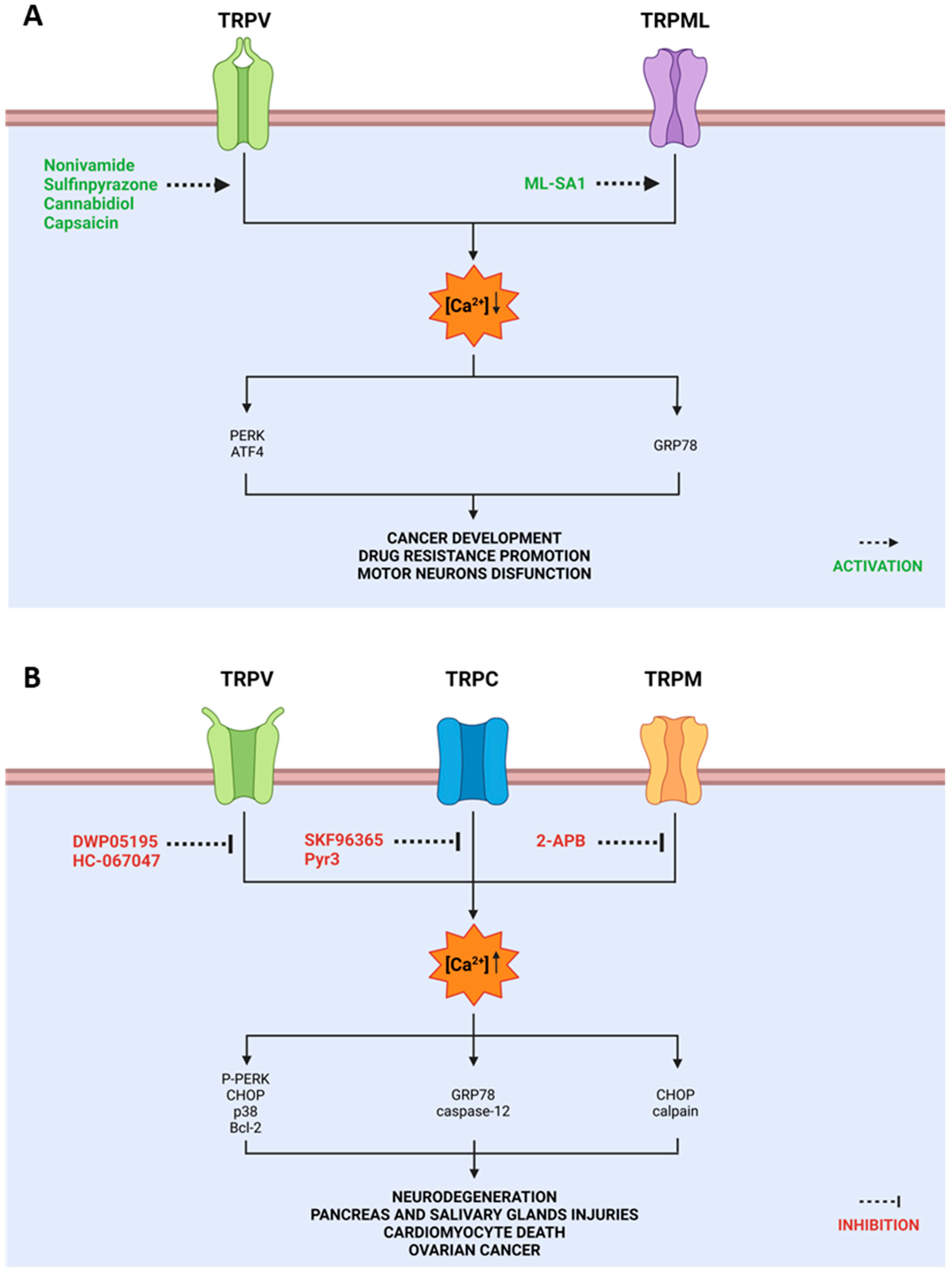
2. TRP and ER Stress
2.1. TRPV in ER Stress
2.2. TRPC in ER Stress
2.3. TRPM in ER Stress
2.4. Other TRP Channels in ER Stress

3. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mickle, A.D.; Shepherd, A.J.; Mohapatra, D.P. Nociceptive TRP Channels: Sensory Detectors and Transducers in Multiple Pain Pathologies. Pharmaceuticals 2016, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.R.; Lee, D.Y.; Chung, E.S.; Oh, U.T.; Kim, S.U.; Jin, B.K. Transient receptor potential vanilloid subtype 1 mediates cell death of mesencephalic dopaminergic neurons in vivo and in vitro. J. Neurosci. 2005, 25, 662–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, K.E.; Gratzke, C.; Hedlund, P. The role of the transient receptor potential (TRP) superfamily of cation-selective channels in the management of the overactive bladder. BJU Int. 2010, 106, 1114–1127. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Kim, J. Emerging role of transient receptor potential (TRP) channels in cancer progression. BMB Rep. 2020, 53, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.; Voeltz, G. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amodio, G.; Pagliara, V.; Moltedo, O.; Remondelli, P. Structural and Functional Significance of the Endoplasmic Reticulum Unfolded Protein Response Transducers and Chaperones at the Mitochondria-ER Contacts: A Cancer Perspective. Front. Cell Dev. Biol. 2021, 26, 641194. [Google Scholar] [CrossRef] [PubMed]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, H.; Nishitoh, H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes 2013, 4, 306–333. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 1, 421–438. [Google Scholar] [CrossRef]
- Kitamura, M. Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am. J. Physiol. Renal Physiol. 2008, 295, F323–F334. [Google Scholar] [CrossRef]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.R.; Chae, H.G. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 2021, 53, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease. Front. Cell Dev. Biol. 2017, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 25, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Valenzuela, V.; Collyer, E.; Armentano, D.; Parsons, G.B.; Court, F.A.; Hetz, C. Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell Death Dis. 2012, 16, e272. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J. Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Amodio, G.; Moltedo, O.; Faraonio, R.; Remondelli, P. Targeting the endoplasmic reticulum unfolded protein response to counteract the oxidative stress-Induced endothelial dysfunction. Oxid. Med. Cell. Longev. 2018, 2018, 4946289. [Google Scholar] [CrossRef]
- Amodio, G.; Moltedo, O.; Fasano, D.; Zerillo, L.; Oliveti, M.; Di Pietro, P.; Faraonio, R.; Barone, P.; Pellecchia, M.T.; De Rosa, A.; et al. PERK-Mediated Unfolded Protein Response Activation and Oxidative Stress in PARK20 Fibroblasts. Front. Neurosci. 2019, 27, 673. [Google Scholar] [CrossRef] [Green Version]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell. Biol. 2001, 153, 1011–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shacham, T.; Patel, C.; Lederkremer, G.Z. PERK Pathway and Neurodegenerative Disease: To Inhibit or to Activate? Biomolecules 2021, 11, 354. [Google Scholar] [CrossRef] [PubMed]
- Maytin, E.V.; Ubeda, M.; Lin, J.C.; Habener, J.F. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp. Cell Res. 2001, 267, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M.; et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 2012, 21, 2245–2262. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Bashir, S.; Banday, M.; Qadri, O.; Bashir, A.; Hilal, N.; Nida, I.F.; Rader, S.; Fazili, K.M. The molecular mechanism and functional diversity of UPR signaling sensor IRE1. Life Sci. 2021, 265, 118740. [Google Scholar] [CrossRef]
- Chen, L.; Xu, S.; Liu, L.; Wen, X.; Xu, Y.; Chen, J.; Teng, J. Cab45S inhibits the ER stress-induced IRE1-JNK pathway and apoptosis via GRP78/BiP. Cell Death Dis. 2014, 8, e1219. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 27, 13935–13940. [Google Scholar] [CrossRef] [Green Version]
- Hitomi, J.; Katayama, T.; Taniguchi, M.; Honda, A.; Imaizumi, K.; Tohyama, M. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci. Lett. 2004, 4, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Schindler, A.J.; Schekman, R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. USA 2009, 20, 17775–17780. [Google Scholar] [CrossRef]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, M.C.; Larburu, N.; Durairaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct. Mol. Biol. 2019, 26, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Deécio, L.E.; Ales, K.C.; Miriam, C. The Role for Endoplasmic Reticulum Stress in Diabetes Mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef] [Green Version]
- Chipurupalli, S.; Samavedam, U.; Robinson, N. Crosstalk Between ER Stress, Autophagy and Inflammation. Front. Med. 2021, 5, 758311. [Google Scholar] [CrossRef] [PubMed]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell. 2019, 111, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Vestuto, V.; Amodio, G.; Pepe, G.; Basilicata, M.G.; Belvedere, R.; Napolitano, E.; Guarnieri, D.; Pagliara, V.; Paladino, S.; Rodriquez, M.; et al. Cocoa Extract Provides Protection against 6-OHDA Toxicity in SH-SY5Y Dopaminergic Neurons by Targeting PERK. Biomedicines 2022, 10, 2009. [Google Scholar] [CrossRef]
- Asik, R.M.; Suganthy, N.; Aarifa, M.A.; Kumar, A.; Szigeti, K.; Mathe, D.; Gulyás, B.; Archunan, G.; Padmanabhan, P. Alzheimer’s Disease: A Molecular View of β-Amyloid Induced Morbific Events. Biomedicines 2021, 9, 1126. [Google Scholar] [CrossRef]
- Benedetti, R.; Montani, M.S.G.; Romeo, M.A.; Arena, A.; Santarelli, R.; D’Orazi, G.; Cirone, M. Role of UPR Sensor Activation in Cell Death–Survival Decision of Colon Cancer Cells Stressed by DPE Treatment. Biomedicines 2021, 9, 1262. [Google Scholar] [CrossRef]
- Ciccarese, F. Cancer Metabolism and Resistance to Cell Death: Novel Therapeutic Perspectives. Biomedicines 2022, 10, 1828. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell. Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 15, 3496–3499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Nichols, P.; Spicer, D.; Groshen, S.; Yu, M.C.; Lee, A.S. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006, 15, 7849–7853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyrko, P.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007, 15, 9809–9816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönthal, A.H. Endoplasmic reticulum stress: Its role in disease and novel prospects for therapy. Scientifica 2012, 2012, 857516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, T.F.; Tatsukawa, H.; Matsuura, T.; Nagatsuma, K.; Hirose, S.; Kojima, S. Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J. Cell. Phys. 2012, 227, 1130–1137. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 1, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, K.M.; Kennedy, D.; Gorman, A.M.; Gupta, S.; Healy, S.J.; Samali, A. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med. 2011, 15, 2025–2039. [Google Scholar] [CrossRef] [Green Version]
- Brundin, P.; Li, J.Y.; Holton, J.L.; Lindvall, O.; Revesz, T. Research in motion: The enigma of Parkinson’s disease pathology spread. Nat. Rev. Neurosci. 2008, 9, 741–745. [Google Scholar] [CrossRef]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 15, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Chami, M.; Checler, F. Alterations of the Endoplasmic Reticulum (ER) Calcium Signaling Molecular Components in Alzheimer’s Disease. Cells 2020, 1, 2577. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; LaFerla, F.M.; Callamaras, N.; Parker, I. Subcellular mechanisms of presenilin-mediated enhancement of calcium signaling. Neurobiol. Dis. 2001, 8, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef]
- Hetz, C.; Glimcher, L.H. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol. Cell 2009, 11, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Daverkausen-Fischer, L.; Pröls, F. Regulation of calcium homeostasis and flux between the endoplasmic reticulum and the cytosol. J. Biol. Chem. 2022, 298, 102061. [Google Scholar] [CrossRef]
- Sukumaran, P.; Schaar, A.; Sun, Y.; Singh, B.B. Functional role of TRP channels in modulating ER stress and Autophagy. Cell Calcium 2016, 60, 123–132. [Google Scholar] [CrossRef]
- Ong, H.L.; de Souza, L.B.; Cheng, K.T.; Ambudkar, I.S. Physiological functions and regulation of TRPC. Handb. Exp. Pharmacol. 2014, 223, 1005–1034. [Google Scholar] [CrossRef] [PubMed]
- Yamakage, M.; Namiki, A. Calcium channels--basic aspects of their structure, function and gene encoding; anesthetic action on the channels--a review. Can. J. Anaesth. 2002, 49, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammels, E.; Parys, J.B.; Missiaen, L.; De Smedt, H.; Bultynck, G. Intracellular Ca2+ storage in health and disease: A dynamic equilibrium. Cell Calcium 2010, 47, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Montell, C. The TRP superfamily of cation channels. Sci. STKE 2005, 22, re3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohacs, T. Regulation of transient receptor potential channels by the phospholipase C pathway. Adv. Biol. Regul. 2013, 53, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Saotome, K.; Singh, A.K.; Yelshanskaya, M.V.; Sobolevsky, A.I. Crystal structure of the epithelial calcium channel TRPV6. Nature 2016, 23, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Vennekens, R.; Owsianik, G.; Nilius, B. Vanilloid transient receptor potential cation channels: An overview. Curr. Pharm. Des. 2008, 14, 18–31. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Liao, Q.; Chen, C.; Yang, X.; Xie, R.; Xu, J. The Role of Transient Receptor Potential Vanilloid 1 in Common Diseases of the Digestive Tract and the Cardiovascular and Respiratory System. Front. Physiol. 2019, 21, 1064. [Google Scholar] [CrossRef]
- Szallasi, A.; Cortright, D.N.; Blum, C.A.; Eid, S.R. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat. Rev. Drug Discov. 2007, 6, 357–372. [Google Scholar] [CrossRef]
- Thomas, K.C.; Sabnis, A.S.; Johansen, M.E.; Lanza, D.L.; Moos, P.J.; Yost, G.S.; Reilly, C.A. Transient receptor potential vanilloid 1 agonists cause endoplasmic reticulum stress and cell death in human lung cells. J. Pharmacol. Exp. Ther. 2007, 321, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.; Prasad, S.; Ravindran, J.; Yadav, V.R.; Aggarwal, B.B. Capsazepine, a TRPV1 antagonist, sensitizes colorectal cancer cells to apoptosis by TRAIL through ROS-JNK-CHOP-mediated upregulation of death receptors. Free Radic. Biol. Med. 2012, 53, 1977–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Chapa, J.J.; Singha, P.K.; Self, K.K.; Sallaway, M.L.; McHardy, S.F.; Hart, M.J.; McGuff, H.S.; Valdez, M.C.; Ruiz, F., 2nd; Polusani, S.R.; et al. The novel capsazepine analog, CIDD-99, significantly inhibits oral squamous cell carcinoma in vivo through a TRPV1-independent induction of ER stress, mitochondrial dysfunction, and apoptosis. J. Oral Pathol. Med. 2019, 48, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Harpe, A.; Beukes, N.; Frost, C.L. CBD activation of TRPV1 induces oxidative signaling and subsequent ER stress in breast cancer cell lines. Biotechnol. Appl. Biochem. 2022, 69, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Lee, K.T.; Lim, M.C.; Choi, J.H. TRPV1 Antagonist DWP05195 Induces ER Stress-Dependent Apoptosis through the ROS-p38-CHOP Pathway in Human Ovarian Cancer Cells. Cancers 2020, 12, 1702. [Google Scholar] [CrossRef] [PubMed]
- Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002, 277, 34287–34294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.J.; Wu, I.J.; Hong, J.Y.; Liu, B.H.; Liang, R.Y.; Yuan, T.M.; Chuang, S.M. Capsaicin-induced TRIB3 upregulation promotes apoptosis in cancer cells. Cancer Manag. Res. 2018, 10, 4237–4248. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.; Liedtke, W.B. Osmomechanical-Sensitive TRPV Channels in Mammals. In Neurobiol. TRP Channels; CRC Press: Boca Raton, FL, USA, 2017; Volume 5. [Google Scholar] [CrossRef]
- Holzer, P. Transient receptor potential (TRP) channels as drug targets for diseases of the digestive system. Pharmacol. Ther. 2011, 131, 142–170. [Google Scholar] [CrossRef]
- Chung, M.K.; Lee, H.; Caterina, M.J. Warm temperatures activate TRPV4 in mouse 308 keratinocytes. J. Biol. Chem. 2003, 278, 32037–32046. [Google Scholar] [CrossRef] [Green Version]
- Birder, L.; Kullmann, F.A.; Lee, H.; Barrick, S.; de Groat, W.; Kanai, A.; Caterina, M. Activation of urothelial transient receptor potential vanilloid 4 by 4alpha-phorbol 12,13-didecanoate contributes to altered bladder reflexes in the rat. J. Pharmacol. Exp. Ther. 2007, 323, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liedtke, W.; Choe, Y.; Martí-Renom, M.A.; Bell, A.M.; Denis, C.S.; Sali, A.; Hudspeth, A.J.; Friedman, J.M.; Heller, S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 2000, 103, 525–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Liu, J.; Wen, X.; Bai, L.; Shao, R.; Bai, J. TRPV4 contributes to ER stress: Relation to apoptosis in the MPP+-induced cell model of Parkinson’s disease. Life Sci. 2020, 261, 118461. [Google Scholar] [CrossRef]
- Shen, J.; Tu, L.; Chen, D.; Tan, T.; Wang, Y.; Wang, S. TRPV4 channels stimulate Ca2+-induced Ca2+ release in mouse neurons and trigger endoplasmic reticulum stress after intracerebral hemorrhage. Brain Res. Bull. 2019, 146, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Wang, Y.; Liu, D.; Liang, Y.; Liu, W.; Huang, Y.; Xie, L.; Cao, W.; Xu, Y.; Chen, L. HC067047 Ameliorates SAE by suppressing endoplasmic reticulum stress and oxidative stress-induced pyroptosis in mice hippocampus. SSRN 2022, 20, 4189328. [Google Scholar] [CrossRef]
- Peng, J.B.; Chen, X.Z.; Berger, U.V.; Weremowicz, S.; Morton, C.C.; Vassilev, P.M.; Brown, E.M.; Hediger, M.A. Human calcium transport protein CaT1. Biochem. Biophys. Res. Commun. 2000, 278, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Peng, J.B.; Tou, L.; Takanaga, H.; Adam, R.M.; Hediger, M.A.; Freeman, M.R. Calcium-selective ion channel, CaT1, is apically localized in gastrointestinal tract epithelia and is aberrantly expressed in human malignancies. Lab. Investig. 2002, 82, 1755–1764. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Li, N.; Wang, Y.; Yu, J.; Mi, J. Calcium channel TRPV6 promotes breast cancer metastasis by NFATC2IP. Cancer Lett. 2021, 28, 150–160. [Google Scholar] [CrossRef]
- Hoenderop, J.G.; Vennekens, R.; Müller, D.; Prenen, J.; Droogmans, G.; Bindels, R.J.; Nilius, B. Function and expression of the epithelial Ca(2+) channel family: Comparison of mammalian ECaC1 and 2. J. Physiol. 2001, 537, 747–761. [Google Scholar] [CrossRef]
- Song, H.; Dong, M.; Zhou, J.; Sheng, W.; Li, X.; Gao, W. Expression and prognostic significance of TRPV6 in the development and progression of pancreatic cancer. Oncol Rep. 2018, 39, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Meng, Z.; Lu, J.; Chen, F.M.; Wong, W.T.; Tse, G.; Zheng, C.; Keung, W.; Tse, K.; Li, R.A.; et al. TRPV6 protects ER stress-induced apoptosis via ATF6α-TRPV6-JNK pathway in human embryonic stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 120, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B.; Penner, R. Store depletion and calcium influx. Physiol. Rev. 1997, 77, 901–930. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.; Plant, T.D.; Obukhov, A.G.; Hofmann, T.; Gudermann, T.; Schultz, G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J. Biol. Chem. 2000, 275, 17517–17526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunert-Keil, C.; Bisping, F.; Krüger, J.; Brinkmeier, H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genom. 2006, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Montell, C. Physiology, phylogeny, and functions of the TRP superfamily of cation channels. Sci. STKE 2001, 2001, re1. [Google Scholar] [CrossRef]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, S.; Sun, Y.; Watt, J.A.; Wang, S.; Lei, S.; Birnbaumer, L.; Singh, B.B. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Investig. 2012, 122, 1354–1367. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, P.; Sun, Y.; Zangbede, F.Q.; da Conceicao, V.N.; Mishra, B.; Singh, B.B. TRPC1 expression and function inhibit ER stress and cell death in salivary gland cells. FASEB Bioadv. 2019, 1, 40–50. [Google Scholar] [CrossRef]
- Da Conceicao, V.N.; Sun, Y.; Zboril, E.K.; De la Chapa, J.J.; Singh, B.B. Loss of Ca2+ entry via Orai-TRPC1 induces ER stress, initiating immune activation in macrophages. J. Cell. Sci. 2019, 133, jcs237610. [Google Scholar] [CrossRef] [Green Version]
- Schaar, A.; Sun, Y.; Sukumaran, P.; Rosenberger, T.A.; Krout, D.; Roemmich, J.N.; Brinbaumer, L.; Claycombe-Larson, K.; Singh, B.B. Ca2+ entry via TRPC1 is essential for cellular differentiation and modulates secretion via the SNARE complex. J. Cell. Sci. 2019, 132, jcs231878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyonaka, S.; Kato, K.; Nishida, M.; Mio, K.; Numaga, T.; Sawaguchi, Y.; Yoshida, T.; Wakamori, M.; Mori, E.; Numata, T.; et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Natl. Acad. Sci. USA 2009, 106, 5400–5405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Lee, K.P.; Yang, D.; Shin, D.M.; Abramowitz, J.; Kiyonaka, S.; Birnbaumer, L.; Mori, Y.; Muallem, S. Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity. Gastroenterology 2011, 140, 2107–2115. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; He, F.F.; Wang, H.; Fang, Z.; Shao, N.; Tian, X.J.; Liu, J.S.; Zhu, Z.H.; Wang, Y.M.; Wang, S.; et al. Calcium entry via TRPC6 mediates albumin overload-induced endoplasmic reticulum stress and apoptosis in podocytes. Cell Calcium 2011, 50, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.C.; Sun, G.B.; Ye, J.X.; Wang, J.; Zhang, M.D.; Sun, X.B. Salvianolic acid B attenuates doxorubicin-induced ER stress by inhibiting TRPC3 and TRPC6 mediated Ca2+ overload in rat cardiomyocytes. Toxicol. Lett. 2017, 276, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lü, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2020, 85, 102111. [Google Scholar] [CrossRef]
- Ferioli, S.; Zierler, S.; Zaißerer, J.; Schredelseker, J.; Gudermann, T.; Chubanov, V. TRPM6 and TRPM7 differentially contribute to the relief of heteromeric TRPM6/7 channels from inhibition by cytosolic Mg2+ and Mg·ATP. Sci. Rep. 2017, 7, 8806. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, M.; Kozak, J.A.; Shimizu, Y.; McLachlin, D.T.; Yamaguchi, H.; Wei, F.Y.; Tomizawa, K.; Matsui, H.; Chait, B.T.; Cahalan, M.D.; et al. Channel function is dissociated from the intrinsic kinase activity and autophosphorylation of TRPM7/ChaK1. J. Biol. Chem. 2005, 280, 20793–20803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perraud, A.L.; Schmitz, C.; Scharenberg, A.M. TRPM2 Ca2+ permeable cation channels: From gene to biological function. Cell Calcium 2003, 33, 519–531. [Google Scholar] [CrossRef]
- Gilliam, J.C.; Wensel, T.G. TRP channel gene expression in the mouse retina. Vision Res. 2011, 51, 2440–2452. [Google Scholar] [CrossRef] [Green Version]
- Maddodi, N.; Setaluri, V. Prognostic significance of melanoma differentiation and trans-differentiation. Cancers 2010, 2, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agosto, M.A.; Anastassov, I.A.; Robichaux, M.A.; Wensel, T.G. A Large Endoplasmic Reticulum-Resident Pool of TRPM1 in Retinal ON-Bipolar Cells. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Togashi, K.; Hara, Y.; Tominaga, T.; Higashi, T.; Konishi, Y.; Mori, Y.; Tominaga, M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006, 25, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Gao, Y.; Wang, C.; Tao, R.; Wu, Y.; Zhan, K.; Liao, M.; Lu, N.; Lu, Y.; Wilcox, C.S.; et al. Nitration of TRPM2 as a Molecular Switch Induces Autophagy During Brain Pericyte Injury. Antioxid. Redox Signal 2017, 27, 1297–1316. [Google Scholar] [CrossRef] [PubMed]
- Ostapchenko, V.G.; Chen, M.; Guzman, M.S.; Xie, Y.F.; Lavine, N.; Fan, J.; Beraldo, F.H.; Martyn, A.C.; Belrose, J.C.; Mori, Y.; et al. The Transient Receptor Potential Melastatin 2 (TRPM2) Channel Contributes to β-Amyloid Oligomer-Related Neurotoxicity and Memory Impairment. J. Neurosci. 2015, 35, 15157–15169. [Google Scholar] [CrossRef] [Green Version]
- Fonfria, E.; Murdock, P.R.; Cusdin, F.S.; Benham, C.D.; Kelsell, R.E.; McNulty, S. Tissue distribution profiles of the human TRPM cation channel family. J. Recept. Signal Transduct. Res. 2006, 26, 159–178. [Google Scholar] [CrossRef]
- Nadler, M.J.; Hermosura, M.C.; Inabe, K.; Perraud, A.L.; Zhu, Q.; Stokes, A.J.; Kurosaki, T.; Kinet, J.P.; Penner, R.; Scharenberg, A.M.; et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 2001, 411, 590–595. [Google Scholar] [CrossRef]
- Szegezdi, E.; Fitzgerald, U.; Samali, A. Caspase-12 and ER-stress-mediated apoptosis: The story so far. Ann. N. Y. Acad. Sci. 2003, 1010, 186–194. [Google Scholar] [CrossRef]
- Huang, Y.; Leng, T.D.; Inoue, K.; Yang, T.; Liu, M.; Horgen, F.D.; Fleig, A.; Li, J.; Xiong, Z.G. TRPM7 channels play a role in high glucose-induced endoplasmic reticulum stress and neuronal cell apoptosis. J. Biol. Chem. 2018, 293, 14393–14406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, X.; Wang, Y.; Li, X.; Huang, C.; Li, J. Inhibition of transient receptor potential melastatin 7 (TRPM7) channel induces RA FLSs apoptosis through endoplasmic reticulum (ER) stress. Clin. Rheumatol. 2014, 33, 1565–1574. [Google Scholar] [CrossRef]
- Zhu, Y.; Men, R.; Wen, M.; Hu, X.; Liu, X.; Yang, L. Blockage of TRPM7 channel induces hepatic stellate cell death through endoplasmic reticulum stress-mediated apoptosis. Life Sci. 2014, 94, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Y.; Shuai, S.; Ding, D.; Li, R.; Luo, R. TRPM8 promotes aggressiveness of breast cancer cells by regulating EMT via activating AKT/GSK-3β pathway. Tumour Biol. 2014, 35, 8969–8977. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Z.; Meng, Z.; Cao, H.; Zhu, G.; Liu, T.; Wang, X. Knockdown of TRPM8 suppresses cancer malignancy and enhances epirubicin-induced apoptosis in human osteosarcoma cells. Int. J. Biol. Sci. 2013, 10, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y.; Ohkubo, T.; Ikebe, T.; Yamazaki, J. Blockade of TRPM8 activity reduces the invasion potential of oral squamous carcinoma cell lines. Int. J. Oncol. 2012, 40, 1431–1440. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Barritt, G.J. TRPM8 in prostate cancer cells: A potential diagnostic and prognostic marker with a secretory function? Endocr. Relat. Cancer 2006, 13, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Di Sarno, V.; Giovannelli, P.; Medina-Peris, A.; Ciaglia, T.; Di Donato, M.; Musella, S.; Lauro, G.; Vestuto, V.; Smaldone, G.; Di Matteo, F.; et al. New TRPM8 blockers exert anticancer activity over castration-resistant prostate cancer models. Eur. J. Med. Chem. 2022, 238, 114435. [Google Scholar] [CrossRef]
- Bidaux, G.; Borowiec, A.S.; Gordienko, D.; Beck, B.; Shapovalov, G.G.; Lemonnier, L.; Flourakis, M.; Vandenberghe, M.; Slomianny, C.; Dewailly, E.; et al. Epidermal TRPM8 channel isoform controls the balance between keratinocyte proliferation and differentiation in a cold-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, E3345–E3354. [Google Scholar] [CrossRef]
- Bidaux, G.; Borowiec, A.S.; Dubois, C.; Delcourt, P.; Schulz, C.; Vanden Abeele, F.; Lepage, G.; Desruelles, E.; Bokhobza, A.; Dewailly, E.; et al. Targeting of short TRPM8 isoforms induces 4TM-TRPM8-dependent apoptosis in prostate cancer cells. Oncotarget 2016, 7, 29063–29080. [Google Scholar] [CrossRef]
- Sun, M.; Goldin, E.; Stahl, S.; Falardeau, J.L.; Kennedy, J.C.; Acierno, J.S., Jr.; Bove, C.; Kaneski, C.R.; Nagle, J.; Bromley, M.C.; et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 2000, 9, 2471–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colletti, G.A.; Kiselyov, K. TRPML1. Adv. Exp. Med. Biol. 2011, 704, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, V.; Petrozziello, T.; Sisalli, M.J.; Boscia, F.; Canzoniero, L.M.T.; Secondo, A. The activation of Mucolipin TRP channel 1 (TRPML1) protects motor neurons from L-BMAA neurotoxicity by promoting autophagic clearance. Sci. Rep. 2019, 9, 10743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.M.; Yu, T.K.; Chu, H.H.; Park, H.S.; Jang, K.Y.; Moon, W.S.; Kang, M.J.; Lee, D.G.; Kim, M.H.; Lee, J.H.; et al. Expression of ER stress and autophagy-related molecules in human non-small cell lung cancer and premalignant lesions. Int. J. Cancer 2012, 131, E362–E370. [Google Scholar] [CrossRef]
- Nguyen, N.D.; Memon, T.A.; Burrell, K.L.; Almestica-Roberts, M.; Rapp, E.; Sun, L.; Scott, A.F.; Rower, J.E.; Deering-Rice, C.E.; Reilly, C.A. Transient Receptor Potential Ankyrin-1 and Vanilloid-3 Differentially Regulate Endoplasmic Reticulum Stress and Cytotoxicity in Human Lung Epithelial Cells After Pneumotoxic Wood Smoke Particle Exposure. Mol. Pharmacol. 2020, 98, 586–597. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vestuto, V.; Di Sarno, V.; Musella, S.; Di Dona, G.; Moltedo, O.; Gomez-Monterrey, I.M.; Bertamino, A.; Ostacolo, C.; Campiglia, P.; Ciaglia, T. New Frontiers on ER Stress Modulation: Are TRP Channels the Leading Actors? Int. J. Mol. Sci. 2023, 24, 185. https://doi.org/10.3390/ijms24010185
Vestuto V, Di Sarno V, Musella S, Di Dona G, Moltedo O, Gomez-Monterrey IM, Bertamino A, Ostacolo C, Campiglia P, Ciaglia T. New Frontiers on ER Stress Modulation: Are TRP Channels the Leading Actors? International Journal of Molecular Sciences. 2023; 24(1):185. https://doi.org/10.3390/ijms24010185
Chicago/Turabian StyleVestuto, Vincenzo, Veronica Di Sarno, Simona Musella, Giorgio Di Dona, Ornella Moltedo, Isabel Maria Gomez-Monterrey, Alessia Bertamino, Carmine Ostacolo, Pietro Campiglia, and Tania Ciaglia. 2023. "New Frontiers on ER Stress Modulation: Are TRP Channels the Leading Actors?" International Journal of Molecular Sciences 24, no. 1: 185. https://doi.org/10.3390/ijms24010185