
Integrated Analysis of Bulk RNA-Seq and Single-Cell RNA-Seq Unravels the Influences of SARS-CoV-2 Infections to Cancer Patients


Abstract
:1. Introduction
2. Results
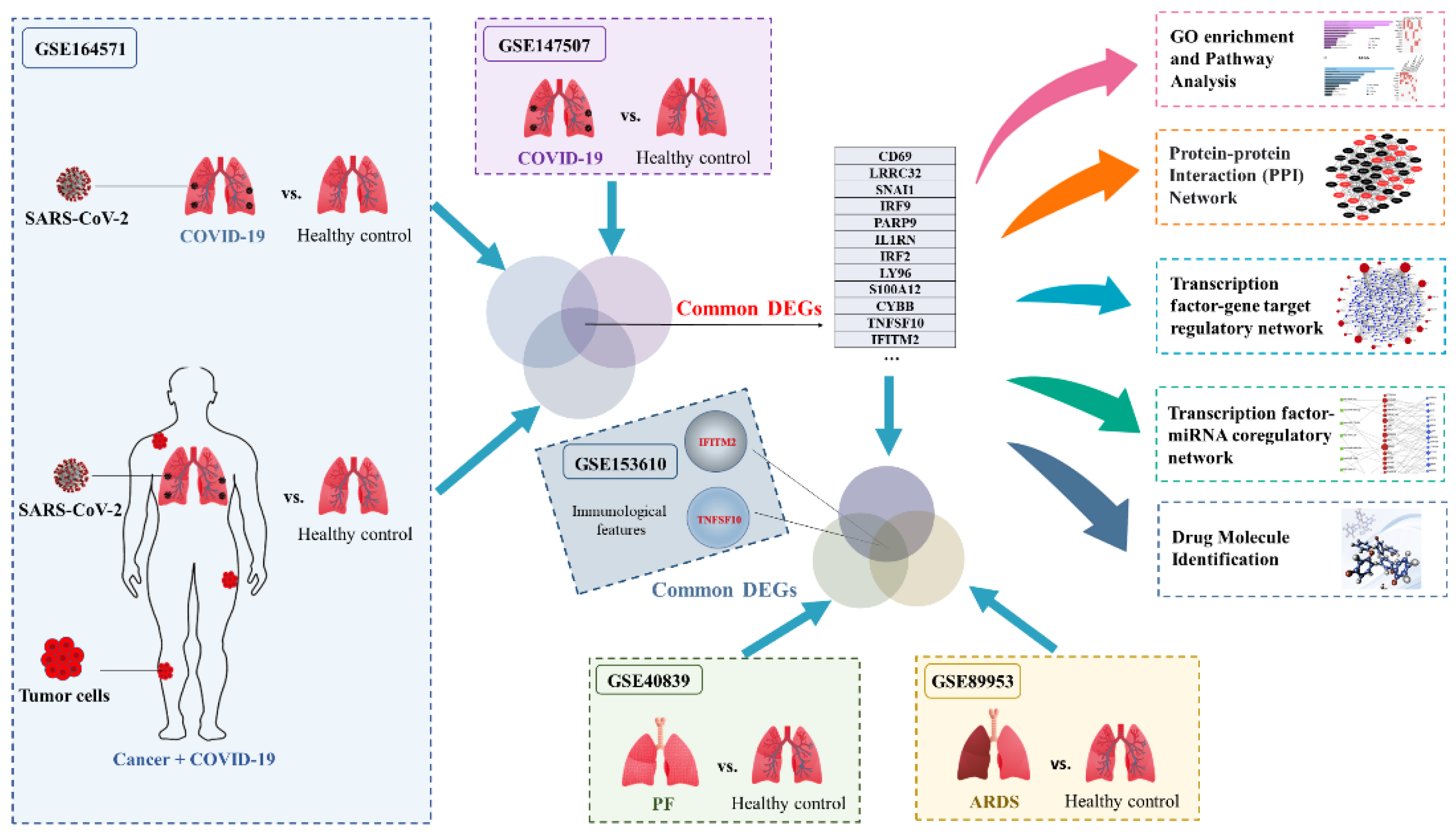
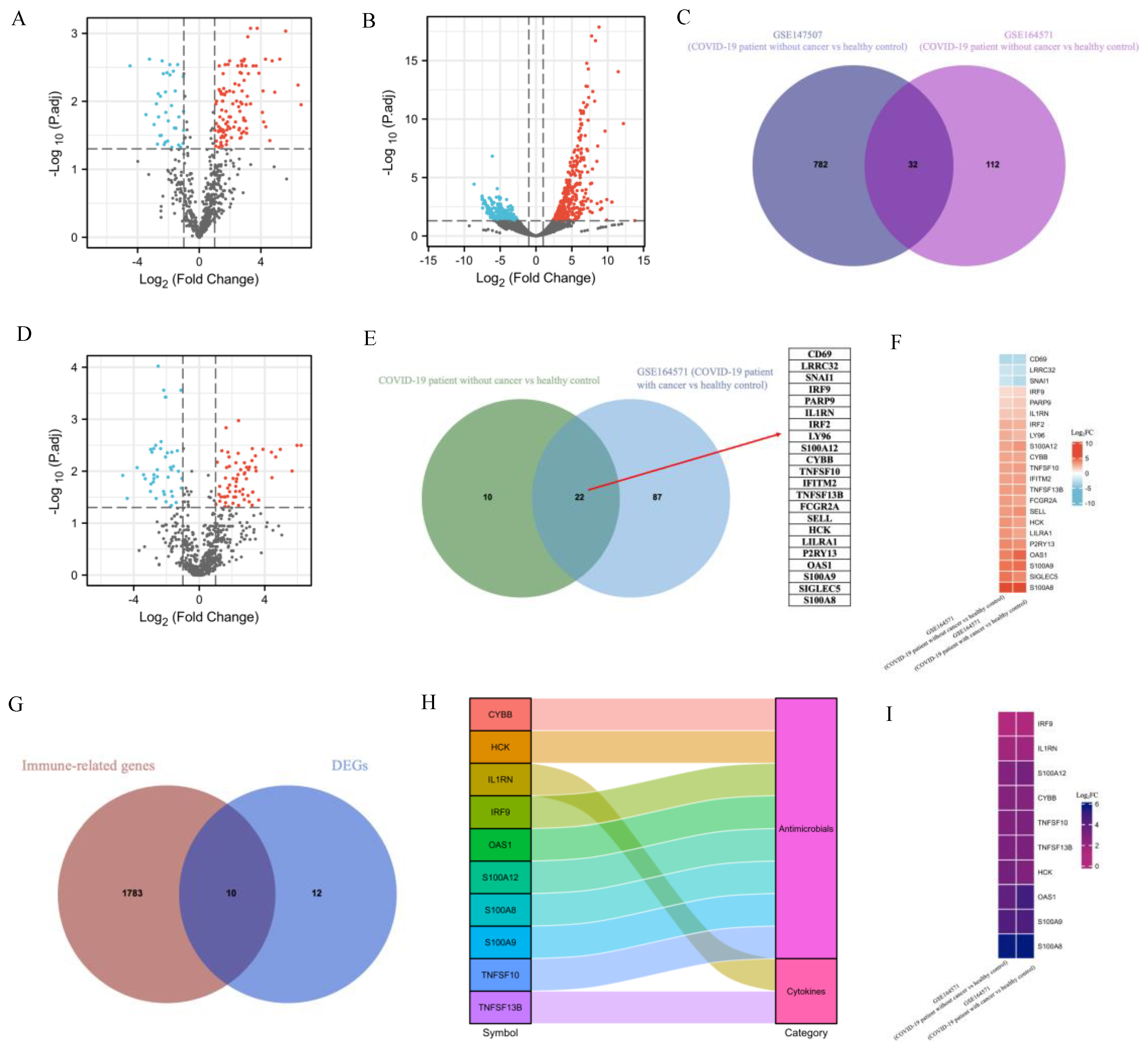
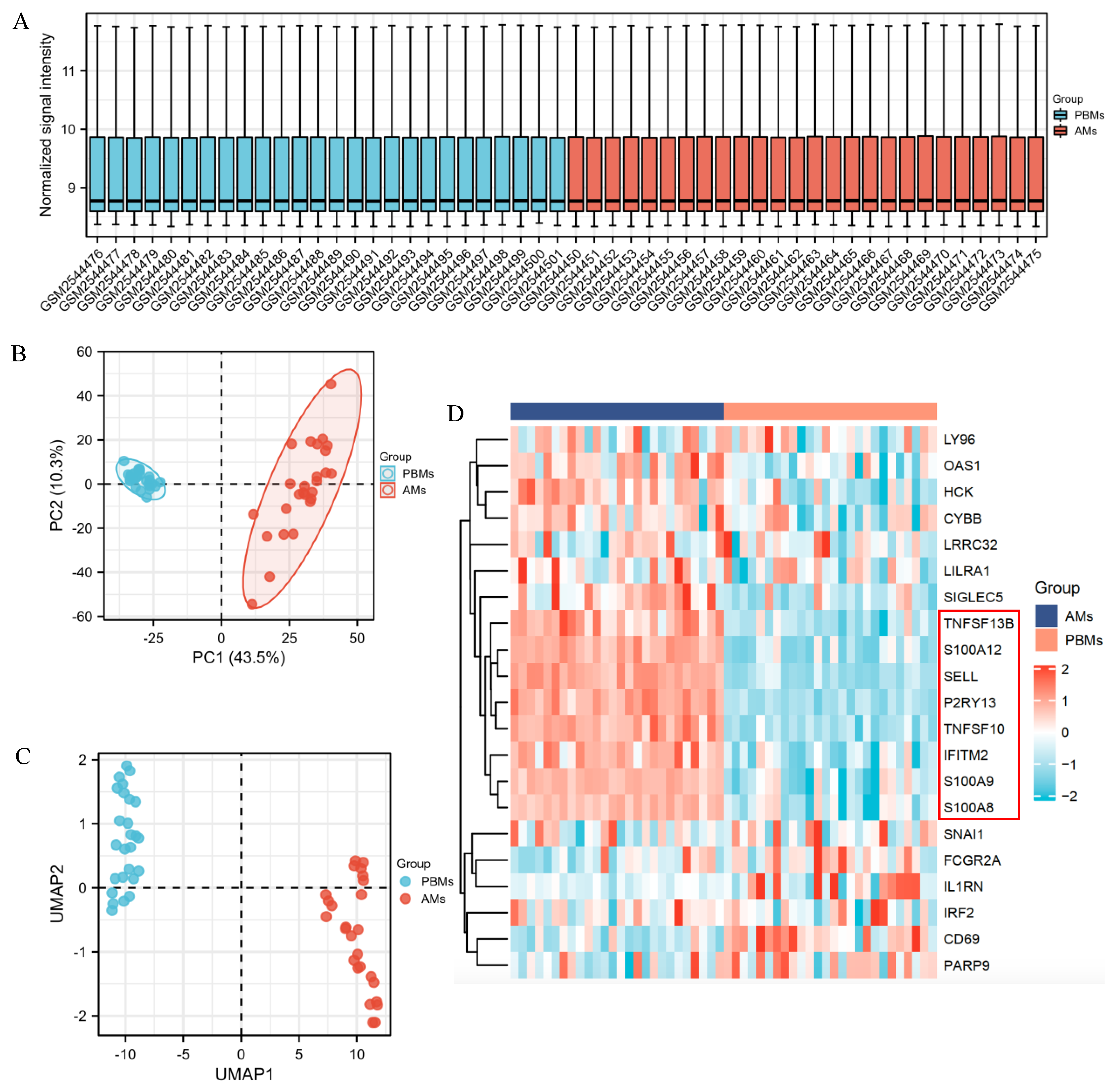
2.1. Transcriptomic Differences between SARS-CoV-2 and Healthy Control
2.2. Identification of DEGs and Screening of Common Genes between COVID-19 Patients with and without Cancer
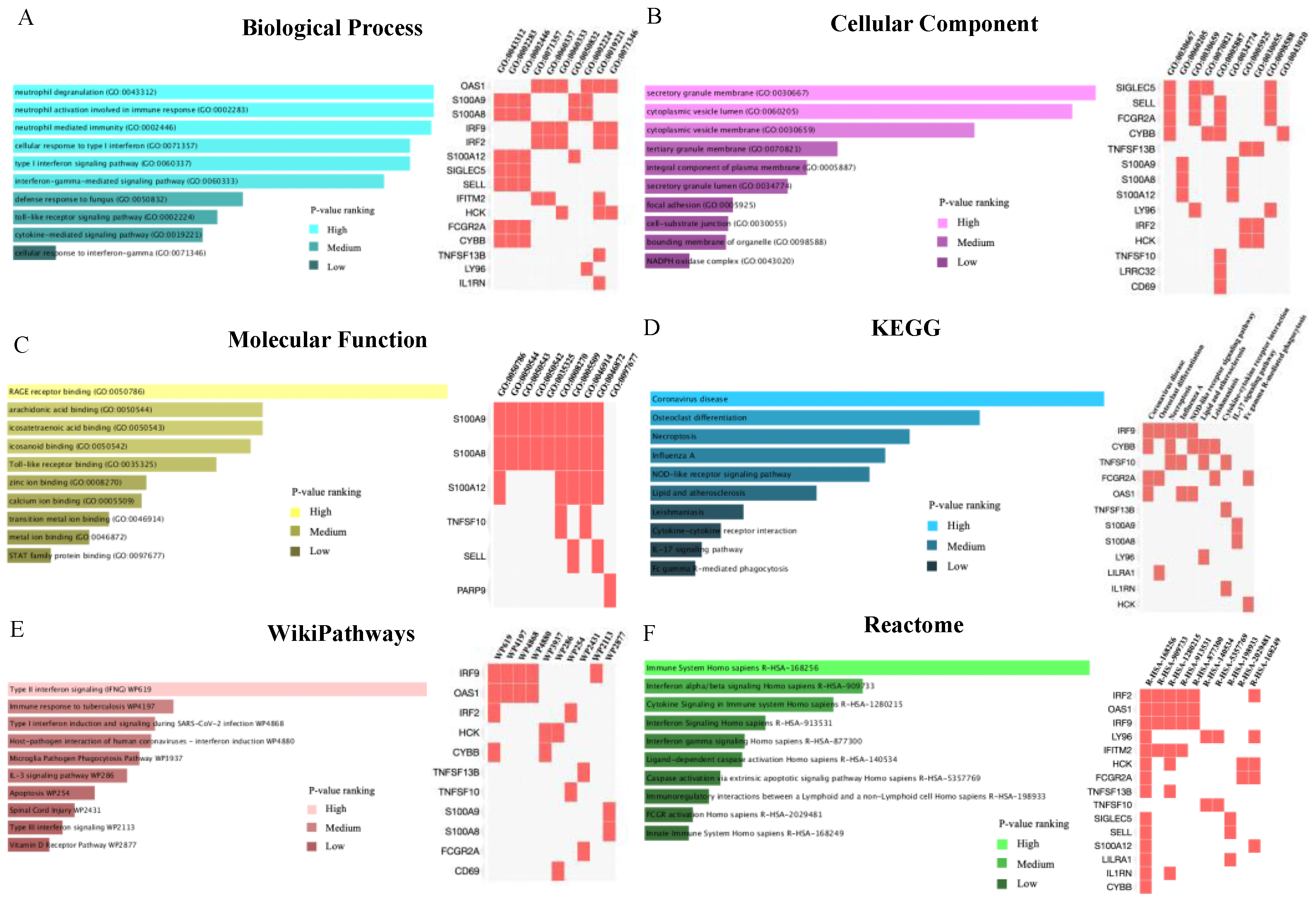
2.3. Common Genes-Based GO and Pathway Analysis
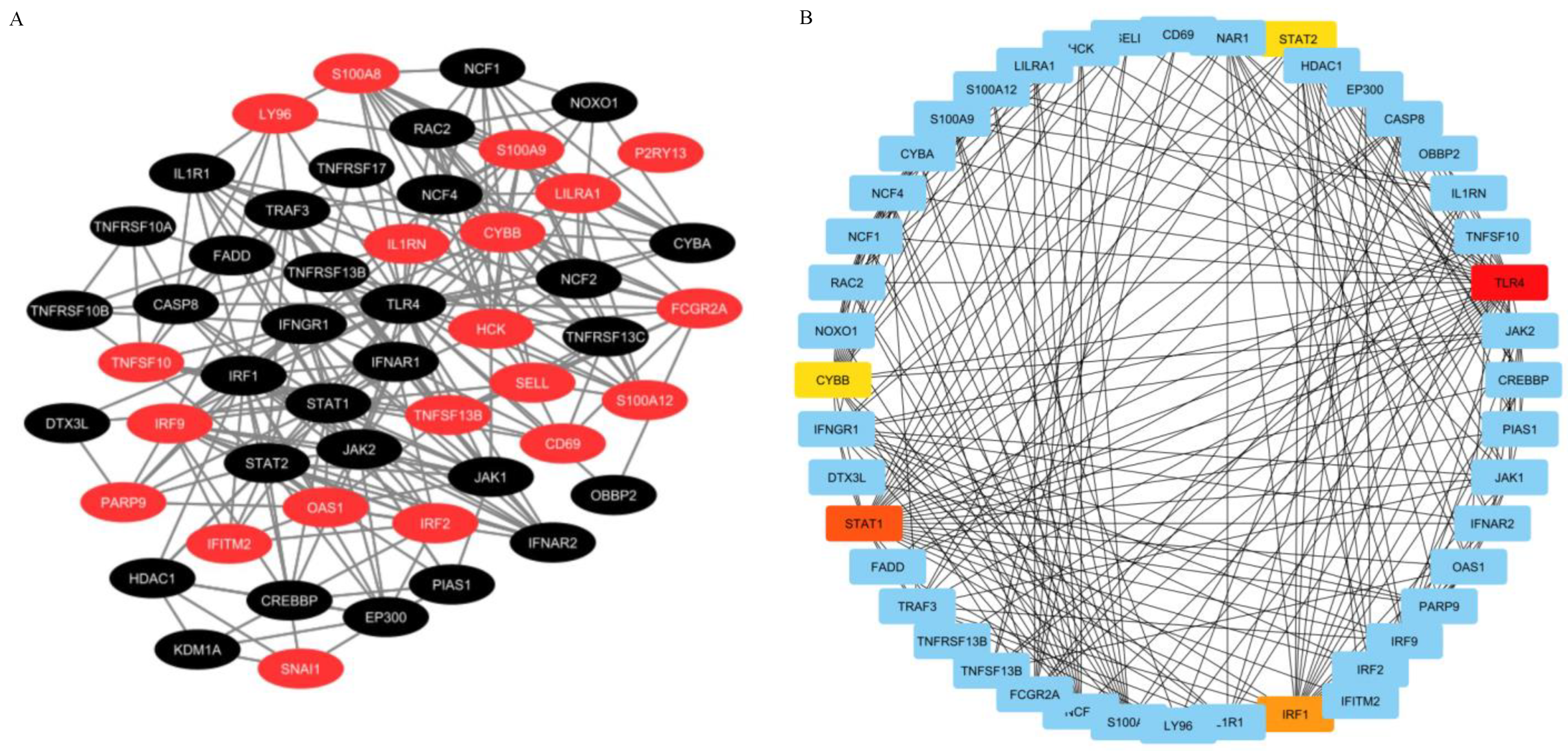
2.4. PPI Network Construction and Hub Genes Extraction
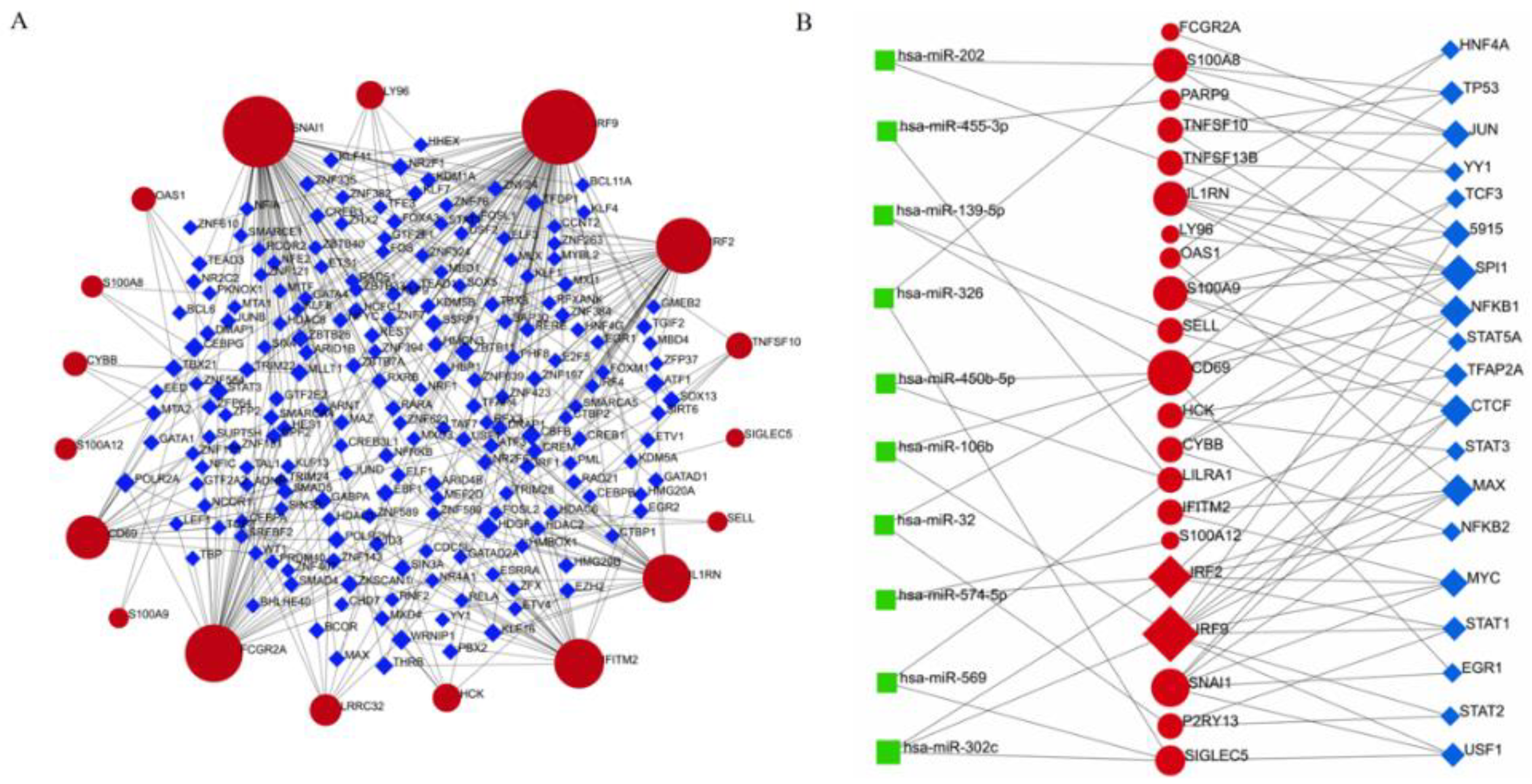
2.5. TF–DEG Interactions and miRNA–DEG–TF Coregulatory Networks
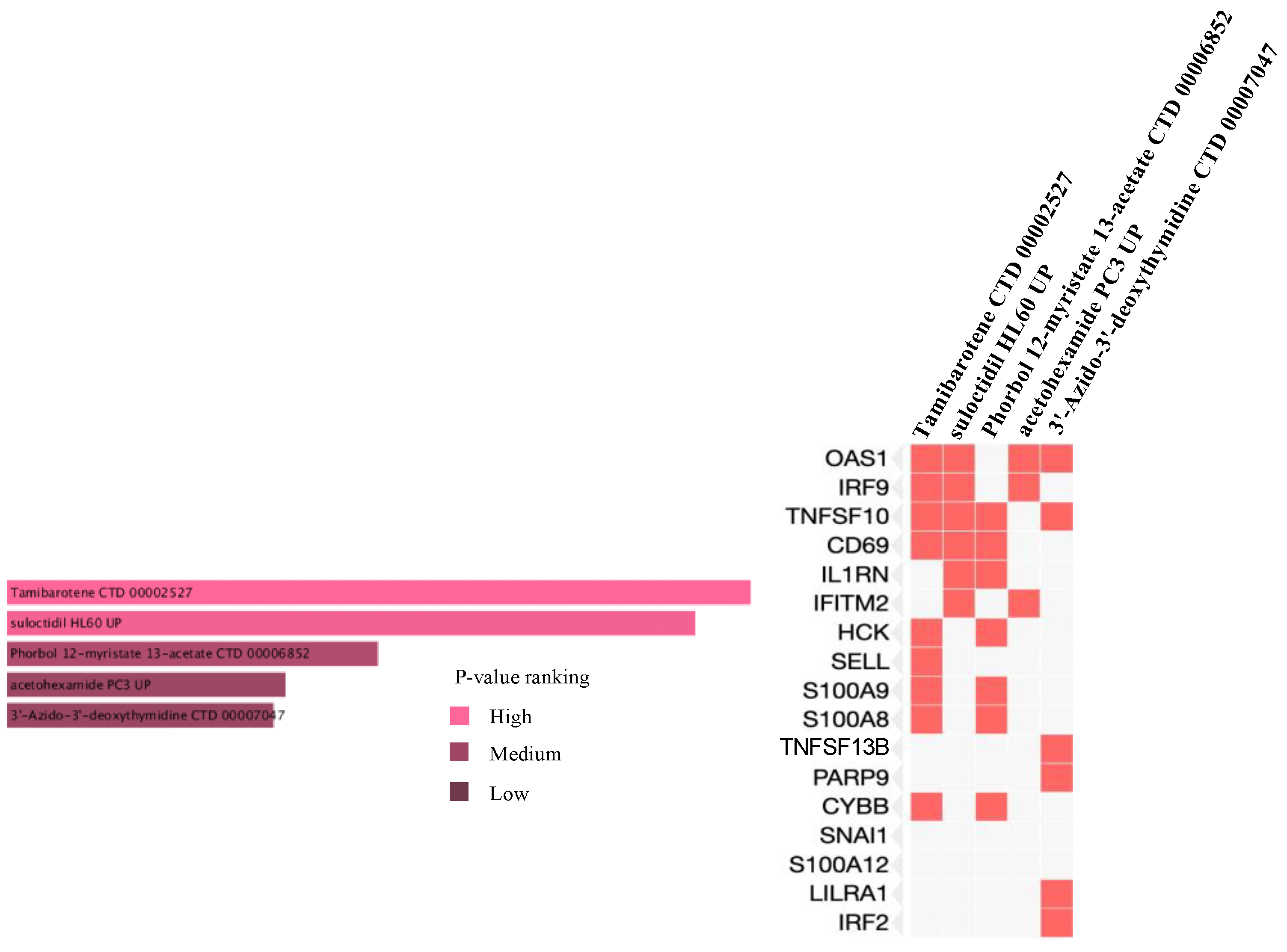
2.6. Prediction of Drug Candidate Molecules
2.7. Identification of DEGs for ARDS Patients
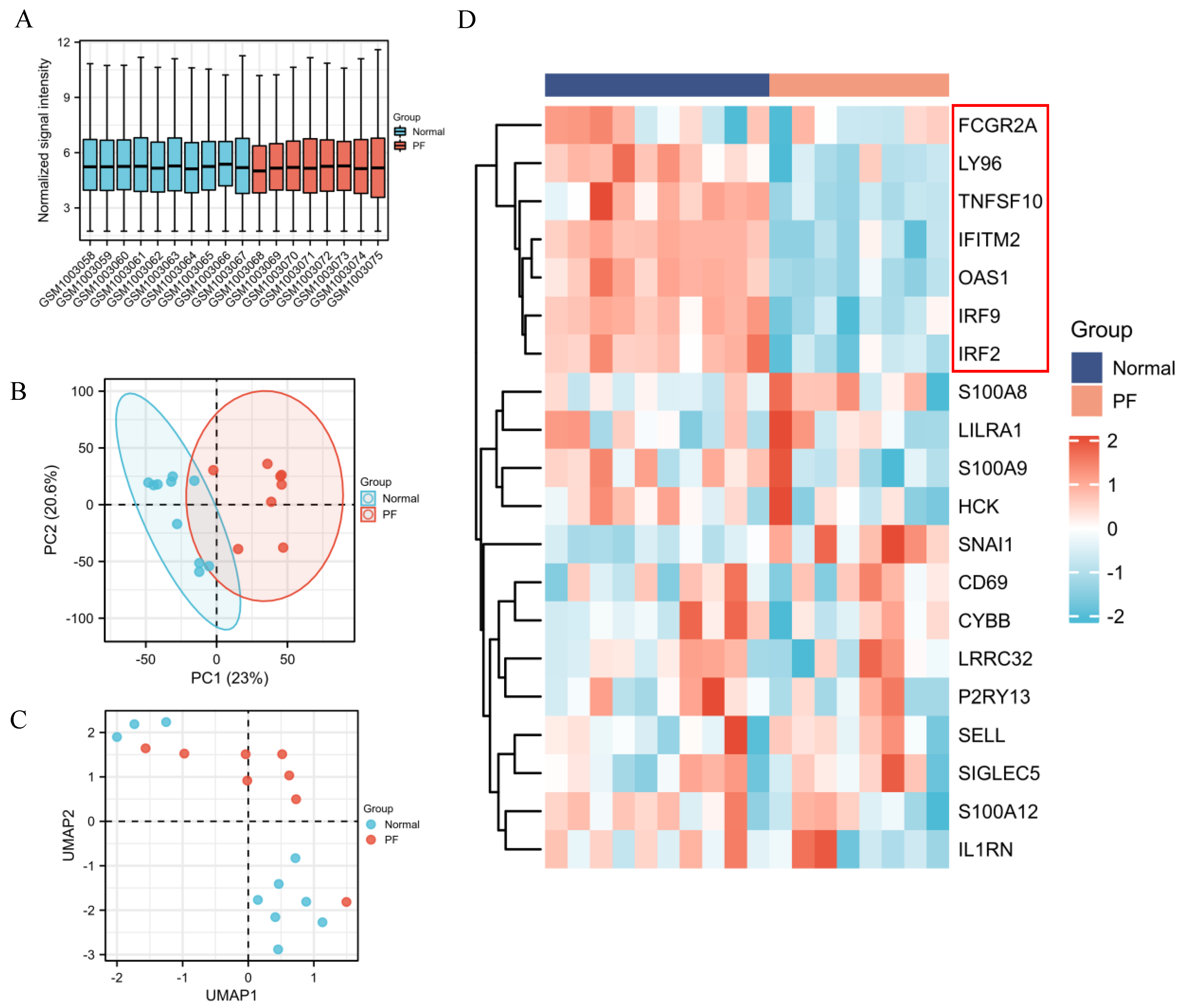
2.8. Identification of DEGs between PF and Healthy Control
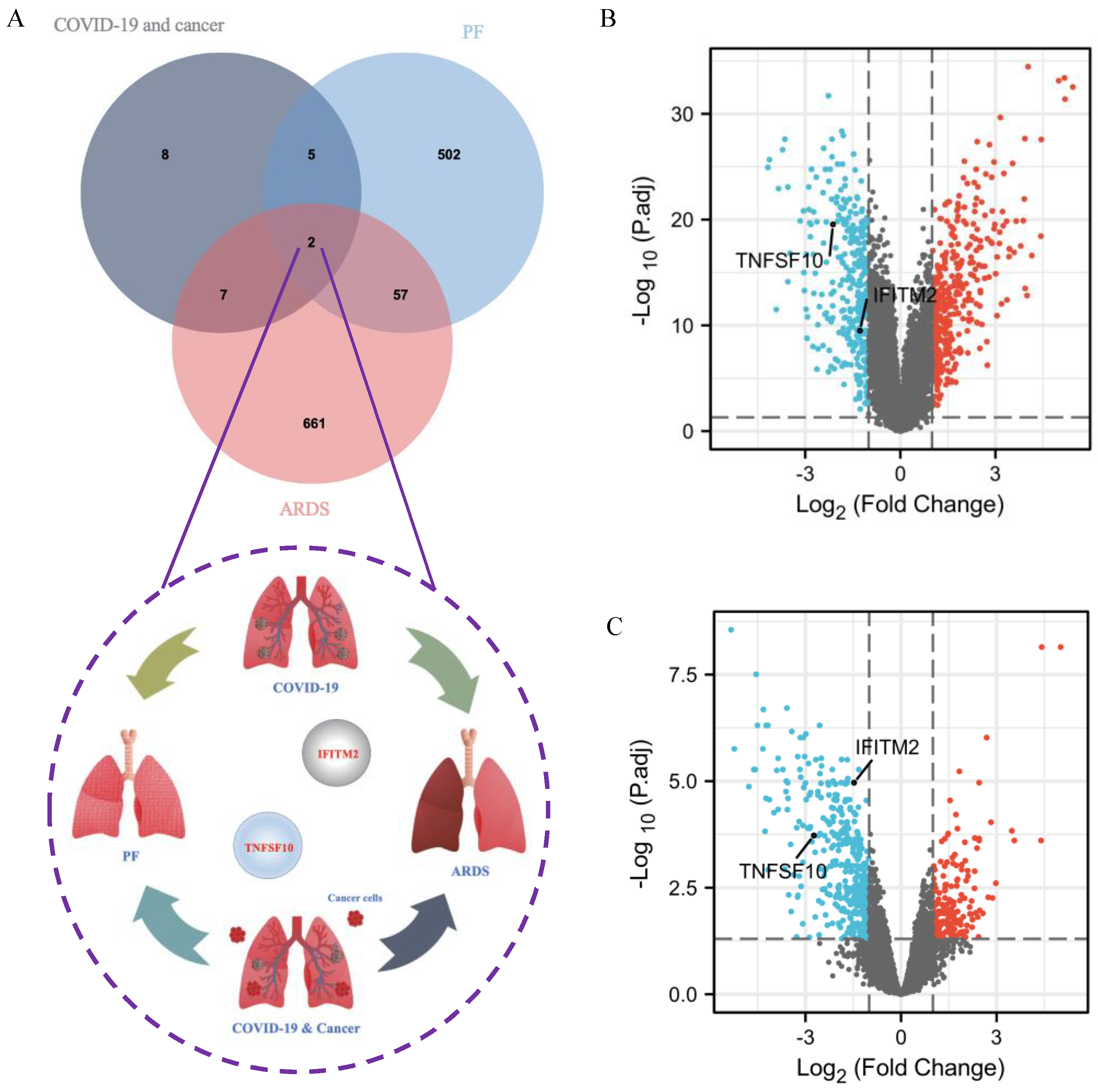
2.9. Identification of Common DEGs among COVID-19 Patients without and with Cancer, ARDS and PF
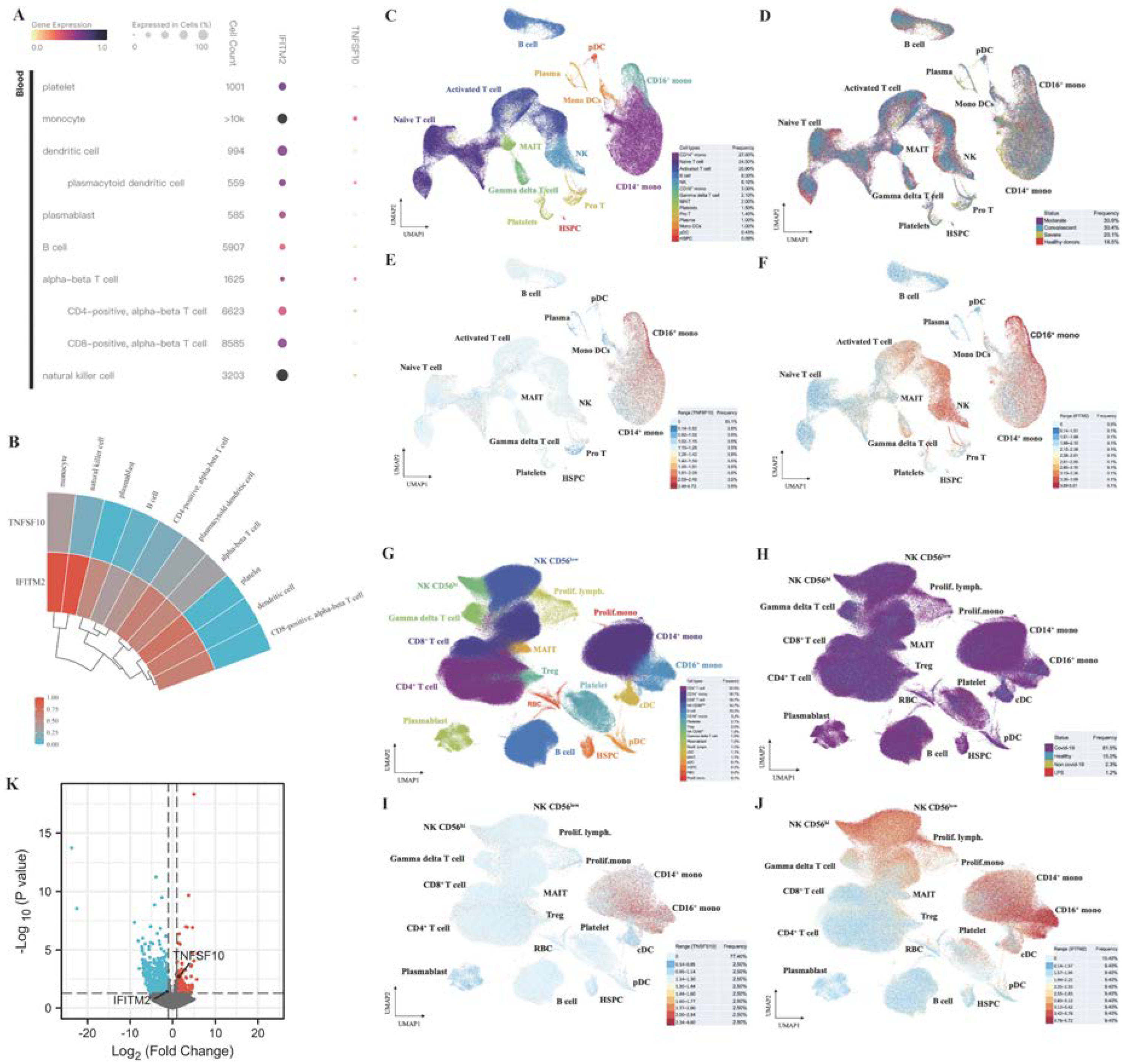
2.10. Single-Cell RNA-Seq Analysis Revealed Immunological Features of TNFSF10 and IFITM2 in COVID-19 Patients

3. Discussion
4. Materials and Methods
4.1. GEO Dataset Used in This Study
4.2. Identification of Common DEGs among COVID-19 without and with Cancer, ARDS and PF
4.3. Acquisition and Classification of Immune-Related Genes
4.4. Gene Ontology (GO) and Pathways Analysis Based on Common DEGs
4.5. Analysis of PPI Network and Hub Genes Identification
4.6. Analysis of TF–DEG Interactions and miRNA–DEG–TF Coregulatory Network
4.7. Prediction of Drug Candidate Molecules
4.8. Visualization of Single-Cell RNA-Seq Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, M.; Liu, D.; Liu, M.; Zhou, F.; Li, G.; Chen, Z.; Zhang, Z.; You, H.; Wu, M.; Zheng, Q.; et al. Patients with Cancer Appear More Vulnerable to SARS-CoV-2: A Multicenter Study during the COVID-19 Outbreak. Cancer Discov. 2020, 10, 783–791. [Google Scholar] [CrossRef]
- Karmouty-Quintana, H.; Thandavarayan, R.A.; Keller, S.P.; Sahay, S.; Pandit, L.M.; Akkanti, B. Emerging Mechanisms of Pulmonary Vasoconstriction in SARS-CoV-2-Induced Acute Respiratory Distress Syndrome (ARDS) and Potential Therapeutic Targets. Int. J. Mol. Sci. 2020, 21, 8081. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; the Northwell COVID-19 Research Consortium; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Vasarmidi, E.; Tsitoura, E.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K.M. Pulmonary fibrosis in the aftermath of the COVID-19 era (Review). Exp. Ther. Med. 2020, 20, 2557–2560. [Google Scholar] [CrossRef]
- Mo, X.; Jian, W.; Su, Z.; Chen, M.; Peng, H.; Peng, P.; Lei, C.; Chen, R.; Zhong, N.; Li, S. Abnormal pulmonary function in COVID-19 patients at time of hospital discharge. Eur. Respir. J. 2020, 55, 2001217. [Google Scholar] [CrossRef]
- Desai, S.R.; Wells, A.U.; Rubens, M.B.; Evans, T.W.; Hansell, D.M. Acute respiratory distress syndrome: CT abnormalities at long-term follow-up. Radiology 1999, 210, 29–35. [Google Scholar] [CrossRef]
- Masclans, J.R.; Roca, O.; Munoz, X.; Pallisa, E.; Torres, F.; Rello, J.; Morell, F. Quality of life, pulmonary function, and tomographic scan abnormalities after ARDS. Chest 2011, 139, 1340–1346. [Google Scholar] [CrossRef]
- Spagnolo, P.; Balestro, E.; Aliberti, S.; Cocconcelli, E.; Biondini, D.; Casa, G.D.; Sverzellati, N.; Maher, T.M. Pulmonary fibrosis secondary to COVID-19: A call to arms? Lancet Respir. Med. 2020, 8, 750–752. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, A.; De Vitis, C.; de Latouliere, L.; di Martino, S.; De Nicola, F.; Goeman, F.; Mottini, C.; Paolini, F.; D’Ascanio, M.; Ricci, A.; et al. Multi-omic approach identifies a transcriptional network coupling innate immune response to proliferation in the blood of COVID-19 cancer patients. Cell Death Dis. 2021, 12, 1019. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.E9. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mi, Y.; Jiang, B.; Chen, X.; Ji, C.; Li, Y.; Zhao, X.; Lai, Y.; Hu, Y.; Tang, A.; et al. Tamibarotene Compared to All-Trans Retinoic Acid (ATRA) As Add-on to Arsenic Trioxide (ATO) in Subjects with Relapsed Acute Promyelocytic Leukemia (APL). Blood 2015, 126, 220. [Google Scholar] [CrossRef]
- Assi, S.; Hajj, H.E.; Hayar, B.; Pisano, C.; Saad, W.; Darwiche, N. Development and Challenges of Synthetic Retinoid Formulations in Cancer. Curr. Drug Deliv. 2022. [Google Scholar] [CrossRef]
- Harker, L.A. Antiplatelet drugs in the management of patients with thrombotic disorders. Semin. Thromb. Hemost. 1986, 12, 134–155. [Google Scholar] [CrossRef]
- Szallasi, Z.; Krsmanovic, L.; Blumberg, P.M. Nonpromoting 12-deoxyphorbol 13-esters inhibit phorbol 12-myristate 13-acetate induced tumor promotion in CD-1 mouse skin. Cancer Res. 1993, 53, 2507–2512. [Google Scholar]
- Siew Bee, N.; Guy, G.R. Two protein kinase C activators, bryostatin-1 and phorbol-12-myristate-13-acetate, have different effects on haemopoietic cell proliferation and differentiation. Cell. Signal. 1992, 4, 405–416. [Google Scholar] [CrossRef]
- Wang, Y.; Biswas, G.; Prabu, S.K.; Avadhani, N.G. Modulation of mitochondrial metabolic function by phorbol 12-myristate 13-acetate through increased mitochondrial translocation of protein kinase Calpha in C2C12 myocytes. Biochem. Pharmacol. 2006, 72, 881–892. [Google Scholar] [CrossRef]
- Boshell, B.R.; Wilensky, A.S.; Barrett, J.C.; Almon, J.V. Acetohexamide: Comparison with other sulfonylurea compounds in the treatment of diabetes mellitus. Clin. Pharmacol. Ther. 1962, 3, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Esnouf, R.M.; Hopkins, A.L.; Jones, E.Y.; Kirby, I.; Keeling, J.; Ross, C.K.; Larder, B.A.; Stuart, D.I.; Stammers, D.K. 3’-Azido-3’-deoxythymidine drug resistance mutations in HIV-1 reverse transcriptase can induce long range conformational changes. Proc. Natl. Acad. Sci. USA 1998, 95, 9518–9523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalski, J.E.; Kurche, J.S.; Schwartz, D.A. From ARDS to pulmonary fibrosis: The next phase of the COVID-19 pandemic? Transl. Res. 2022, 241, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Morrell, E.D.; Radella, F., 2nd; Manicone, A.M.; Mikacenic, C.; Stapleton, R.D.; Gharib, S.A.; Wurfel, M.M. Peripheral and Alveolar Cell Transcriptional Programs Are Distinct in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2018, 197, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Wang, X.M.; Xing, X.; Xu, Z.; Zhang, C.; Song, J.W.; Fan, X.; Xia, P.; Fu, J.L.; Wang, S.Y.; et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020, 21, 1107–1118. [Google Scholar] [CrossRef]
- Stephenson, E.; Reynolds, G.; Botting, R.A.; Calero-Nieto, F.J.; Morgan, M.D.; Tuong, Z.K.; Bach, K.; Sungnak, W.; Worlock, K.B.; Yoshida, M.; et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med. 2021, 27, 904–916. [Google Scholar] [CrossRef]
- You, B.; Ravaud, A.; Canivet, A.; Ganem, G.; Giraud, P.; Guimbaud, R.; Kaluzinski, L.; Krakowski, I.; Mayeur, D.; Grellety, T.; et al. The official French guidelines to protect patients with cancer against SARS-CoV-2 infection. Lancet Oncol. 2020, 21, 619–621. [Google Scholar] [CrossRef] [Green Version]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal. Transduct. Target. Ther. 2020, 5, 84. [Google Scholar] [CrossRef]
- Violi, F.; Oliva, A.; Cangemi, R.; Ceccarelli, G.; Pignatelli, P.; Carnevale, R.; Cammisotto, V.; Lichtner, M.; Alessandri, F.; De Angelis, M.; et al. Nox2 activation in Covid-19. Redox Biol. 2020, 36, 101655. [Google Scholar] [CrossRef]
- Lopez-Martinez, R.; Albaiceta, G.M.; Amado-Rodriguez, L.; Cuesta-Llavona, E.; Gomez, J.; Garcia-Clemente, M.; Vazquez-Coto, D.; Alvarez, V.; Coto, E. The FCGR2A rs1801274 polymorphism was associated with the risk of death among COVID-19 patients. Clin. Immunol. 2022, 236, 108954. [Google Scholar] [CrossRef]
- Bravo Garcia-Morato, M.; Calvo Apalategi, A.; Bravo-Gallego, L.Y.; Blazquez Moreno, A.; Simon-Fuentes, M.; Garmendia, J.V.; Mendez Echevarria, A.; Del Rosal Rabes, T.; Dominguez-Soto, A.; Lopez-Granados, E.; et al. Impaired control of multiple viral infections in a family with complete IRF9 deficiency. J. Allergy Clin. Immunol. 2019, 144, 309–312.e10. [Google Scholar] [CrossRef] [PubMed]
- Wickenhagen, A.; Sugrue, E.; Lytras, S.; Kuchi, S.; Noerenberg, M.; Turnbull, M.L.; Loney, C.; Herder, V.; Allan, J.; Jarmson, I.; et al. A prenylated dsRNA sensor protects against severe COVID-19. Science 2021, 374, eabj3624. [Google Scholar] [CrossRef] [PubMed]
- Chiappalupi, S.; Salvadori, L.; Vukasinovic, A.; Donato, R.; Sorci, G.; Riuzzi, F. Targeting RAGE to prevent SARS-CoV-2-mediated multiple organ failure: Hypotheses and perspectives. Life Sci. 2021, 272, 119251. [Google Scholar] [CrossRef]
- Pandey, N.; Chauhan, A.; Jain, N. TLR4 Polymorphisms and Expression in Solid Cancers. Mol. Diagn. Ther. 2018, 22, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Yuan, S.; Cao, J.; Tang, K.; Qiu, Y.; Seow, H.C.; Man, R.C.H.; Shao, Z.; Huang, Y.; Liang, R.; et al. Inhaled Dry Powder Formulation of Tamibarotene, a Broad-Spectrum Antiviral against Respiratory Viruses Including SARS-CoV-2 and Influenza Virus. Adv. Ther. 2021, 4, 202100059. [Google Scholar] [CrossRef]
- Kanai, F.; Obi, S.; Fujiyama, S.; Shiina, S.; Tamai, H.; Mochizuki, H.; Koike, Y.; Imamura, J.; Yamaguchi, T.; Saida, I.; et al. An open-label phase I/II study of tamibarotene in patients with advanced hepatocellular carcinoma. Hepatol. Int. 2014, 8, 94–103. [Google Scholar] [CrossRef]
- Takeshita, A.; Asou, N.; Atsuta, Y.; Sakura, T.; Ueda, Y.; Sawa, M.; Dobashi, N.; Taniguchi, Y.; Suzuki, R.; Nakagawa, M.; et al. Tamibarotene maintenance improved relapse-free survival of acute promyelocytic leukemia: A final result of prospective, randomized, JALSG-APL204 study. Leukemia 2019, 33, 358–370. [Google Scholar] [CrossRef]
- Winstone, H.; Lista, M.J.; Reid, A.C.; Bouton, C.; Pickering, S.; Galao, R.P.; Kerridge, C.; Doores, K.J.; Swanson, C.M.; Neil, S.J.D. The Polybasic Cleavage Site in SARS-CoV-2 Spike Modulates Viral Sensitivity to Type I Interferon and IFITM2. J. Virol. 2021, 95, e02422-20. [Google Scholar] [CrossRef]
- Xu, L.; Zhou, R.; Yuan, L.; Wang, S.; Li, X.; Ma, H.; Zhou, M.; Pan, C.; Zhang, J.; Huang, N.; et al. IGF1/IGF1R/STAT3 signaling-inducible IFITM2 promotes gastric cancer growth and metastasis. Cancer Lett. 2017, 393, 76–85. [Google Scholar] [CrossRef]
- Yang, N.; Liu, Z.; Pang, S.; Wu, J.; Liang, J.; Sun, L. Predicative value of IFITM2 in renal clear cell carcinoma: IFITM2 is associated with lymphatic metastasis and poor clinical outcome. Biochem. Biophys. Res. Commun. 2021, 534, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.J.; Zhang, J.; Hardeman, A.; Liu, M.; Karginova, O.; Romero, R.; Khramtsova, G.F.; Zheng, Y.; Huo, D.; Olopade, O.I. An Enhancer Variant Associated with Breast Cancer Susceptibility in Black Women Regulates TNFSF10 Expression and Antitumor Immunity in Triple-Negative Breast Cancer. Hum. Mol. Genet. 2022, ddac168. [Google Scholar] [CrossRef] [PubMed]
- Burgess, H.M.; Depledge, D.P.; Thompson, L.; Srinivas, K.P.; Grande, R.C.; Vink, E.I.; Abebe, J.S.; Blackaby, W.P.; Hendrick, A.; Albertella, M.R.; et al. Targeting the m(6)A RNA modification pathway blocks SARS-CoV-2 and HCoV-OC43 replication. Genes Dev. 2021, 35, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Hui, H.; Bray, B.; Gonzalez, G.M.; Zeller, M.; Anderson, K.G.; Knight, R.; Smith, D.; Wang, Y.; Carlin, A.F.; et al. METTL3 regulates viral m6A RNA modification and host cell innate immune responses during SARS-CoV-2 infection. Cell Rep. 2021, 35, 109091. [Google Scholar] [CrossRef]
- Smyth, M.J.; Hayakawa, Y.; Takeda, K.; Yagita, H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat. Rev. Cancer 2002, 2, 850–861. [Google Scholar] [CrossRef]
- Masselli, E.; Vaccarezza, M.; Carubbi, C.; Pozzi, G.; Presta, V.; Mirandola, P.; Vitale, M. NK cells: A double edge sword against SARS-CoV-2. Adv. Biol. Regul. 2020, 77, 100737. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, L. The CD14+ CD16+ blood monocytes: Their role in infection and inflammation. J. Leukoc. Biol. 2007, 81, 584–592. [Google Scholar] [CrossRef]
- Szaflarska, A.; Baj-Krzyworzeka, M.; Siedlar, M.; Weglarczyk, K.; Ruggiero, I.; Hajto, B.; Zembala, M. Antitumor response of CD14+/CD16+ monocyte subpopulation. Exp. Hematol. 2004, 32, 748–755. [Google Scholar] [CrossRef]
- Zhang, D.; Guo, R.; Lei, L.; Liu, H.; Wang, Y.; Wang, Y.; Qian, H.; Dai, T.; Zhang, T.; Lai, Y.; et al. Frontline Science: COVID-19 infection induces readily detectable morphologic and inflammation-related phenotypic changes in peripheral blood monocytes. J. Leukoc. Biol. 2021, 109, 13–22. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudie, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Andorf, S.; Gomes, L.; Dunn, P.; Schaefer, H.; Pontius, J.; Berger, P.; Desborough, V.; Smith, T.; Campbell, J.; et al. ImmPort: Disseminating data to the public for the future of immunology. Immunol. Res. 2014, 58, 234–239. [Google Scholar] [CrossRef]
- Brunson, J. ggalluvial: Layered Grammar for Alluvial Plots. J. Open Source Softw. 2020, 5, 2017. [Google Scholar] [CrossRef]
- Subramanian, A.; Kuehn, H.; Gould, J.; Tamayo, P.; Mesirov, J.P. GSEA-P: A desktop application for Gene Set Enrichment Analysis. Bioinformatics 2007, 23, 3251–3253. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- King, O.D.; Foulger, R.E.; Dwight, S.S.; White, J.V.; Roth, F.P. Predicting gene function from patterns of annotation. Genome Res. 2003, 13, 896–904. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Melius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Sikic, M.; Tomic, S.; Vlahovicek, K. Prediction of protein-protein interaction sites in sequences and 3D structures by random forests. PLoS Comput. Biol. 2009, 5, e1000278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.P.; Wu, C.; Miao, H.; Wu, H. RegNetwork: An integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database 2015, 2015, bav095. [Google Scholar] [CrossRef] [Green Version]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug signatures database for gene set analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef] [Green Version]
- Speir, M.L.; Bhaduri, A.; Markov, N.S.; Moreno, P.; Nowakowski, T.J.; Papatheodorou, I.; Pollen, A.A.; Raney, B.J.; Seninge, L.; Kent, W.J.; et al. UCSC Cell Browser: Visualize Your Single-Cell Data. Bioinformatics 2021, 37, 4578–4580. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Name of Drugs | DrugBank Accession Number [14] | Chemical Formula | Chemical Structure | Background |
|---|---|---|---|---|---|
| 1 | Tamibarotene | DB04942 | C22H25NO3 |  | Tamibarotene is a novel synthetic retinoid for acute promyelocytic leukaemia (APL) [15]. Tamibarotene is currently approved in Japan for treatment of recurrent APL and is undergoing clinical trials in the United States [16]. |
| 2 | Suloctidil | DB13340 | C20H35NOS |  | A peripheral vasodilator that was formerly used in the management of peripheral and cerebral vascular disorders [17]. |
| 3 | Phorbol 12–myristate 13–acetate | / | C36H56O8 |  | It has a role as a protein kinase C agonist, an antineoplastic agent, a reactive oxygen species generator, a plant metabolite, a mitogen, a carcinogenic agent and an apoptosis inducer [18,19,20]. |
| 4 | Acetohexamide | DB00414 | C15H20N2O4S |  | A sulfonylurea hypoglycemic agent that is metabolized in the liver to 1-hydrohexamide [21]. |
| 5 | 3′-Azido–3′-deoxythymidine | DB00495 | C10H13N5O4 |  | A dideoxynucleoside compound in which the 3’-hydroxy group on the sugar moiety has been replaced by an azido group. This modification prevents the formation of phosphodiester linkages which are needed for the completion of nucleic acid chains. The compound is a potent inhibitor of HIV replication, acting as a chain-terminator of viral DNA during reverse transcription. It improves immunologic function, partially reverses the HIV-induced neurological dysfunction and improves certain other clinical abnormalities associated with AIDS. Its principal toxic effect is dose-dependent suppression of bone marrow, resulting in anemia and leukopenia [22]. |
| Group | Serial Number | Sex | Age | Neoplastic Disease | Degree of Severity | Sample Source | Anticancer Treatment | Days between COVID-19 First Positive Swap and Blood Collection |
|---|---|---|---|---|---|---|---|---|
| Healthy Donors | HD Y | F | 54 | N.A. | N.A. | PBMCs | N.A. | N.A. |
| HD 7 | M | 51 | N.A. | N.A. | PBMCs | N.A. | N.A. | |
| COVID-19 patients without cancer | Sand-003 | M | 60 | N.A. | Critical | PBMCs | N.A. | 37 |
| Sand-004 | F | 69 | N.A. | Critical | PBMCs | N.A. | 57 | |
| Sand-007 | F | 88 | N.A. | Moderate | PBMCs | N.A. | 37 | |
| Sand-010 | M | 65 | N.A. | Mild | PBMCs | N.A. | 2 | |
| Sand-100 | M | 68 | N.A. | Severe | PBMCs | N.A. | 39 | |
| COVID-19 patients with cancer | Sand-005 | M | 69 | Clear cell renal cell carcinoma (CCRCC) | Severe | PBMCs | No treatment (neo-diagnosis) | 37 |
| Sand-006 | M | 74 | Chronic Lymphatic Leukemia (CLL) | Critical | PBMCs | No treatment (neo-diagnosis) | 42 | |
| Sand-008 | M | 70 | Lung cancer | Severe | PBMCs | No treatment | 24 | |
| Sand-009 | F | 74 | Gastrointestinal Cancer | Mild | PBMCs | No treatment (neo-diagnosis) | 2 | |
| Sand-011 | M | 69 | Classical mixed cellularity Hodgkin Lymphoma | Severe | PBMCs | No treatment (neo-diagnosis) | 55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Qin, Y.; Fu, Y.; Gao, Z.; Deng, Y. Integrated Analysis of Bulk RNA-Seq and Single-Cell RNA-Seq Unravels the Influences of SARS-CoV-2 Infections to Cancer Patients. Int. J. Mol. Sci. 2022, 23, 15698. https://doi.org/10.3390/ijms232415698
Chen Y, Qin Y, Fu Y, Gao Z, Deng Y. Integrated Analysis of Bulk RNA-Seq and Single-Cell RNA-Seq Unravels the Influences of SARS-CoV-2 Infections to Cancer Patients. International Journal of Molecular Sciences. 2022; 23(24):15698. https://doi.org/10.3390/ijms232415698
Chicago/Turabian StyleChen, Yu, Yujia Qin, Yuanyuan Fu, Zitong Gao, and Youping Deng. 2022. "Integrated Analysis of Bulk RNA-Seq and Single-Cell RNA-Seq Unravels the Influences of SARS-CoV-2 Infections to Cancer Patients" International Journal of Molecular Sciences 23, no. 24: 15698. https://doi.org/10.3390/ijms232415698